
Abstract
The intolerable burden of malaria has for too long plagued humanity and the prospect of eradicating malaria is an optimistic, but reachable, target in the 21st century. However, extensive knowledge is needed about the spatial structure of mosquito populations in order to develop effective interventions against malaria transmission. We hypothesized that the microbiota associated with a mosquito reflects acquisition of bacteria in different environments. By analyzing the whole-body bacterial flora of An. gambiae mosquitoes from Burkina Faso by 16 S amplicon sequencing, we found that the different environments gave each mosquito a specific bacterial profile. In addition, the bacterial profiles provided precise and predicting information on the spatial dynamics of the mosquito population as a whole and showed that the mosquitoes formed clear local populations within a meta-population network. We believe that using microbiotas as proxies for population structures will greatly aid improving the performance of vector interventions around the world.
Similar content being viewed by others

Introduction
As a part of a holistic approach to controlling the disease, the eradication of malaria requires a strong intervention against the vectors transmitting malaria. Today, impregnated bednets are efficient barriers of night-time malaria transmission. However, the anthropophilic malaria mosquitoes have shifted their feeding patterns to circumvent the bednet barriers, whilst an increasing pesticide resistance in the mosquitoes also reduces the effectivity of bednets and indoor residual spraying1. Under these prevailing conditions it is anticipated that new strategies must be explored which eliminate the parasites in the mosquitoes themselves. Control strategies involving genetic modification of mosquitoes (transgenesis) and genetic modification of their gut bacteria (paratransgenesis) both build on the premises that once released, the modified organism is maintained and spreads by itself through the vector population2,3. In paratransgenesis, the delivery of modified bacteria can be either through larval breeding sites or artificial sugar sources, but fundamental knowledge of where malaria mosquitoes acquire their bacteria is lacking. For transgenesis, the prerequisite for a successful intervention is good knowledge of malaria mosquito life history including dispersal distances, formation of local- and metapopulations and rates of exchange between populations.
Based on the hypothesis that the microbiota associated with a mosquito can be seen as a shadow cast by life-history events, where different environments leave their mark by contributing to the flora associated with the insect, we collected An. gambiae adult mosquitoes from three villages in Burkina Faso (Fig. 1). We analyzed the whole-body bacterial flora of the mosquitoes using 16 S amplicon sequencing yielding an average of 17,300 (SD 13,161.83) sequences per mosquito (see Methods and Supplementary Fig. S1). After selecting the mosquitoes with at least 7500 sequences based on rarefaction-curves analysis (Supplementary Fig. S2), we found that the mosquito samples harbored a wide range of bacterial taxa and display clear individual differences (Fig. 2). Horn clustering analysis of the samples showed that the mosquitoes clustered according to village with very few exceptions (Supplementary Fig. S3). The tightest clustering appeared between female mosquitoes from the same village, but clustering was equally common between gravid and non-blood-fed mosquitoes within those villages. NMDS score plots show apparent clustering of mosquitoes according to where the malaria mosquitoes were captured (Fig. 3), indicating a strong association between the microbiota composition and village of capture. The sub populations in the different villages are clearly separated despite being within flight distance from each other, but a couple of samples from the village VK3 have a bacterial flora more similar to the bacteria associated with mosquitoes of villages VK5 and VK7. This indicates that there is some interaction between the villages and therefore suggests the existence of local populations within a meta-population network.

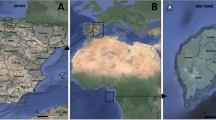
Rice field is marked in green. VK villages in dark blue are study sites. Black lines between villages indicate roads. The figure was made in Microsoft PowerPoint 2010.

Sequences are designated to order or families. M Male. FBF Female blood-fed. FNBF Female non-blood-fed.

The mosquitoes clustered together have a more similar microbiota and show a strong correlation to village of origin. All OTUs of each sample are utilized for the clustering. The mosquitoes were collected from randomly selected houses in village VK5 at 15 May 2012 and in villages VK3 and VK7 at 16 May 2012. M Male. FBF Female blood-fed. FNBF Female non-blood-fed.
In a supervised model, random forest classification showed 90% successful classification (i.e. 26 out of 29 individual samples; Fig. 4), thus confirming clustering and association between microbiota and village of capture and furthermore providing powerful predictive capabilities of class (village) assignment based on microbiota composition. Moreover, the use of the rdCV (repeated double cross validation) procedure provides a population of classification probability estimates per observation, providing additional depth of information regarding the certainty of classification compared to point estimates. Aside from the fact that most observations are correctly classified, some observations should be commented upon: i) The majority of samples (22 out of 29) had probability distributions practically without overlap between locations, thus indicating individuals with a full life history of exposure to location-specific microbiota and as a consequence unambiguous prediction results; ii) Two such samples from VK7 (VK7-21 and 22) were clearly classified as VK3. Although the causality of this discrepancy is not known, our hypothesis is that this clear case of misclassification corresponds to individuals from VK3 having recently flown in to VK7, thus not having had the time to acquire the OTU signature of VK7; iii) Four samples (VK3-41 and VK7-28, 31–35 and 36–40) showed substantial overlap between the probability distributions of VK3 and VK7 and three samples (VK3-47 and VK5-2 and 16–20) similarly between all three locations. We hypothesize that these findings relate to individuals either with longer flying distances or older individuals with longer flight history having therefore been exposed to several location-specific microbiota, and; iv) There is no evidence for a difference between males and females in terms of classification accuracy. Although the accuracy of data on males is potentially decreased by the pooling of individuals to ensure a sufficient quantity of DNA, the high correct classification rate indicates such a high degree of location fidelity that pooling from a practical perspective seems to not be an issue. It should be noted that predictive models where the low number of observations for practical purposes do not allow separation of data into training, validation and testing sets are easily over-fitted to spurious “false positive” variables, as is the case for the current data set4. However, in the present case, permutation analysis clearly showed both model validity (p = 2.4 * 10−8; Supplementary Fig. S4) and that the rdCV procedure effectively minimized statistical overfitting by comparing the estimated H0 mean (19.4 misclassifications) to the predicted H0 mean (i.e. 2/3 * 29 observations ≈ 19.3 misclassifications).

Multiple random forest modelling was performed within a repeated double cross validation scheme to minimize the risk of statistical overfitting (see method). Classification probability per submodel (n = 200) is color coded per village (VK3: green; VK5: orange; VK7: blue) and the global classification probability per individual and village marked in larger size and similar color. Successful classification was confirmed for 26 out of 29 individual samples (90%) and misclassified individuals are marked by a black circle. Of special interest are two individuals (VK7-21 and VK7-22) which although captured in VK7 possibly appertain to VK3 (p ≥ 0.58), thus indicating the potential to correctly identify migrating individuals.
The most important determinants of the random forest classification are in the order of importance the genera Massilia, Wolbachia, Shewanella and Acinetobacter (Supplementary Table S1). These four genera are likely acquired at different time points during the mosquito life history. The first time point from which we obtain information is the egg stage. Wolbachia is maternally inherited via the egg and was recently discovered for the first time in An. gambiae mosquitoes, from Burkina Faso5. In our data set, Wolbachia displayed a pronounced difference between the villages. It was found in all our specimens from VK5 and in several of the VK5 mosquitoes at a high abundance (36–47%), but in contrast at a low frequency and abundance in both VK3 and VK7 (Fig. 5). The mosquitoes in this study were captured wild in the adult stage; therefore, it is very unlikely that they all stem from a single mother. The predominance for Wolbachia in VK5 therefore suggests a high degree of village fidelity that extends back in time to the previous generation. This would imply that effective malaria interventions in one village would not be rapidly undermined by the invasion of malaria mosquitoes from a neighboring village. In the paper by Baldini et al.5, the Wolbachia obtained from An. gambiae M and S forms are reported to belong to a novel strain/supergroup of Wolbachia. This statement is based on the analysis of concatenated sequences from whole-genome shotgun metagenomic sequencing. For us to be able to compare with their data, we have used the W-spec 16 S sequences found in the supplementary data of Baldini et al.5 together with our data and previously published sequences. We find that both our W-spec sequences and those of Baldini et al.5 cluster together with the A and B supergroups (Fig. 6); thus, our analysis does not support the proposition of a new Anopheles-specific Wolbachia strain. One should note however, that for a final designation of supergroup in Wolbachia from the Anopheles mosquitoes in Burkina Faso, phylogenetic analyses of sequences from Wolbachia wsp or fbpA would be required. Unfortunately, neither we, nor Baldini et al.5 were able to obtain sequences from these genes and thus the final designation of the Wolbachia supergroup will await future work. Our data corroborate only one of the two Wolbachia strains found by Baldini et al.5 (Fig. 6), which could suggest the existence of multiple Wolbachia infections, but it would require extensive sampling over time to understand whether indeed two populations of Wolbachia are present in the An. gambiae M form.

A high abundance of Wolbachia in village VK5 is seen. The positions of the samples are determined by the whole bacterial flora (identical to fig. 3), but the color is dependent on the number of Wolbachia sequences per sample. Yellow color indicates no sequences.

Maximum Likelihood phylogenetic tree was conducted with 1000 bootstraps. The tree with the highest log likelihood (−300.4647) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. KJ sequences are from Baldini et al. (11); the KP sequence is from the current study. The location of each village is written after the GenBank sequence for the mosquito data. For the WSPEC from the Wolbachia genomes, the last capital letter denotes the Wolbachia supergroup.
The second time point from which we obtain information is the larval stage. The genera Massilia and Shewanella possibly reflect larval breeding sites since both Massilia and Shewanella form biofilms in fresh water6,7, a common food source for mosquito larvae. Indeed, Shewanella was one out of twelve genera isolated from midguts of Iranian Anopheles larvae8 and was also abundant in egg masses in non-biting midges (Chironomidae), the most common fresh-water insect9.
The third time point is connected to the adult food source (for females predominantly during early adulthood). Both male and female mosquitoes use nectar as a sugar source and in the early life as adult mosquitoes both sexes search for nectar sources only. A predominant genus of bacteria in nectar is the Acinetobacter where the abundance were between 49% and 90% in tropical plants according to 454 sequencing of 16 S amplicons10. Specific analysis of Acinetobacter revealed it to be present in almost all mosquitoes with the highest abundance in males (Supplementary Table S2), which is as expected since they only feed on sugar sources. The variable abundance of Acinetobacter in different mosquitoes may reflect the time between a nectar meal and capture or the intensity of feeding on a specific flower species. The most abundant Acinetobacter species show no difference between males and females, but that the mosquitoes in VK5 share a distinct set of Acinetobacter different from what is found in the villages VK3 and VK7 (permutation-based ANOVA: p = 1.7 * 10−5; Fig. 7). The data may reflect different nectar sources in the villages since Acinetobacter species often are plant-specific10,11. It also implies that the mosquitoes have predominantly fed on nectar in the vicinity of where they were captured and thus supports the hypothesis of village fidelity suggested by the data obtained from Wolbachia sequences.

The mosquitoes clustered together have a more similar microbiota and for VK5 show a strong correlation to village of origin. All OTUs belonging to Acinetobacter in each sample are utilized for the clustering. M Male. FBF Female blood-fed. FNBF Female non-blood-fed.
We have also looked at what could be regarded as the last important life history event for a female mosquito, namely the blood meal. The similarity between gravid and non-blood-fed females in our data set indicates that the bacterial flora associated with the blood-host-seeking experience and blood feeding does not have a major impact on the general microbiota associated with gravid females (Fig. 3). The blood meal, once acquired, leads to rapid proliferation of bacteria12,13,14, but favors a limited number of bacterial species that can utilize the blood and at the same time survive the oxidative stress that follows blood degradation15. However, our data show that this rapid shift in gut flora composition is no longer strongly affecting the overall microbiota a few days after the blood meal, indicating that the gut flora may revert to a pre-blood meal composition after the blood meal has been digested. The volatiles produced by skin bacteria play an important role in the guidance of female An. gambiae to humans16; it is not clear, however, if any bacteria also are picked up from the skin during feeding. We therefore specifically analyzed the presence of the three genera of human skin bacteria most strongly attracting An. gambiae females: Bacillus, Brevibacterium and Corynebacterium16, but found no correlation to sex/diet (Supplementary Fig. S5).
When analyzing the bacteria on a genus level, we find that the most abundant genera, making up more than 40% of all sequences identified to genus level, are Thorsellia, Wolbachia, Massilia and Acinetobacter (Supplementary Table S3). Of these, the most abundant is Thorsellia, which was originally isolated in Kenya from An. gambiae17 and has since then been shown to be the dominant species in different Kenyan populations of An. gambiae mosquitoes15,18. Briones et al.15 consistently isolated T. anophelis from the water surface micro-layer (SML), i.e., where mosquito larvae feed, as well as in 40% of the adults, while Wang et al.15 found that almost 70% of the bacteria in young adults belonged to Thorsellia. The Thorsellia sequences obtained in Burkina Faso correspond well to the three species of Thorsellia isolated from Kenya19,20, but also suggest the existence of other Thorsellia species (Supplementary Fig. S6).
The rate and direction at which insects move, spread and interact is key information for vector control strategies. From a monitoring point of view it can provide vital information on the movement patterns of individual insects that may carry disease between communities. For control measures aimed at altering the vector competence of insect populations, knowledge about insect movement is a fundamental prerequisite. A recent example is the very successful malaria intervention on the island Zanzibar where intensified usage of impregnated bednets and adequate drug treatment has brought malaria down by 80% and child mortality by 50%21. Whether this kind of intervention is also applicable on the mainland depends very much on the dispersal of malaria mosquitoes between communities. Our data suggest that neighboring villages function as metapopulations with restricted overlap and thus act almost as functional islands on the mainland. If this is true for other regions on the mainland, an intensified village-by-village malaria intervention might be more appropriate than was previously considered.
By genetic means such as microsatellites, it is possible to analyze meta-populations on a generational scale22. However, research on the spatial dynamics of mosquitoes over shorter time frames have required mark-release-recapture experiments where a large number of mosquitoes are radioactively labeled or marked with fluorescent dye before release. This technique is plagued by a plethora of technical problems and massive releases of vectors of disease bring ethical issues. For example, in Burkina Faso 21,000 An. gambiae females were released and only around 1% were recaptured in the same village and about 0.2% in the neighboring villages23. In contrast, by analyzing insect-bacterial associations, we are now able to study ecological distances within a much shorter time frame, using a limited number of specimens and without performing any releases. The generality of microbiotas as proxies for analysis of metapopulations and dispersal has large potential in animal research and shows that our lives are strongly interconnected with the bacteria surrounding us.
Conclusion
In conclusion, this study shows that the microbiota from whole-body mosquitoes is mirroring the environment in which the mosquitoes live and that based on the microbiota, the mosquitoes can be assigned to different populations. The most important determinants for village connection represent three important life history events. The presence of Wolbachia reflects the locality of females (mothers) from the previous generation, Shewanella and Massilia sequences are probably obtained in breeding sites during the larval stage, and Acinetobacter sequences suggest that the nectar sources for the adults differ. However, in order to draw any strong conclusions, precise information about environment-specific bacteria is needed and thus will be the target for future studies.
Methods
Study sites and mosquito collection
The collection sites are located in the western part of Burkina Faso in the large rice-growing area Vallée du Kou situated about 30 km North-West of Bobo-Dioulasso (Fig. 1). The villages of VK3 and VK5 are surrounded by rice fields, while VK7 has rice fields in the South and Savannah in the North. Because of the irrigation system, rice fields form permanent mosquito breeding sites for the An. gambiae M form (An. coluzzii24). There is also maize, roots and mixed vegetable cropping in the area. Mosquitoes were collected inside houses on two consecutive days (15 and 16 May, 2012) with mouth aspiration and stored in individual eppendorf tubes until further processing. To avoid that mosquitoes contaminated each other, they were handled separately one at a time. From January to June, the An. gambiae local population is exclusively composed of the M form. We verified that the mosquitoes collected belonged to the An. gambiae M form by PCR-RFLP according to Fanello et al.25. In total, 60 mosquitoes were collected from three villages. To ensure enough bacterial DNA, the physically smaller males were pooled with five males each before DNA extraction and PCR. Thus a total of 36 samples were used.
DNA and amplicon preparation
DNA from whole mosquitoes was prepared with the Promega Wizard Genomic DNA Purification Kit according to the manufacturer’s instructions. Bacterial 16 S rDNA (Escherichia coli position 341–805) were amplified by using general bacterial primers 341F (CCTACGGGNGGCWGCAG) and 805R (GACTACHVGGGTATCTAATCC)26. This primer pair matches approximately 90% of all good-quality bacterial sequences and covers all phyla in the Ribosomal Database Project release 10.25. Each DNA sample was individually PCR-amplified with Ready to go PCR beads (GE Health Care) by initial denaturation at 95 °C for 5 min followed by 35 cycles of [40 s at 95 °C, 40 s at 53 °C and 1 min at 72 °C] followed by a final 7-min extension at 72 °C. In a second PCR was added 1 of 50 flanking barcode sequence pairs to run samples in parallel27 using the same conditions as above, but only for 10 cycles of iteration.
Library preparation
Sequencing libraries were prepared from 0.2 μg of PCR-product according to the TruSeq DNA sample preparation guide #15005180 revC using reagents from the TruSeq DNA sample prep kit set A and set B v2 (Illumina). Briefly, the DNA fragments were end-repaired followed by purification using AMPure XP beads (Beckman Coulter). An A-base was added to the blunt ends of the DNA fragments and adapters and index tags for sequencing were ligated, followed by purification using AMPure XP beads. The DNA-fragments were amplified for 10 cycles of PCR, followed by purification using AMPure XP beads (Beckman Coulter). The quality of the library was evaluated using the Fragment analyzer from Advanced Analyticals and the DNF-910 dsDNA reagent kit. The adapter-ligated fragments were quantified by qPCR using the Library quantification kit for Illumina (KAPA Biosystems) on a StepOnePlus instrument (Applied Biosystems/Life technologies) prior to cluster generation and sequencing.
MiSeq sequencing
A 14 pM solution of DNA was subjected to cluster generation and paired-end sequencing with 300 bp read length on the MiSeq system (Illumina Inc.) using the v3 chemistry according to the manufacturer’s protocols. Base calling was done on the instrument by RTA 1.18.42 and the resulting .bcl files were demultiplexed and converted to fastq format with tools provided by CASAVA 1.8.4 (Illumina Inc.), allowing for one mismatch in the index sequence. Additional statistics on sequence quality were compiled with an in-house script from the fastq-files, RTA and CASAVA output files. Sequencing was performed by the SNP&SEQ Technology Platform in Uppsala, Sweden www.sequencing.se.
Bioinformatics analysis
Sequences were processed using the illumitag pipeline as described in Sinclair et al.27. In short, the paired-end reads were merged using PANDAseq28, and filtered based on their Phred scores. Multiplexing-barcodes were cut-off, and chimeras removed. Chimeras and OTU (Operational Taxonomic Unit) clustering at 97% sequence similarity was done using UPARSE29. The taxonomical annotation of the found OTUs was performed by CREST using the SILVAmod-database30. A total of 1.8M reads were obtained and after processing27 it led to a final number of 670 k reads left for analysis. The sequences were distributed as shown in Supplementary Fig. S1. Based on the analysis of rarefaction curves (Supplementary Fig. S2), seven samples were removed from the analysis for having less than 7500 reads giving a final number of 560 k reads (on average 19,300 reads/sample). The remaining read counts for the remaining 29 samples were then rarefied to the lowest number of reads per sample (8452 for sample VK7_NBF1).
NMDS plots and similarity heatmaps
Data analysis was done using the R-software (version 3.0.2). The heatmap trees were generated using the hierarchical clustering function from the R-software. The sample-wise horn-similarity heatmap was generated using the ‘vegdist’ function from the R-package ‘vegan’ (version 2.0–10), and the OTU- and taxonomical co-ocurrence heatmaps using simple pearson correlation. Non-metric dimensional scaling (NMDS) was performed using the ‘metaMDS’ function from the R-package ‘vegan’ and the horn’s dissimilarity index. Permutation-based ANOVA has been done with the ‘adonis’ function of the ‘vegan’ package with 106 permutations.
Predictive modeling
To provide predictive results of classification of mosquitoes according to their microbiome makeup, a supervised multivariate algorithm was developed using the R-software (version 3.2.0). In this algorithm, the ‘randomForest’ function from the ‘randomForest’ package (version 4.6–10) was used as classifying method within an in-house developed repeated double cross validation (rdCV) scheme4,31 to reduce the probability of statistical overfitting. Furthermore, the random forest was tuned over number of OTUs within the inner CV loop to provide an unbiased selection of the most informative OTUs, thereby optimising model performance32. In brief, tuning was performed by iteratively performing the inner validation loop using successively fewer OTUs, where in each step of the inner loop the 10% least informative OTUs (decided by average ranking of inner segment models) were removed. The double CV loop was repeated (n = 200) to achieve a population of class prediction probabilities per observation, thus enhancing the information content of prediction analysis. Permutation analysis was performed to test overall model validity and degree of overfitting by calculating the cumulative probability of actual model misclassification within a t-distributed H0 population. The H0 population was in turn populated by number of misclassification from models (n = 400) in which classifications labels were randomly drawn without replacement. Student’s t-distribution assumption was assessed by visually inspecting the H0 population histogram (Supplementary Fig. S4).
Wolbachia 16 S rDNA amplification
For Wolbachia PCR detection on selected samples, primers specific for Wolbachia 16 S rDNA (W-SpecF 5′-CATACCTATTCGAAGGGATAG-3′ and W-SpecR 5′-AGCTTCGAGTGAAACCAATTC-3′) were used as for universal 16 S rDNA amplification with the following conditions: initial denaturation at 95 °C for 2 min followed by 2 cycles of [2 min at 95 °C, 1 min at 60 °C and 1 min at 72 °C] followed by 35 cycles of [30 s at 95 °C, 1 min at 60 °C and 45 s at 72 °C] followed by a final 5-min extension at 72 °C33. Positive bands were cloned and sequenced.
Molecular phylogenetic analyses
The evolutionary histories of Wolbachia (and Thorsellia) were inferred by using the Maximum Likelihood method based on the Tamura-Nei model34 with 1000 bootstraps. The trees with the highest log likelihood (−300.4647 for Wolbachia and −1088.7983 for Thorsellia) are shown in Fig. 6 and Supplementary Fig. S6, respectively. The percentage of trees in which the associated taxa clustered together is shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. A discrete Gamma distribution was used to model evolutionary rate differences among sites [5 categories (+G, parameter = 0.4473 for Wolbachia; +G, parameter = 0.1992, for Thorsellia)]. The trees are drawn to scale, with branch lengths measured in the number of substitutions per site. The analyses involved 20 Wolbachia nucleotide sequences and 10 Thorsellia nucleotide sequences, respectively. All positions containing gaps and missing data were eliminated. There were a total of 120 Wolbachia positions and 420 Thorsellia positions, respectively in the final datasets. Evolutionary analyses were conducted in MEGA635.
Additional Information
How to cite this article: Buck, M. et al. Bacterial associations reveal spatial population dynamics in Anopheles gambiae mosquitoes. Sci. Rep. 6, 22806; doi: 10.1038/srep22806 (2016).
References
Gatton, M. L. et al. The importance of mosquito behavioural adaptations to malaria control in Africa. Evolution 67, 1218–1230 (2013).
Terenius, O., Marinotti, O., Sieglaff, D. & James, A. A. Molecular genetic manipulation of vector mosquitoes. Cell Host Microbe 4, 417–423 (2008).
Riehle, M. A. & Jacobs-Lorena, M. Using bacteria to express and display anti-parasite molecules in mosquitoes: current and future strategies. Insect Biochem. Mol. Biol. 35, 699–707 (2005).
Westerhuis, J. A. et al. Assessment of PLSDA cross validation. Metabolomics 4, 81–89 (2008).
Baldini, F. et al. Evidence of natural Wolbachia infections in field populations of Anopheles gambiae . Nature Comm. 5, 3985 (2014).
Liu, R. et al. Diversity of bacteria and mycobacteria in biofilms of two urban drinking water distribution systems. Canadian J. Microbiol. 58, 261–270 (2012).
Cheng, Z., Meng, X., Wang, H., Chen, M. & Li, M. Isolation and characterization of broad spectrum coaggregating bacteria from different water systems for potential use in bioaugmentation. PLoS One 9, e94220 (2014).
Chavshin, A. R. et al. Identification of bacterial microflora in the midgut of the larvae and adult of wild caught Anopheles stephensi: A step toward finding suitable paratransgenesis candidates. Acta Trop. 121, 129–134 (2012).
Halpern, M., Landsberg, O., Raats, D. & Rosenberg, E. Culturable and VBNC Vibrio cholerae: interactions with chironomid egg masses and their bacterial population. Microb. Ecol. 53, 285–293 (2007).
Fridman, S., Izhaki, I., Gerchman, Y. & Halpern, M. Bacterial communities in floral nectar. Environ. Microbiol. Rep. 4, 97–104 (2012).
Alvarez-Perez, S., Lievens, B., Jacquemyn, H. & Herrera, C. M. Acinetobacter nectaris sp nov and Acinetobacter boissieri sp nov., isolated from floral nectar of wild Mediterranean insect-pollinated plants. Int. J. Syst. Evol. Microbiol. 63, 1532–1539 (2013).
Chavshin, A. R. et al. Escherichia coli expressing a green fluorescent protein (GFP) In Anopheles stephensi: a preliminary model for paratransgenesis. Symbiosis 60, 17–24 (2013).
Pumpuni, C. B., Beier, M. S., Nataro, J. P., Guers, L. D. & Davis, J. R. Plasmodium falciparum: inhibition of sporogonic development in Anopheles stephensi by gram-negative bacteria. Exp. Parasitol. 77, 195–199 (1993).
Gaio Ade, O. et al. Contribution of midgut bacteria to blood digestion and egg production in Aedes aegypti (Diptera: Culicidae) (L.). Parasit. Vectors 4, 105 (2011).
Wang, Y., Gilbreath, T. M., 3rd, Kukutla, P., Yan, G. & Xu, J. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6, e24767 (2011).
Verhulst, N. O. et al. Differential attraction of malaria mosquitoes to volatile blends produced by human skin bacteria. PLoS One 5, e15829 (2010).
Lindh, J. M., Terenius, O. & Faye, I. 16 S rRNA gene-based identification of midgut bacteria from field-caught Anopheles gambiae sensu lato and A. funestus mosquitoes reveals new species related to known insect symbionts. Appl. Environ. Microbiol. 71, 7217–7223 (2005).
Briones, A. M., Shililu, J., Githure, J., Novak, R. & Raskin, L. Thorsellia anophelis is the dominant bacterium in a Kenyan population of adult Anopheles gambiae mosquitoes. ISME J. 2, 74–82 (2008).
Kämpfer, P. et al. Proposal of Thorsellia kenyensis sp. nov. and Thorsellia kandunguensis sp. nov., isolated from larvae of Anopheles arabiensis, as members of the family Thorselliaceae fam. nov. Int. J. Syst. Evol. Microbiol. 65, 444–451 (2015).
Kämpfer, P. et al. Thorsellia anophelis gen. nov., sp. nov., a new member of the Gammaproteobacteria. Int. J. Syst. Evol. Microbiol. 56, 335–338 (2006).
Bhattarai, A. et al. Impact of artemisinin-based combination therapy and insecticide-treated nets on malaria burden in Zanzibar. PLoS Med. 4, e309 (2007).
Ugelvig, L. V., Andersen, A., Boomsma, J. J. & Nash, D. R. Dispersal and gene flow in the rare, parasitic Large Blue butterfly Maculinea arion . Mol. Ecol. 21, 3224–3236 (2012).
Costantini, C. et al. Density, survival and dispersal of Anopheles gambiae complex mosquitoes in a west African Sudan savanna village. Med. Vet. Entomol. 10, 203–219 (1996).
Coetzee, M. et al. Anopheles coluzzii and Anopheles amharicus, new members of the Anopheles gambiae complex. Zootaxa 3619, 246–274 (2013).
Fanello, C., Santolamazza, F. & della Torre, A. Simultaneous identification of species and molecular forms of the Anopheles gambiae complex by PCR-RFLP. Med. Vet. Entomol. 16, 461–464 (2002).
Herlemann, D. P. R. et al. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 5, 1571–1579 (2011).
Sinclair, L., Osman, O. A., Bertilsson, S. & Eiler, A. Microbial community composition and diversity via 16 S rRNA gene amplicons: evaluating the Illumina platform. PLoS One 10, e0116955 (2015).
Masella, A. P., Bartram, A. K., Truszkowski, J. M., Brown, D. G. & Neufeld, J. D. PANDAseq: PAired-eND Assembler for Illumina sequences. BMC Bioinform. 13 (2012).
Edgar, R. C. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10, 996 (2013).
Lanzen, A. et al. CREST - Classification Resources for Environmental Sequence Tags. PLoS One 7 (2012).
Filzmoser, P., Liebmann, B. & Varmuza, K. Repeated double cross validation. J. Chemometrics 23, 160–171 (2009).
Hanhineva, K. et al. Discovery of urinary biomarkers of whole grain rye intake in free-living subjects using nontargeted LC-MS metabolite profiling. Mol. Nutr. Food Res. 59, 2315–2325 (2015).
Werren, J. H. & Windsor, D. M. Wolbachia infection frequencies in insects: evidence of a global equilibrium? Proc. Biol. Sci/The Royal Society 267, 1277–1285 (2000).
Tamura, K. & Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial-DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526 (1993).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Acknowledgements
The amplicon data have been deposited in GenBank under the accession number PRJNA265916. The Wolbachia 16 S sequence has been deposited in GenBank under the accession number KP089991. We thank Louise Malmgren, Klas Andersson, Ouédraogo Karim, Yaro Gibril, Bamogo Félix, Sanou David for mosquito collection, Stefan Bertilsson for scientific and technical support and Jan Bengtsson and Tomas Pärt for comments on the manuscript. The authors declare no competing interests. This study was supported by the Swedish Research Council and an SLU strategic grant for excellent research for young researchers (to OT).
Author information
Authors and Affiliations
Contributions
O.T. and R.H. designed the study; L.K.J.N. performed experiments, and R.D. supervised mosquito collections. M.B. analyzed the 16 S rRNA metagenomics data. C.B. performed supervised predictive modelling, unbiased OTU selection and contributed to the discussion on inference thereof. O.T. and R.H. wrote the manuscript. All authors read, commented and approved the manuscript in its final form.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Buck, M., Nilsson, L., Brunius, C. et al. Bacterial associations reveal spatial population dynamics in Anopheles gambiae mosquitoes. Sci Rep 6, 22806 (2016). https://doi.org/10.1038/srep22806
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22806
This article is cited by
-
Isolation, characterization and functional analysis of a bacteriophage targeting Culex pipiens pallens resistance-associated Aeromonas hydrophila
Parasites & Vectors (2024)
-
Holobiont perspectives on tripartite interactions among microbiota, mosquitoes, and pathogens
The ISME Journal (2023)
-
Bacterial communities in carnivorous pitcher plants colonize and persist in inquiline mosquitoes
Animal Microbiome (2022)
-
Interspecies microbiome transplantation recapitulates microbial acquisition in mosquitoes
Microbiome (2022)
-
Microbial Diversity of Adult Aedes aegypti and Water Collected from Different Mosquito Aquatic Habitats in Puerto Rico
Microbial Ecology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
