
Summary:
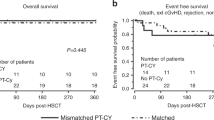
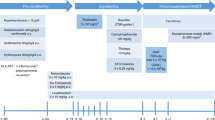
Since 1988, 24 children have undergone haematopoietic stem cell transplantation (HSCT) for severe sickle cell disease (SCD) in our unit, 13 being grafted after having been exposed to hydroxyurea (HU) to control SCD-related complications. Different pre-transplant conditioning regimens were given over time: Bu14/Cy200 in six patients (group 1), Bu16/Cy200/antithymocyte globulin (ATG) in five (group 2) and Bu16/Cy200/ATG with HU prior to HSCT in 13 (group 3). The aim of this study is to compare the outcome after HSCT of these groups of patients, which differ according to pre-transplant drug exposure. Overall, 20 of the 24 transplanted children had stable engraftment and have remained free of SCD-related symptoms after HSCT; 19 of them are currently alive and cured of SCD. In group 1 (HU−, ATG−), we observed one unexplainable late death, one absent engraftment, one late rejection and one mixed stable chimerism. In group 2 (HU−, ATG+), we observed the absence of engraftment in two patients and one early rejection. In group 3 (HU+, ATG+), we observed no cases of either absent engraftment, mixed stable chimerism or late rejection. In our experience, pre-transplant treatment with HU seems to be associated with a lower incidence of rejection/absent engraftment in severe SCD patients. These results need to be confirmed with a larger number of patients.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

References
Platt OS, Brambilla DJ, Rosse WF et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med 1994; 330: 1639–1644.
Ohene-Frempong K, Weiner SJ, Sleeper LA et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood 1998; 91: 288–294.
Platt OS, Thorington BD, Brambilla DJ et al. Pain in sickle cell disease: rates and risk factors for early death. N Engl J Med 1991; 325: 11–16.
Vichinsky EP, Neumayr LD, Earles AN et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med 2000; 342: 1855–1865.
Vermylen C, Fernandez-Robles E, Ninane J, Cornu G . Bone marrow transplantation in five children with sickle cell anaemia. Lancet 1988; i: 1427–1428.
Bernaudin F, Vernant JP, Vilmer E et al. Results of myeloblative allogenic stem cell transplant (SCT) for severe sickle cell disease (SCD) in France. Blood 2002; 11 (Suppl. 1a): 5a (abstr. 4).
Vermylen C, Cornu G, Ferster A et al. Haematopoïetic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant 1998; 22: 1–6.
Walters MC, Storb R, Patience M et al. Impact of bone marrow transplantation for symptomatic sickle cell disease: an interim report. Multicenter investigation of bone marrow transplantation for sickle cell disease. Blood 2000; 15: 1918–1924.
Amado RG, Schiller GJ . Nonmyeloablative approaches to the treatment of sickle hemoglobinopathies. Semin Oncol 2000; 27: 82–89.
Fixler J, Vichinsky E, Walters MC . Stem cell transplantation for sickle cell disease: can we reduce the toxicity? Pediatr Pathol Mol Med 2001; 20: 73–86.
Iannone F, Casella JF, Fuchs EJ et al. Failure of a minimally toxic non-myeloblative regimen to establish stable donor engraftment after transplantation for sickle cell anemia and β-thalassemia. Blood 2002; 11 (Suppl. 1a): 46a (abstr. 161).
Walters MC, Nienhuis AW, Vichinsky E . Novel therapeutic approaches in sickle cell disease. Hematology 2002; 1: 10–34.
Locatelli F, Rocha V, Reed W et al. Related umbilical cord blood transplantation in patients with thalassemia and sickle cell disease. Blood 2003; 15: 2137–2143.
Charache S, Terrin ML, Moore RD et al. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med 1995; 332: 1317–1322.
Charache S, Barton FB, Moore et al. Hydroxyurea in sickle cell anemia: clinical utility of a myelosuppressive ‘switching’ agent. Medicine 1996; 75: 300–326.
De Montalambert M, Belloy M, Bernaudin F et al. Three year follow-up of hydroxyurea treatment in severely ill children with sickle cell disease. The French Study Group on Sickle Cell Disease. J Pediatr Hematol Oncol 1997; 19: 313–318.
De Montalembert M, Begue P, Bernaudin F et al. Preliminary report of a toxicity study of hydroxyurea in sickle cell disease. French Study Group on Sickle Cell disease. Arch Dis Child 1999; 81: 437–439.
Ferster A, Vermylen C, Cornu G et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996; 88: 1960–1964.
Ferster A, Tahriri P, Vermylen C et al. Five years of experience with hydroxyurea in children and young adults with sickle cell disease. Blood 2001; 97: 3628–3632.
Hoppe C, Vichinsky E, Quirolo K et al. Use of hydroxyurea in children ages 2–5 years with sickle cell disease. J Pediatr Hematol Oncol 2000; 22: 330–334.
Jayabose S, Tugal O, Sandoval C et al. Clinical and hematologic effects of hydroxyurea in children with sickle cell anemia. J Pediatr 1996; 129: 559–565.
Kinney TR, Helms RW, O'Branski EE et al. Safety of hydroxyurea in children with sickle cell anemia: results of the HUG-KIDS study, a phase I/II trial. Pediatric Hydroxyrea Group. Blood 1999; 94: 1550–1554.
Ohene-Frempong K, Smith-Whitley K . Use of hydroxyurea in children with sickle cell disease: what comes next? Semin Hematol 1997; 34 (Suppl. 3): 30–41.
Steinberg MH, Barton F, Castro O et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia. JAMA 2003; 289: 1645–1651.
Van Besien K, Koshy M, Anderson-Shaw L et al. Allogeneic stem cell transplantation for sickle cell disease. A study of patients’ decisions. Blood 2001; 28: 545–549.
Walters MC, Sullivan KM, Bernaudin F et al. Neurologic complications after allogeneic marrow transplantation for sickle cell anemia. Blood 1995; 85: 879–884.
Davies SC . Bone marrow transplant for sickle cell disease – the dilemma [review]. Blood 1993; 7: 4–9.
Gaziev D, Polchi P, Lucarelli G et al. Second marrow transplants for graft failure in patients with thalassemia. Bone Marrow Transplant 1999; 24: 1299–1306.
Acknowledgements
This work was supported by grants from the Fonds National des Recherches Scientifiques (FNRS) no. 7.4525.98; no. 7.4503.00; no. 7.4501.02.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brachet, C., Azzi, N., Demulder, A. et al. Hydroxyurea treatment for sickle cell disease: impact on haematopoietic stem cell transplantation's outcome. Bone Marrow Transplant 33, 799–803 (2004). https://doi.org/10.1038/sj.bmt.1704443
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1704443
Keywords
This article is cited by
-
A survey on patient perception of reduced-intensity transplantation in adults with sickle cell disease
Bone Marrow Transplantation (2007)
-
Randomized trial of two different conditioning regimens for bone marrow transplantation in thalassemia – the role of busulfan pharmacokinetics in determining outcome
Bone Marrow Transplantation (2005)
