
Abstract
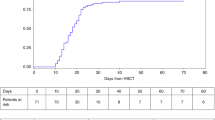
We reviewed outcomes after allogeneic hematopoietic cell transplantation (HCT) in 35 children with Chediak–Higashi syndrome (CHS). Twenty-two patients had a history of the life-threatening accelerated phase of CHS before HCT and 11 were in accelerated phase at transplantation. Thirteen patients received their allograft from an human leukocyte antigen (HLA)-matched sibling, 10 from an alternative related donor and 12 from an unrelated donor. Eleven recipients of HLA-matched sibling donor, three recipients of alternative related donor and eight recipients of unrelated donor HCT are alive. With a median follow-up of 6.5 years, the 5-year probability of overall survival is 62%. Mortality was highest in those with accelerated phase disease at transplantation and after alternative related donor HCT. Only four of 11 patients with active disease at transplantation are alive. Seven recipients of alternative related donor HCT had active disease at transplantation and this may have influenced the poor outcome in this group. Although numbers are limited, HCT appears to be effective therapy for correcting and preventing hematologic and immunologic complications of CHS, and an unrelated donor may be a suitable alternative for patients without an HLA-matched sibling. Early referral and transplantation in remission after accelerated phase disease may improve disease-free survival.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

References
Barak Y, Nir E . Chediak–Higashi syndrome. Am J Pediatr Hematol Oncol 1987; 9: 42–55.
Pettit RE, Berdal KG . Chediak–Higashi syndrome. Neurologic appearance. Arch Neurol 1984; 41: 1001–1002.
Grossi CE, Crist WM, Abo T, Velardi A, Cooper MD . Expression of the Chediak–Higashi lysosomal abnormality in human peripheral blood lymphocyte subpopulations. Blood 1985; 65: 837–844.
Certain S, Barrat F, Pastural E, Le Deist F, Goyo-Rivas J, Jabado N et al. Protein truncation test of LYST reveals heterogenous mutations in patients with Chediak–Higashi syndrome. Blood 2000; 95: 979–983.
Zarzour W, Kleta R, Frangoul H, Suwannarat P, Jeong A, Kim SY et al. Two novel CHS1 (LYST) mutations: clinical correlations in an infant with Chediak–Higashi syndrome. Mol Genet Metab 2005; 85: 125–132.
Davis WC, Douglas SD . Defective granule formation and function in the Chediak–Higashi syndrome in man and animals. Semin Hematol 1972; 9: 431–450.
Abo T, Roder JC, Abo W, Cooper MD, Balch CM . Natural killer (HNK-1+) cells in Chediak–Higashi patients are present in normal numbers but are abnormal in function and morphology. J Clin Invest 1982; 70: 193–197.
Rubin CM, Burke BA, McKenna RW, McClain KL, White JG, Nesbit ME et al. The accelerated phase of Chediak–Higashi syndrome. An expression of the virus-associated hemophagocytic syndrome? Cancer 1985; 56: 524–530.
Aslan Y, Erduran E, Gedik Y, Mocan H, Yildiran A . The role of high dose methylprednisolone and splenectomy in the accelerated phase of Chediak–Higashi syndrome. Acta Haematol 1996; 96: 105–107.
Fukuda M, Morimoto T, Ishida Y, Suzuki Y, Murakami Y, Kida K et al. Improvement of peripheral neuropathy with oral prednisolone in Chediak–Higashi syndrome. Eur J Pediatr 2000; 159: 300–301.
Filipovich AH, Shapiro RS, Ramsay NK, Kim T, Blazar B, Kersey J et al. Unrelated donor bone marrow transplantation for correction of lethal congenital immunodeficiencies. Blood 1992; 80: 270–276.
Fischer A, Landais P, Friedrich W, Gerritsen B, Fasth A, Porta F et al. Bone marrow transplantation (BMT) in Europe for primary immunodeficiencies other than severe combined immunodeficiency: a report from the European Group for BMT and the European Group for Immunodeficiency. Blood 1994; 83: 1149–1154.
Diamond HR, Souza MH, Silva ML, Tabak DG, Ferman S, M-Silva VM et al. Natural killer cell activity in a patient with Chediak–Higashi syndrome submitted to bone marrow transplantation. Pediatr Hematol Oncol 1995; 12: 399–402.
Mottonen M, Lanning M, Saarinen UM . Allogeneic bone marrow transplantation in Chediak–Higashi syndrome. Pediatr Hematol Oncol 1995; 12: 55–59.
Haddad E, Le Deist F, Blanche S, Benkerrou M, Rohrlich P, Vilmer E et al. Treatment of Chediak–Higashi syndrome by allogenic bone marrow transplantation: report of 10 cases. Blood 1995; 85: 3328–3333.
Przepiorka D, Weisdorf D, Martin P, Klingemann HG, Beatty P, Hows J et al. 1994 Consensus conference on acute GVHD grading. Bone Marrow Transplant 1995; 15: 825–828.
Flowers ME, Kansu E, Sullivan KM . Pathophysiology and treatment of graft-versus-host disease. Hematol Oncol Clin North Am 1999; 13: 1091–1112, viii-ix.
Klein JP, Moeschberger ML . Survival Analysis: Techniques of Censored and Truncated Data. Springer-Verlag: Berlin, 2003.
Tardieu M, Lacroix C, Neven B, Bordigoni P, de Saint Basile G, Blanche S et al. Progressive neurologic dysfunctions 20 years after allogeneic bone marrow transplantation for Chediak–Higashi syndrome. Blood 2005; 106: 40–42.
Acknowledgements
This study was supported by Public Health Service Grant U24-CA76518-08 from the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and the National Heart, Lung and Blood Institute.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eapen, M., DeLaat, C., Baker, K. et al. Hematopoietic cell transplantation for Chediak–Higashi syndrome. Bone Marrow Transplant 39, 411–415 (2007). https://doi.org/10.1038/sj.bmt.1705600
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1705600
Keywords
This article is cited by
-
Successful use of emapalumab in refractory hemophagocytic lymphohistiocytosis in a child with Chédiak–Higashi syndrome: a case report
Journal of Medical Case Reports (2023)
-
Assisted reproduction mediated resurrection of a feline model for Chediak-Higashi syndrome caused by a large duplication in LYST
Scientific Reports (2020)
-
The neuropsychological phenotype of Chediak-Higashi disease
Orphanet Journal of Rare Diseases (2019)
