
Abstract
The MET tyrosine kinase receptor is a high-affinity receptor for hepatocyte growth factor/scatter factor (HGF/SF). HGF/SF-MET system is necessary for embryonic development, and aberrant MET signalling favours tumorigenesis and metastasis. MET is a prototype of tyrosine kinase receptor, which is able to counteract apoptosis through the initiation of a survival signal involving notably the PI3K–Akt pathway. Paradoxically, the MET receptor is also able to promote apoptosis when activated by HGF/SF or independently of ligand stimulation. The molecular mechanisms underlying this uncommon response have been recently investigated and revealed dual antiapoptotic or proapoptotic property of MET according to the cell type or stress conditions. Although the involvement of MET in the regulation of integrated biological responses mostly took into account its efficient antiapoptotic function, its proapoptotic responses could also be important for regulation of the survival/apoptosis balance and play a role during the development or tumour progression.
Similar content being viewed by others


Main
During development, survival/apoptosis balance plays a major role in morphogenesis and tissue sculpting. In a mature organism, this balance is essential for tissue homoeostasis. Conversely, resistance to apoptosis, leading to more efficient survival, is associated with tumour initiation and progression. Tyrosine kinase receptors are major actors involved in the regulation of this balance. Indeed, in response to growth factors, they initiate intracellular signals that positively regulate cell survival in concert with multiple biological responses such as differentiation, proliferation or motility.
The MET tyrosine kinase receptor and its cognate ligand hepatocyte growth factor/scatter factor (HGF/SF) are known to induce the efficient survival of many cell types submitted to various apoptotic inducers, including serum starvation, death receptor activation or genotoxic treatment (Table 1). Interestingly, it has also been evidenced that in response to HGF/SF, or more recently independently of its ligand, the MET receptor is remarkably able to induce cell death of various cell lines (Table 1). These opposite observations cloud over the portrait of the MET receptor as the HGF/SF-MET signalling cannot be restricted to a survival response. This dual antiapoptotic and proapoptotic property of the MET tyrosine kinase receptor is unconventional and could be involved in the differential regulation of the survival/apoptosis balance according to the cellular context.
Multiple Biological Responses Triggered by HGF/SF-MET
The MET receptor was originally identified as an oncogene, through the fusion of its intracellular kinase domain to the dimerization domain of TPR.1, 2 Both the kinase activity of MET and the dimerization domain of TPR are necessary to acquire a transforming activity. MET is a tyrosine kinase receptor that consists of an exclusively extracellular α-chain and a β-chain shared between extracellular and intracellular compartments. The intracellular region of the β-chain contains a juxtamembrane, a catalytic tyrosine kinase and a C-terminal domain.3
HGF and SF, independently discovered through their respective properties to induce the growth of hepatocyte cell cultures and scattering of epithelial cells, are indeed the same factor.4, 5, 6, 7 HGF/SF is a disulphide-linked α- and β-chain heterodimer with homology to the proteinases of the plasminogen family. It mediates multiple biological responses by activation of its cognate tyrosine kinase receptor MET.
The biological functions of MET and HGF/SF have been specially evaluated in genetically modified mice. Targeted disruption of either the hgf/sf or the met locus leads to middle-stage embryonic lethality, demonstrating their essential role in embryogenesis. These mutant mice display notable defects in placental development, severe size reduction of the liver, absence of limb muscle and defects in limb innervations by motor and sensory neurons.8, 9, 10, 11 These phenotypes corroborate with the pattern of HGF/SF and MET expression during development.
Strong evidence from genetically modified mice depicts MET as a survival receptor. Indeed, strong size reduction of the liver observed in HGF/SF- and MET-deficient mice results from hepatocyte apoptosis.8, 9, 10 In addition, although disruption of the MET receptor in adult liver using Cre/loxP-mediated gene targeting was not detrimental to hepatocyte functions under physiological conditions, their adaptive responses to injury were dramatically affected. Indeed, hepatocytes derived from MET-deficient mice were hypersensitive to Fas-induced apoptosis12, and regeneration of the liver was impaired after partial hepatectomy.13 Inversely, transgenic expression in the liver of constitutively active MET, which consists of intracellular kinase domain of the receptor, renders hepatocytes resistant to Fas-induced apoptosis.14 This demonstrates that HGF/SF signalling is crucial for hepatocyte survival both during embryonic development and in adult.
Upon HGF/SF binding, the MET receptor is dimerized and its tyrosine kinase activity stimulated by autophosphorylation of the receptor.15 Two phosphotyrosine residues located in the non catalytic C-terminal tail of the receptor have been identified as multifunctional docking site able to interact with several cytoplasmic signal transducers.16, 17 Genetic analyses confirmed the crucial involvement of this multifunctional binding site as knock-in mice expressing the MET receptor, in which these two tyrosine residues have been mutated, recapitulate the phenotype observed in the knockout mutants.18 In addition, engineering of knock-in mice, expressing various mutated MET receptors able to recruit a restricted panel of signalling proteins, displays specific defects, demonstrating that the recruitment of specific signalling molecules is essential for proper biological responses during development.19 The biological responses induced by the couple HGF/SF-MET were also evaluated on cell cultures. Mostly, these responses corroborate with the phenotype observed in genetically modified mice. Indeed, the ligand-activated MET stimulates proliferation, scattering, invasion, morphogenesis and survival of epithelial cells, acts as an angiogenic factor and displays chemoattractant and neurotrophic activities.
Survival Signalling Induced by Ligand-Activated MET
Many studies searched to uncover which signalling is involved in the survival response triggered by HGF/SF-MET. Consistent with results obtained with other tyrosine kinase receptors, PI3K–Akt signalling plays a central role in the antiapoptotic responses induced by activated MET in numerous cell types and in response to various apoptotic inducers.20, 21, 22, 23, 24, 25, 26, 27
The MET receptor is able to recruit PI3K directly through the two phosphorylated tyrosine residues of the C-terminal multisubstrate docking site16 or indirectly through GAB1.28 The GAB1 adaptor is also recruited by the C-terminal docking site and, as a consequence, is able to recruit signalling proteins through phosphorylated tyrosine residues. The PI3K pathway is involved in several biological responses mediated by activated MET, including morphogenesis and survival. Interestingly, it has been shown that GAB1 regulates orientation of PI3K to these different responses. In particular, the overexpression of GAB1 inhibits survival in response to HGF/SF concomitantly to inhibition of sustained activation of Akt,28 whereas it promotes morphogenesis induced by HGF/SF-MET.29, 30 This suggests that survival response triggered by HGF/SF is favoured by the direct binding of PI3K to MET, which leads to sustained activation of Akt, whereas its recruitment to GAB1 orients the response to morphogenesis. Furthermore, the involvement of the RAS–ERK signalling pathway in HGF/SF-dependent survival has also been evidenced in several cell types. However, in most cases, the PI3K–Akt signalling is also implicated, suggesting a cooperation of both pathways to promote survival.31, 32, 33
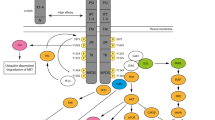
The apoptotic process is notably regulated by proteins of the Bcl family, which are involved in cytochrome c release from mitochondria, and formation of the apoptosome, leading to activation of caspase-9. In many studies, survival responses induced by HGF/SF were correlated with an increased expression of the antiapoptotic Bcl-xL and Bcl-2 proteins, which inhibit mitochondrial-dependent apoptosis.34, 35, 36, 37, 38 Moreover, HGF/SF induced gene expression of the antiapoptotic factor TRAF-2 and cIAP, which are then involved in the HGF/SF-dependent survival response. This regulation involves the transcription factor NF-κB, and the initial activation of PI3K–Akt signalling.39 Furthermore, in response to HGF/SF, the serine–threonine kinase Akt activated downstream of PI3K phosphorylates the proapoptotic protein BAD, leading to its inactivation and thereby preventing apoptosis.40 Interestingly, the HGF/SF survival response mediated by Bcl-xL and Bcl-2 expression requires several hours of stimulation before stress induction to allow gene expression. In contrast, survival mediated through BAD phosphorylation is a more dynamic survival process as it does not require pre-stimulation by HGF/SF40 (Figure 1).

Survival signalling downstream of MET. The survival response triggered by hepatocyte growth factor/scatter factor (HGF/SF) involves activation of the PI3K-Akt pathway initiated by direct recruitment of PI3K to C-terminal docking site of MET, while its indirect recruitment through GAB1 does not lead to cell survival. Akt is notably able to inactivate by phosphorylation the Bcl-2 family BAD protein, which inhibits mitochondrial-dependent apoptosis. RAS-ERK and NF-κB signalling pathways initiated by MET positively regulate transcription of Bcl-2 and Bcl-xL involved in inhibition of mitochondrial apoptosis
It has recently been shown that survival response induced by PI3K–Akt pathway downstream of MET also acts through regulation of p53 activity. Indeed, in primary embryonic hepatocytes and during mouse liver development, HGF/SF regulates transcription of p53 inhibitor MDM2 resulting in cell survival.41
The Dark Side of HGF/SF: Mechanisms of HGF-Induced Apoptosis
Despite the survival role of HGF/SF largely depicted previously, it has been established that it can mediate cytotoxicity and induce cell death of various cell lines. Indeed, HGF/SF was first described as a cytotoxic factor, through its antiproliferative property in different tumour cell lines, with the discovery of a fibroblast-derived tumour cytotoxic factor (F-TCF) identified as the human HGF42, 43, 44, 45 (Table 1). These results were confirmed in other carcinoma cells where HGF/SF can inhibit tumour cell growth both in vitro and in vivo.73 The antiproliferative effect of HGF/SF on transformed cells was explained as a combination between an apoptotic and a cytostatic mechanism.66 Proapoptotic property of HGF/SF is also associated to cell state as HGF/SF was shown to induce death of rat ovarian surface epithelial cells when extracellular matrix or intercellular contacts were disrupted.74 Cell death induced by HGF/SF seems to be related to apoptosis as HGF/SF induced caspase-3 activation in Sarcoma-180 cells and induced apoptosis through extrinsic pathway involving caspase-8, cleavage of BID and cytochrome c release in HIF-1 deficient hepatocarcinoma cell lines.62, 67 Protein kinase C, JNK1 induction, reactive oxygen species production or matrix metalloproteinase induction have also been involved in the apoptotic effect of HGF/SF in other cell lines.60, 63, 64, 75
Interestingly, the proapoptotic property of HGF/SF has positive consequences during liver or lung regeneration after injuries.60, 61 Liver and pulmonary fibrosis are characterized by a loss of liver and lung epithelial cells, replaced by interstitial myofibroblasts with deposit of extracellular matrix proteins. It has been shown that HGF/SF inhibited proliferation and induced apoptotic cell death of liver and pulmonary myofibroblasts, concomitantly with metalloprotease induction and extracellular matrix disappearance, which may be involved in a process of reducing fibrosis. In this model, the proapoptotic and antiapoptotic properties of HGF/SF coexist and could cooperate. Indeed, both the antiapoptotic effect of HGF/SF on hepatocytes and the proapoptotic effect of HGF/SF on liver myofibroblasts are required for a complete resolution of fibrosis in a cirrhotic liver.76, 77
Functional Interaction of Fas and MET: an Explanation of Dual Effect on Cell Survival and Apoptosis
The upstream signalling leading to activation of apoptosis through ligand-activated MET is poorly understood. However, it has been shown that MET interacts with the death receptor Fas, which contributes to the regulation of survival/apoptosis balance.68 Indeed, the extracellular domain of MET associates with Fas, which prevents Fas interaction to its ligand or Fas self-aggregation. The functional consequence of this interaction is an increase of the resistance to Fas-mediated cell death. In contrast, strong HGF/SF stimulation was found to liberate Fas from its interaction to MET, which sensitizes cells to FasL-mediated apoptosis. The authors proposed that the proapoptotic response to HGF/SF on some cell lines might be a consequence of Fas/MET dissociation depending on HGF concentration and FasL expression (Figure 2). The biological relevance was tested with transgenic mice expressing the extracellular domain of MET in the liver. These mice displayed resistance to Fas-dependent hepatic apoptosis and liver failure, consequently of Fas sequestration by the MET extracellular domain. Similar MET–Fas interaction was found in endothelial HUVEC cells, which could provide resistance to apoptosis.78 Therefore, these studies established a novel paradigm through which growth factor receptors may operate to protect cells from death receptors.

Fas and MET interaction for cell survival. Association between MET and the death receptor Fas prevents Fas interaction to its ligand and Fas self-aggregation, promoting increase of the resistance to Fas-mediated cell death. hepatocyte growth factor/scatter factor (HGF/SF) stimulation impairs MET/Fas association, which sensitize cell to Fas-dependent apoptosis
Proteolytic Remodelling of MET by Caspases
Ligand-activated MET influences the fate of the cell through activation of proapoptotic or antiapoptotic signalling. Conversely, the apoptotic process also influences the fate of MET in the absence of ligand as the receptor is a functional target of activated caspases. Following stress induction, the receptor is first cleaved at the aspartic acid 1374 (mouse sequence) within a consensual DNID caspase site located in the extreme C-terminal tail. This initial cleavage removes only five amino acids of MET but favours a second cleavage that occurs at the aspartic acid 1000 within an ESVD motif located in the juxtamembrane domain. These sequential cleavages generate a 40-kDa fragment named p40 MET.79, 80 This fragment contains the catalytic tyrosine kinase domain of MET and is not further anchored to the membrane. p40 MET is generated under apoptotic conditions in various MET-expressing cell lines and primary embryonic hepatocytes and cortical neurons. Importantly, these cleavages do not inactivate the receptor but convert it to a proapoptotic factor as ectopic expression of p40 MET induced cell death concomitantly with activation of caspase-3.79 In addition, the mutation of the C-terminal caspase site, which inhibits generation of the p40 MET proapoptotic fragment, prevents cell death induced by mild stress conditions.80 Therefore, caspase-dependent cleavage of MET converts the survival receptor to a proapoptotic molecule that is involved in apoptosis amplification (Figure 3).

MET is a functional target of caspases. In absence of ligand and upon stress induction, MET is sequentially cleaved by caspases. The first cleavage occurs at the aspartic acid 1374 of the C-terminal tail, leading to generation of a shortened receptor. This cleavage favours a second one at the aspartic acid 1000 of the juxtamembrane region, leading to the generation of a 40 kDa fragment named p40 MET. This fragment displays proapoptotic activity involved in cell death amplification
The tyrosine kinase activity of p40 MET is essential for its proapoptotic biological property. Although this observation is surprising, MET activity is not restricted to survival response as ligand-activated receptor is also able to induce apoptosis. This suggests that the catalytic activity of MET is oriented differently to survival or cell death not only according to the cell type but also according to caspase-dependent cleavage.
Although many caspase substrates are cleaved at several sites,81 the hierarchical organization of these cleavages is poorly defined. The RasGAP protein is cleaved at several sites by caspase-3, with the first cleavage occurring early during the apoptotic process and thus favouring the second one.82, 83 The first cleavage allows the generation of an antiapoptotic fragment, whereas the second cleavage converts it to smaller proapoptotic fragments, supporting the model in which RasGAP functions as a sensor of caspase activity to determine whether a cell will survive or not.84 In the case of MET, its sequential cleavages lead to the generation of an initial shortened MET molecule, cleaved only in its C-terminal region. Although, following this first cleavage, the juxtamembrane cleavage leads to the generation of p40 MET, the shortened MET receptor becomes the predominantly expressed form of MET at the membrane during apoptosis.80 This shortened MET receptor could acquire specific functions that could also be involved in the regulation of the survival/apoptosis balance mediated by the MET tyrosine kinase receptor.
Is MET on the Way to Dependence?
The proapoptotic function mediated by the MET receptor in the absence of its ligand, which contrasts with its antiapoptotic function in response to HGF/SF, is the hallmark of the emerging family of dependence receptors. Indeed, while a receptor is considered as inactive until bound by its ligand, it has been shown that some of them are able to induce cell death in the absence of the ligand. Therefore, the expression of these receptors leads to a dependence towards their ligand for cell survival (see Bredesen et al85). This mechanism has been well described, for instance, for DCC, UNC5H, Patched or p75NTR. This mechanism allows a better understanding of biological processes, in which a survival soluble factor and its transmembrane receptor are involved in the regulation of survival/apoptosis balance.
It can be speculated that, during development, a state of dependence created by MET towards its ligand will occur with physiological consequences. For instance, HGF/SF and MET are involved in innervation of the limb muscles, with MET being expressed by spinal motor neurons and HGF/SF in the limb bud. It has been demonstrated that the complementary expression of the receptor and its ligand is involved in the guidance of the motor neurons from the neural tube to the limb bud.11, 86 In this case, the proapoptotic property of MET could create a state of dependence on HGF/SF involved in the correct innervations of the limb. The future challenge will be to unravel the physiological relevance of the proapoptotic responses induced by the caspase-dependent fragment of MET, especially during development.
Several tyrosine kinase receptors, including RET, ALK, EGFR and ErbB2, are also cleaved by caspases during the apoptotic process, and for most of them the generated fragments are able to induce apoptosis.87, 88, 89, 90 In addition, it has been demonstrated that full-length RET and ALK receptors display proapoptotic properties in the absence of their ligand and upon caspase processing. These receptors are then considered as potential dependence receptors that could also be involved in the regulation of apoptosis in a physiological context.
MET, Apoptosis and Cancer
In numerous tumours, HGF/SF and/or MET are overexpressed, thereby leading to constitutive activation of MET signalling.91 Moreover, the activating mutations of MET have been identified in a hereditary form of papillary renal carcinoma, which directly implicates MET in human cancer.92 Transgenic mice expressing HGF/SF or MET, or knock-in mice expressing MET receptor carrying activating mutations observed in human cancer, developed various types of tumours.93, 94, 95 Indeed, inappropriate MET signalling can induce proliferation, invasion and angiogenesis, the so-called invasive growth that contributes to malignant growth.96 In addition to invasive growth, the tumour progression depends on impaired programmed cell death. In this line, the survival responses induced by the activated receptor are engaged in the MET-dependent transformation. Indeed, cells expressing the activated mutant forms of MET found in human cancer are more resistant to apoptosis.97 Conversely, in MET-dependent transformed cells, the silencing of the receptor induced apoptotic cell death and reduction of tumour growth.98, 99
The mechanism of dependence through caspase cleavages can have important consequences on tumour progression. For instance, expression of netrin-1, the ligand of the dependence receptor DCC, in the digestive tract of mice favours development of cancer in this tissue. In this case, it is possible that the DCC receptor, incorrectly activated by its ligand, does not promote apoptosis, thereby creating a favourable situation for malignant transformation. Similarly, caspase cleavages of MET in the absence of ligand, could favour apoptosis and prevent tumour formation. Inversely, the loss of the proapoptotic property of MET, for instance following aberrant expression of the ligand could promote tumorigenesis. In this hypothesis, HGF/SF-MET-dependent tumour formation would not be exclusively dependent on positive responses such as invasive growth or survival but could also be a consequence of loss of negative responses such as apoptosis.
Potential therapeutic strategies aiming at inhibiting aberrant MET signalling are extensively explored. For instance, ligand/receptor binding can be inhibited by subregions of HGF/SF or MET, acting as antagonists or decoys. Anti-MET or anti-HGF neutralizing antibodies can prevent MET stimulation by its ligand. Furthermore, MET activity can be prevented by small molecule inhibitors or dominant negative forms of MET, which target the catalytic activity of the receptor. MET can also be inhibited at the level of its expression using specific siRNA or ribozyme (see Zhang et al100). The goal of all these strategies is to target MET signalling, which is in most cell types involved in invasive growth and antiapoptotic responses. However, in some transformed cell lines, ligand-activated MET induced apoptosis, suggesting that in these cases activation of MET signalling would prevent malignant transformation. Interestingly, it has been shown that HGF/SF increased ovarian carcinoma cell sensitivity to apoptosis and tumour regression in mice, induced by paclitaxel and cisplatin, the two front-line anticancer agents used in ovarian cancer therapy.71, 72 HGF/SF was then proposed to be used to improve response to chemotherapy in a set of human ovarian carcinomas expressing MET. Therefore, according to proapoptotic or antiapoptotic responses triggered by ligand-activated MET in targeted tumours, adaptive strategies could be considered: either inhibition of MET signalling when invasive growth and survival are induced, or inversely activation of MET signalling by HGF/SF when apoptotic responses are induced.
Abbreviations
- ALK:
-
anaplastic lymphoma kinase
- BAD:
-
Bcl-2 antagonist of cell death
- BID:
-
BH3-interacting domain death agonist
- Cre:
-
cyclization recombination
- DCC:
-
deleted in colorectal carcinoma
- ERK:
-
extracellular regulated kinase
- EGFR:
-
epidermal growth factor receptor
- ErbB2:
-
erythroblastic leukaemia viral oncogene homologue 2
- GAB1:
-
GRB2-associated binding protein 1
- HGF/SF:
-
hepatocyte growth factor/scatter factor
- HIF-1:
-
hypoxia-inducible factor 1
- JNK1:
-
c-Jun N-terminal protein kinase 1
- loxP:
-
locus of X-over P1
- NF-κB:
-
nuclear factor of kappa light polypeptide gene enhancer in B cells
- PI3K:
-
phosphoinositide-3 kinase
- RasGAP:
-
Ras GTPase activating protein
- RET:
-
rearranged during transfection
- TPR:
-
translocated promoter region
- UNC5H:
-
unc-5 homologue
References
Cooper CS, Park M, Blair DG, Tainsky MA, Huebner K, Croce CM et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature 1984; 311: 29–33.
Park M, Dean M, Cooper CS, Schmidt M, O'Brien SJ, Blair DG et al. Mechanism of MET oncogene activation. Cell 1986; 45: 895–904.
Park M, Dean M, Kaul K, Braun MJ, Gonda MA, Vande Woude G . Sequence of MET protooncogene cDNA has features characteristic of the tyrosine kinase family of growth-factor receptors. Proc Natl Acad Sci USA 1987; 84: 6379–6383.
Nakamura T, Nishizawa T, Hagiya M, Seki T, Shimonishi M, Sugimura A et al. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989; 342: 440–443.
Stoker M, Gherardi E, Perryman M, Gray J . Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987; 327: 239–242.
Naldini L, Weidner KM, Vigna E, Gaudino GK, Bardelli A, Ponzetto C et al. Scatter factor and hepatocyte growth factor are undistinguishable ligands for the MET receptor. EMBO J 1991; 10: 2867–2878.
Weidner KM, Arakaki N, Hartmann G, Vandekerckhove J, Weingart S, Rieder H et al. Evidence for the identity of human scatter factor and human hepatocyte growth factor. Proc Natl Acad Sci USA 1991; 88: 7001–7005.
Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C . Essential role for the c-MET receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995; 376: 768–771.
Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995; 373: 699–702.
Uehara Y, Minowa O, Mori C, Shlota K, Kuno J, Noda T et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995; 373: 702–705.
Maina F, Hilton MC, Ponzetto C, Davies AM, Klein R . Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev 1997; 11: 3341–3350.
Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS . Hepatocyte growth factor/c-MET signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci USA 2004; 101: 4477–4482.
Borowiak M, Garratt AN, Wustefeld T, Strehle M, Trautwein C, Birchmeier C . Met provides essential signals for liver regeneration. Proc Natl Acad Sci USA 2004; 101: 10608–10613.
Amicone L, Spagnoli FM, Spath G, Giordano S, Tommasini C, Bernardini S et al. Transgenic expression in the liver of truncated Met blocks apoptosis and permits immortalization of hepatocytes. EMBO J 1997; 16: 495–503.
Longati P, Bardelli A, Ponzetto C, Naldini L, Comoglio PM . Tyrosines1234–1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994; 9: 49–57.
Ponzetto C, Bardelli A, Zhen Z, Maina F, dalla Zonca P, Giordano S et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 1994; 77: 261–271.
Weidner KM, Dicesare S, Sachs M, Brinkmann V, Behrens J, Birchmeier W . Interaction between GAB1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature 1996; 384: 173–176.
Maina F, Casagranda F, Audero E, Simeone A, Comoglio PM, Klein R et al. Uncoupling of Grb2 from the Met receptor in vivo reveals complex roles in muscle development. Cell 1996; 87: 531–542.
Maina F, Pante G, Helmbacher F, Andres R, Porthin A, Davies AM et al. Coupling Met to specific pathways results in distinct developmental outcomes. Mol Cell 2001; 7: 1293–1306.
Fan S, Ma YX, Wang JA, Yuan RQ, Meng Q, Cao Y et al. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3′ kinase. Oncogene 2000; 19: 2212–2223.
Bowers DC, Fan S, Walter KA, Abounader R, Williams JA, Rosen EM et al. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res 2000; 60: 4277–4283.
Xiao GH, Jeffers M, Bellacosa A, Mitsuuchi Y, Vande Woude GF, Testa JR . Anti-apoptotic signaling by hepatocyte growth factor/Met via the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways. Proc Natl Acad Sci USA 2001; 98: 247–252.
Mildner M, Eckhart L, Lengauer B, Tschachler E . Hepatocyte growth factor/scatter factor inhibits UVB-induced apoptosis of human keratinocytes but not of keratinocyte-derived cell lines via the phosphatidylinositol 3-kinase/AKT pathway. J Biol Chem 2002; 277: 14146–14152.
Derksen PW, de Gorter DJ, Meijer HP, Bende RJ, van Dijk M, Lokhorst HM et al. The hepatocyte growth factor/Met pathway controls proliferation and apoptosis in multiple myeloma. Leukemia 2003; 17: 764–774.
Ozaki M, Haga S, Zhang HQ, Irani K, Suzuki S . Inhibition of hypoxia/reoxygenation-induced oxidative stress in HGF-stimulated antiapoptotic signaling: role of PI3-K and Akt kinase upon rac1. Cell Death Differ 2003; 10: 508–515.
Takeuchi K, Ito F . Suppression of adriamycin-induced apoptosis by sustained activation of the phosphatidylinositol-3′-OH kinase-Akt pathway. J Biol Chem 2004; 279: 892–900.
Schulze-Bergkamen H, Brenner D, Krueger A, Suess D, Fas SC, Frey CR et al. Hepatocyte growth factor induces Mcl-1 in primary human hepatocytes and inhibits CD95-mediated apoptosis via Akt. Hepatology 2004; 39: 645–654.
Fan S, Ma YX, Gao M, Yuan RQ, Meng Q, Goldberg ID et al. The multisubstrate adapter GAB1 regulates hepatocyte growth factor (scatter factor)-c-Met signaling for cell survival and DNA repair. Mol Cell Biol 2001; 21: 4968–4984.
Maroun CR, Holgado-Madruga M, Royal I, Naujokas MA, Fournier TM, Wong AJ et al. The GAB1 PH domain is required for localization of GAB1 at sites of cell-cell contact and epithelial morphogenesis downstream from the MET receptor tyrosine kinase. Mol Cell Biol 1999; 19: 1784–1799.
Maroun CR, Moscatello DK, Naujokas MA, Holgado-Madruga M, Wong AJ, Park M . A conserved inositol phospholipid binding site within the pleckstrin homology domain of the GAB1 docking protein is required for epithelial morphogenesis. J Biol Chem 1999; 274: 31719–31726.
Ma H, Calderon TM, Fallon JT, Berman JW . Hepatocyte growth factor is a survival factor for endothelial cells and is expressed in human atherosclerotic plaques. Atherosclerosis 2002; 164: 79–87.
Zeng Q, Chen S, You Z, Yang F, Carey TE, Saims D et al. Hepatocyte growth factor inhibits anoikis in head and neck squamous cell carcinoma cells by activation of ERK and Akt signaling independent of NFkappa B. J Biol Chem 2002; 277: 25203–25208.
Reveneau S, Paumelle R, Deheuninck J, Leroy C, De-Launoit Y, Fafeur V . Inhibition of JNK by HGF/SF prevents apoptosis induced by TNF-alpha. Ann N Y Acad Sci 2003; 1010: 100–103.
Fan S, Wang JA, Yuan RQ, Rockwell S, Andres J, Zlatapolskiy A et al. Scatter factor protects epithelial and carcinoma cells against apoptosis induced by DNA-damaging agents. Oncogene 1998; 17: 131–141.
Kosai K, Matsumoto K, Nagata S, Tsujimoto Y, Nakamura T . Abrogation of Fas-induced fulminant hepatic failure in mice by hepatocyte growth factor. Biochem Biophys Res Commun 1998; 244: 683–690.
Yamamoto K, Morishita R, Hayashi S, Matsushita H, Nakagami H, Moriguchi A et al. Contribution of Bcl-2, but not Bcl-xL and Bax, to antiapoptotic actions of hepatocyte growth factor in hypoxia-conditioned human endothelial cells. Hypertension 2001; 37: 1341–1348.
Nakagami H, Morishita R, Yamamoto K, Taniyama Y, Aoki M, Yamasaki K et al. Hepatocyte growth factor prevents endothelial cell death through inhibition of bax translocation from cytosol to mitochondrial membrane. Diabetes 2002; 51: 2604–2611.
Wang X, Zhou Y, Kim HP, Song R, Zarnegar R, Ryter SW et al. Hepatocyte growth factor protects against hypoxia/reoxygenation-induced apoptosis in endothelial cells. J Biol Chem 2004; 279: 5237–5243.
Fan S, Gao M, Meng Q, Laterra JJ, Symons MH, Coniglio S et al. Role of NF-kappaB signaling in hepatocyte growth factor/scatter factor-mediated cell protection. Oncogene 2005; 24: 1749–1766.
Liu Y . Hepatocyte growth factor promotes renal epithelial cell survival by dual mechanisms. Am J Physiol 1999; 277: F624–633.
Moumen A, Patane S, Porras A, Dono R, Maina F . Met acts on MDM2 via mTOR to signal cell survival during development. Development 2007; 134: 1443–1451.
Higashio K, Shima N, Goto M, Itagaki Y, Nagao M, Yasuda H et al. Identity of a tumor cytotoxic factor from human fibroblasts and hepatocyte growth factor. Biochem Biophys Res Commun 1990; 170: 397–404.
Shima N, Nagao M, Ogaki F, Tsuda E, Murakami A, Higashio K . Tumor cytotoxic factor/hepatocyte growth factor from human fibroblasts: cloning of its cDNA, purification and characterization of recombinant protein. Biochem Biophys Res Commun 1991; 180: 1151–1158.
Tajima H, Matsumoto K, Nakamura T . Hepatocyte growth factor has potent anti-proliferative activity in various tumor cell lines. FEBS Lett 1991; 291: 229–232.
Higashio K, Shima N . Tumor cytotoxic activity of HGF-SF. EXS 1993; 65: 351–368.
Revoltella RP, Borney F, Dal Canto B, D'Urso CM . Apoptosis of serum-free C2.8 mouse embryo hepatocytic cells caused by hepatocyte growth factor deprivation. Cytotechnology 1993; 13: 13–19.
Morita M, Watanabe Y, Akaike T . Protective effect of hepatocyte growth factor on interferon-gamma- induced cytotoxicity in mouse hepatocytes. Hepatology 1995; 21: 1585–1593.
Bardelli A, Longati P, Albero D, Goruppi S, Schneider C, Ponzetto C et al. HGF receptor associates with the anti-apoptotic protein BAG-1 and prevents cell death. EMBO J 1996; 15: 6205–6212.
de Souza Jr M, Razvickas CV, Goncalves EA, Okano IR, Camargo SM, Monte JC et al. The role of growth factors on renal tubular cells submitted to hypoxia and deprived of glucose. Ren Fail 2003; 25: 341–353.
Mizui M, Isaka Y, Takabatake Y, Mizuno S, Nakamura T, Ito T et al. Electroporation-mediated HGF gene transfer ameliorated cyclosporine nephrotoxicity. Kidney Int 2004; 65: 2041–2053.
Okada M, Sugita K, Inukai T, Goi K, Kagami K, Kawasaki K et al. Hepatocyte growth factor protects small airway epithelial cells from apoptosis induced by tumor necrosis factor-alpha or oxidative stress. Pediatr Res 2004; 56: 336–344.
Kitta K, Day RM, Ikeda T, Suzuki YJ . Hepatocyte growth factor protects cardiac myocytes against oxidative stress-induced apoptosis. Free Radic Biol Med 2001; 31: 902–910.
Nakagami H, Morishita R, Yamamoto K, Taniyama Y, Aoki M, Matsumoto K et al. Mitogenic and antiapoptotic actions of hepatocyte growth factor through ERK, STAT3, and AKT in endothelial cells. Hypertension 2001; 37: 581–586.
Kannan R, Jin M, Gamulescu MA, Hinton DR . Ceramide-induced apoptosis: role of catalase and hepatocyte growth factor. Free Radic Biol Med 2004; 37: 166–175.
Dash PR, Whitley GS, Ayling LJ, Johnstone AP, Cartwright JE . Trophoblast apoptosis is inhibited by hepatocyte growth factor through the Akt and beta-catenin mediated up-regulation of inducible nitric oxide synthase. Cell Signal 2005; 17: 571–580.
Radhakrishnan N, Bhaskaran M, Singhal PC . Hepatocyte growth factor modulates H(2)O(2)-induced mesangial cell apoptosis through induction of heme oxygenase-1. Nephron Physiol 2005; 101: p92–p98.
Ishihara N, Takagi N, Niimura M, Takagi K, Nakano M, Tanonaka K et al. Inhibition of apoptosis-inducing factor translocation is involved in protective effects of hepatocyte growth factor against excitotoxic cell death in cultured hippocampal neurons. J Neurochem 2005; 95: 1277–1286.
Skibinski G, Skibinska A, James K . Hepatocyte growth factor (HGF) protects c-MET-expressing Burkitt's lymphoma cell lines from apoptotic death induced by DNA damaging agents. Eur J Cancer 2001; 37: 1562–1569.
Leiriao P, Albuquerque SS, Corso S, Van Gemert GJ, Sauerwein RW, Rodriguez A et al. HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cell Microbiol 2005; 7: 603–609.
Mizuno S, Matsumoto K, Li MY, Nakamura T . HGF reduces advancing lung fibrosis in mice: a potential role for MMP-dependent myofibroblast apoptosis. FASEB J 2005; 19: 580–582.
Kim WH, Matsumoto K, Bessho K, Nakamura T . Growth inhibition and apoptosis in liver myofibroblasts promoted by hepatocyte growth factor leads to resolution from liver cirrhosis. Am J Pathol 2005; 166: 1017–1028.
Arakaki N, Kazi JA, Kazihara T, Ohnishi T, Daikuhara Y . Hepatocyte growth factor/scatter factor activates the apoptosis signaling pathway by increasing caspase-3 activity in sarcoma 180 cells. Biochem Biophys Res Commun 1998; 245: 211–215.
Gohda E, Okauchi H, Iwao M, Yamamoto I . Induction of apoptosis by hepatocyte growth factor/scatter factor and its augmentation by phorbol esters in Meth A cells. Biochem Biophys Res Commun 1998; 245: 278–283.
Arakaki N, Kajihara T, Arakaki R, Ohnishi T, Kazi JA, Nakashima H et al. Involvement of oxidative stress in tumor cytotoxic activity of hepatocyte growth factor/scatter factor. J Biol Chem 1999; 274: 13541–13546.
Shima N, Itagaki Y, Nagao M, Yasuda H, Morinaga T, Higashio K . A fibroblast-derived tumor cytotoxic factor/F-TCF (hepatocyte growth factor/HGF) has multiple functions in vitro. Cell Biol Int Rep 1991; 15: 397–408.
Conner EA, Wirth PJ, Kiss A, Santoni-Rugiu E, Thorgeirsson SS . Growth inhibition and induction of apoptosis by HGF in transformed rat liver epithelial cells. Biochem Biophys Res Commun 1997; 236: 396–401.
Matteucci E, Modora S, Simone M, Desiderio MA . Hepatocyte growth factor induces apoptosis through the extrinsic pathway in hepatoma cells: favouring role of hypoxia-inducible factor-1 deficiency. Oncogene 2003; 22: 4062–4073.
Wang X, DeFrances MC, Dai Y, Pediaditakis P, Johnson C, Bell A et al. A mechanism of cell survival: sequestration of Fas by the HGF receptor Met. Mol Cell 2002; 9: 411–421.
Matteucci E, Castoldi R, Desiderio MA . Hepatocyte growth factor induces pro-apoptotic genes in HepG2 hepatoma but not in B16-F1 melanoma cells. J Cell Physiol 2001; 186: 387–396.
Tacchini L, De Ponti C, Matteucci E, Follis R, Desiderio MA . Hepatocyte growth factor-activated NF-kappaB regulates HIF-1 activity and ODC expression, implicated in survival, differently in different carcinoma cell lines. Carcinogenesis 2004; 25: 2089–2100.
Rasola A, Anguissola S, Ferrero N, Gramaglia D, Maffe A, Maggiora P et al. Hepatocyte growth factor sensitizes human ovarian carcinoma cell lines to paclitaxel and cisplatin. Cancer Res 2004; 64: 1744–1750.
Bardella C, Dettori D, Olivero M, Coltella N, Mazzone M, Di Renzo MF . The therapeutic potential of hepatocyte growth factor to sensitize ovarian cancer cells to cisplatin and paclitaxel in vivo. Clin Cancer Res 2007; 13: 2191–2198.
Shiota G, Rhoads DB, Wang TC, Nakamura T, Schmidt EV . Hepatocyte growth factor inhibits growth of hepatocellular carcinoma cells. Proc Natl Acad Sci USA 1992; 89: 373–377.
Lail-Trecker MR, Peluso CE, Peluso JJ . Hepatocyte growth factor disrupts cell contact and stimulates an increase in type 3 inositol triphosphate receptor expression, intracellular calcium levels, and apoptosis of rat ovarian surface epithelial cells. Endocrine 2000; 12: 303–314.
Conner EA, Teramoto T, Wirth PJ, Kiss A, Garfield S, Thorgeirsson SS . HGF-mediated apoptosis via p53/bax-independent pathway activating JNK1. Carcinogenesis 1999; 20: 583–590.
Matsuda Y, Matsumoto K, Ichida T, Nakamura T . Hepatocyte growth factor suppresses the onset of liver cirrhosis and abrogates lethal hepatic dysfunction in rats. J Biochem (Tokyo) 1995; 118: 643–649.
Ueki T, Kaneda Y, Tsutsui H, Nakanishi K, Sawa Y, Morishita R et al. Hepatocyte growth factor gene therapy of liver cirrhosis in rats. Nat Med 1999; 5: 226–230.
Smyth LA, Brady HJ . cMet and Fas receptor interaction inhibits death-inducing signaling complex formation in endothelial cells. Hypertension 2005; 46: 100–106.
Tulasne D, Deheuninck J, Lourenco FC, Lamballe F, Ji Z, Leroy C et al. Proapoptotic function of the MET tyrosine kinase receptor through caspase cleavage. Mol Cell Biol 2004; 24: 10328–10339.
Foveau B, Leroy C, Ancot F, Deheuninck J, Ji Z, Fafeur V et al. Amplification of apoptosis through sequential caspase cleavage of the MET tyrosine kinase receptor. Cell Death Differ 2007; 14: 752–764.
Fischer U, Janicke RU, Schulze-Osthoff K . Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 2003; 10: 76–100.
Yang JY, Widmann C . Antiapoptotic signaling generated by caspase-induced cleavage of RasGAP. Mol Cell Biol 2001; 21: 5346–5358.
Yang JY, Michod D, Walicki J, Murphy BM, Kasibhatla S, Martin SJ et al. Partial cleavage of RasGAP by caspases is required for cell survival in mild stress conditions. Mol Cell Biol 2004; 24: 10425–10436.
Yang JY, Michod D, Walicki J, Widmann C . Surviving the kiss of death. Biochem Pharmacol 2004; 68: 1027–1031.
Bredesen DE, Mehlen P, Rabizadeh S . Receptors that mediate cellular dependence. Cell Death Differ 2005; 12: 1031–1043.
Ebens A, Brose K, Leonardo ED, Hanson Jr MG, Bladt F, Birchmeier C et al. Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron 1996; 17: 1157–1172.
Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M et al. The RET proto-oncogene induces apoptosis: a novel mechanism for Hirschsprung disease. EMBO J 2000; 19: 4056–4063.
Mourali J, Benard A, Lourenco FC, Monnet C, Greenland C, Moog-Lutz C et al. Anaplastic lymphoma kinase is a dependence receptor whose proapoptotic functions are activated by caspase cleavage. Mol Cell Biol 2006; 26: 6209–6222.
Bae SS, Choi JH, Oh YS, Perry DK, Ryu SH, Suh PG . Proteolytic cleavage of epidermal growth factor receptor by caspases. FEBS Lett 2001; 491: 16–20.
Tikhomirov O, Dikov M, Carpenter G . Identification of proteolytic fragments from ErbB-2 that induce apoptosis. Oncogene 2005; 24: 3906–3913.
Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF . Met, metastasis, motility and more. Nat Rev Mol Cell Biol 2003; 4: 915–925.
Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997; 16: 68–73.
Takayama H, LaRochelle WJ, Sharp R, Otsuka T, Kriebel P, Anver M et al. Diverse tumorigenesis associated with aberrant development in mice overexpressing hepatocyte growth factor/scatter factor. Proc Natl Acad Sci USA 1997; 94: 701–706.
Wang R, Ferrell LD, Faouzi S, Maher JJ, Bishop JM . Activation of the Met receptor by cell attachment induces and sustains hepatocellular carcinomas in transgenic mice. J Cell Biol 2001; 153: 1023–1034.
Graveel C, Su Y, Koeman J, Wang LM, Tessarollo L, Fiscella M et al. Activating Met mutations produce unique tumor profiles in mice with selective duplication of the mutant allele. Proc Natl Acad Sci USA 2004; 101: 17198–17203.
Comoglio PM, Boccaccio C . Scatter factors and invasive growth. Semin Cancer Biol 2001; 11: 153–165.
Giordano S, Maffe A, Williams TA, Artigiani S, Gual P, Bardelli A et al. Different point mutations in the MET oncogene elicit distinct biological properties. FASEB J 2000; 14: 399–406.
Shinomiya N, Gao CF, Xie Q, Gustafson M, Waters DJ, Zhang YW et al. RNA interference reveals that ligand-independent MET activity is required for tumor cell signaling and survival. Cancer Res 2004; 64: 7962–7970.
Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE et al. Lung cancer cell lines harboring MET gene amplification are dependent on MET for growth and survival. Cancer Res 2007; 67: 2081–2088.
Zhang YW, Graveel C, Shinomiya N, Vande Woude GF . Met decoys: will cancer take the bait? Cancer Cell 2004; 6: 5–6.
Acknowledgements
We would like to thank Fabienne Lamballe for helpful comments. This work was supported by the CNRS, INSERM, the Institute Pasteur of Lille, the University of Lille I, and by grants from the ‘Association pour la recherche sur le cancer’ and the ‘Ligue contre le Cancer, comité Nord’. BF was supported by a ‘Institut Pasteur/Région Nord- Pas de Calais’ fellowship.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by H Ichijo
Rights and permissions
About this article
Cite this article
Tulasne, D., Foveau, B. The shadow of death on the MET tyrosine kinase receptor. Cell Death Differ 15, 427–434 (2008). https://doi.org/10.1038/sj.cdd.4402229
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4402229
Keywords
This article is cited by
-
The multiple paths towards MET receptor addiction in cancer
Oncogene (2018)
-
c-Met expression and activity in urogenital cancers – novel aspects of signal transduction and medical implications
Cell Communication and Signaling (2017)
-
Necrosis- and apoptosis-related Met cleavages have divergent functional consequences
Cell Death & Disease (2015)
-
Sustained endoplasmic reticulum stress inhibits hepatocyte proliferation via downregulation of c-Met expression
Molecular and Cellular Biochemistry (2014)
-
MicroRNA-34a is dispensable for p53 function as teratogenesis inducer
Archives of Toxicology (2014)
