
Abstract
Alzheimer’s disease is characterized by the misfolding and self-assembly of the amyloidogenic protein amyloid-β (Aβ). The aggregation of Aβ leads to diverse oligomeric states, each of which may be potential targets for intervention. Obtaining insight into Aβ oligomers at the atomic level has been a major challenge to most techniques. Here, we use magic angle spinning recoupling 1H-1H NMR experiments to overcome many of these limitations. Using 1H-1H dipolar couplings as a NMR spectral filter to remove both high and low molecular weight species, we provide atomic-level characterization of a non-fibrillar aggregation product of the Aβ1-40 peptide using non-frozen samples without isotopic labeling. Importantly, this spectral filter allows the detection of the specific oligomer signal without a separate purification procedure. In comparison to other solid-state NMR techniques, the experiment is extraordinarily selective and sensitive. A resolved 2D spectra could be acquired of a small population of oligomers (6 micrograms, 7% of the total) amongst a much larger population of monomers and fibers (93% of the total). By coupling real-time 1H-1H NMR experiments with other biophysical measurements, we show that a stable, primarily disordered Aβ1-40 oligomer 5–15 nm in diameter can form and coexist in parallel with the well-known cross-β-sheet fibrils.
Similar content being viewed by others
Introduction
Alzheimer’s disease (AD) is a fatal neurological disorder affecting more than five million people in the United States today; a figure that is expected to increase three-fold by 2050 if therapeutics remain inadequate1. Although the exact cause of AD remains undetermined, many signs point to the involvement of the aggregation of the amyloid-β (Aβ) peptide at some stage2,3. The aggregation of Aβ leads to the formation of senile plaques found in patients with AD, the main constituent of which is the Aβ peptide in its fibrillar form4. However, attempts at pharmaceutical intervention aimed at targeting Aβ aggregation has been complicated by the myriad forms that aggregates of Aβ can adopt, many of which remain poorly characterized3,5,6.
Much effort has been undertaken in the way of understanding the structural details of the monomeric7,8,9 and fibrillar10,11,12,13 forms of Aβ both computationally and experimentally; however, there are only few existing structural models of the intermediates formed along the misfolding pathway of Aβ14,15,16,17. Unfortunately, these are the species currently believed to be most critical for pathogenesis in Alzheimer’s and other amyloid related neurodegenerative diseases3. Most of the models that do exist bear a close structural resemblance to the fiber end-product15,18,19,20, with few exceptions21. However, considerable evidence from lower-resolution techniques like CD suggests some (but not all) of the most toxic oligomers may have a considerably different structure that may be closer to the Aβ monomer than the Aβ fiber22,23,24,25.
The transient nature and high heterogeneity of amyloid oligomers present significant challenges for high-resolution structural studies. Oligomers of a specific conformation are difficult to isolate, which has thus far severely limited high-resolution characterization of Aβ. Moreover, oligomer structures of Aβ1-40 and Aβ1-42 have not been obtained by crystallography (only a low resolution structure of a specific Aβ oligomer obtained by powder X-ray diffraction and modeling exists)26, nor have they been obtained for oligomers of other amyloidogenic proteins, except for αB-crystallin27.
Previous studies have shown Aβ1-40’s ability to form unordered, globular aggregates with little to no secondary structure22,24,25,28. Not only are these oligomers a critical step in the aggregation of Aβ, but they also exhibit a high degree of cytotoxicity22,24,25. A time dependent 19F NMR study showed that such oligomers can persist even after prolonged incubation (>50 days) and the formation of amyloid fibrils29; however, none of these studies demonstrated structural details beyond low-resolution measurements. To characterize the disordered globular oligomers, we chose experimental conditions similar to these previous studies using low salt concentrations and neutral pH, yet here we also apply agitation during seeded fibrillization. This method of preparation results in the formation of an aggregated Aβ sample comprised mostly of fibrils with a small population of a predominantly disordered oligomeric species of Aβ1-40. Remarkably, this method of preparation yielded disordered oligomers reproducibly at almost 10% of the total peptide concentration without perturbative methods, such as chemical- or photo-crosslinking, freezing, amino acid substitution, or any other type of protein engineering to stabilize the oligomer.
Using magic angle spinning (MAS) NMR spectroscopy on these samples, we are able to resolve structural details otherwise unobservable by other biophysical measurements. We do so by bridging the gap in the limitations imposed on solution- and solid-state NMR methods; a molecular weight limit in solution NMR and problems with sensitivity in solid-state NMR. These limitations are overcome by taking advantage of both solution- and solid-state NMR techniques: performing solid-state NMR experiments on a liquid sample with “solid” characteristics. A similar approach was taken with a solution sample of Aβ by monitoring the formation and kinetics of large aggregates sedimenting out of solution30. However, this technique used 13C detection requiring large amounts of sample and expensive, isotopic labeling.
Here, we employ a RFDR (radio frequency driven dipolar recoupling)31 -based 2D 1H/1H chemical shift correlation experiment to overcome these limitations. High-resolution structural features of high molecular weight oligomers are difficult to characterize directly by solution NMR; however, the RFDR-based 2D 1H/1H experiment enables the specific detection of this oligomeric species over fibrillar and monomeric Aβ1-40 due to the line-narrowing afforded by slow MAS and the reintroduction of residual 1H-1H dipolar couplings by RFDR. Previously, the applicability of RFDR for the study of such “soft” solid systems was used to investigate the structural and motional characteristics of a resin-bound peptide32, a micelle-associated cytochrome b533 and membrane-bound peptides34,35,36. Here, we demonstrate the utility of 1H/1H RFDR for the specific structural characterization of a minority population of a stable, disordered Aβ1-40 oligomer containing sparse secondary structure, at low abundance, directly from an unlabeled sample in coexistence with amyloid fibrils without further purification or filtration. To the best of our knowledge, this is the first instance in which 1H-1H dipolar couplings have been used for structural studies of an amyloid oligomer, which may provide a general method to study intermediate size oligomers of the type believed to play a crucial role in amyloid pathology3.
Results and discussion
Aβ1-40 can form a disordered oligomer in parallel with β-sheet fibrils
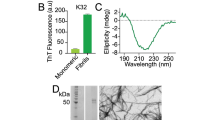
Since intermediate size (<100 kDa) oligomers have been a prime target of both pharmaceutical and scientific research3, we first sought to find a condition that allowed the reproducible generation of oligomers of this type. Our previous study utilizing real-time observation of a single-site 19F-label at the Met-35 of Aβ1-40, found that Aβ1-40 oligomers are observed and stabilized well into the late stages of the aggregation process29. In agreement with Suzuki et al.29, we find the coexistence of oligomers and fibers in our samples; however, the oligomers formed under our shaking conditions are not of the β-sheet type. A CD spectrum (Fig. 1a, solid) shows an intensity minimum at 225 nm indicating the bulk sample is predominantly β-sheet. Using size-cutoff filtration with a Spin-X 0.22 μm filter, we could isolate the oligomers and any residual monomers from the fibrils of Aβ1-40. A noticeably different CD spectrum is apparent in the filtrate, which shows a strong minimum near 198 nm indicative of a predominantly disordered structure (Fig. 1a, blue) very similar to that of early aggregation states of Aβ (Fig. 1a, red). We further confirmed the lack of β-sheet content of the filtered Aβ1-40 oligomers using the amyloid-specific dye, ThT (Fig. 1b). Similar to the freshly dissolved Aβ1-40 sample, the oligomers isolated by spin-X filtration display essentially no fluorescence in its ThT emission profile, indicating a β-sheet conformation is not present (Fig. 1b). Conversely, an intense ThT signal is present in the isolated fibril fraction. Measuring the concentration of the spin-X filtrate by the bicinchoninic acid (BCA) protein concentration assay (Thermo Scientific) revealed 17.0 ± 6.0 μM or only 7.3 + 2.6% of the total Aβ1-40 concentration (231 μM) was not fibrillar. Together, the ThT, CD and BCA results confirm the sample primarily consists of amyloid fibrils with a minority population of a largely disordered, relatively low MW species.

Biophysical characterization of Aβ1-40 disordered oligomers.
(a) CD spectra, (b) ThT fluorescence and (c) bis-ANS fluorescence of fibrillar (black), the spin-X-isolated oligomer (blue) and freshly dissolved (red) samples of Aβ1-40. In panel c, the emission spectrum of bis-ANS in solution is shown in grey. (d) Distributions of the hydrodynamic radii of the freshly dissolved Aβ1-40 (red) and the spin-X-isolated Aβ1-40 oligomer (blue) determined by DLS. Representative AFM and TEM images of the Aβ1-40 oligomers (e) and fibrils (f). Scale bars in both images are 100 nm. All experiments were performed in 10 mM sodium phosphate buffer, pH 7.4 at 25 °C.
Since the CD spectra of the spin-X filtrate resemble CD spectra of monomeric Aβ, it is possible that the filtered sample consists of residual monomeric Aβ1-40. We first tested this possibility with the molecular probe 4,4′-Dianilino-1,1′-Binaphthyl-5,5′-Disulfonic Acid (bis-ANS). The spectral properties and the quantum yield of bis-ANS are highly sensitive to polarity, thus upon binding to hydrophobic surfaces bis-ANS becomes appreciably more fluorescent. A unique feature of bis-ANS is its ability to selectively identify different aggregation states of Aβ1-40 through distinct emission spectra (Fig. 1c)22,37,38. The emission spectra of bis-ANS after isolation and separation of the fibrils from disordered oligomers is shown in Fig. 1c, along with the bis-ANS signal from a freshly dissolved Aβ1-40 sample prepared without incubation or seeding. The emission spectrum of bis-ANS in the spin-X-isolated oligomers (Fig. 1c, blue) is significantly more blue-shifted than that of the emission spectrum observed from the fibril fraction (Supplementary Fig. 1). Also in Fig. 1c, the bis-ANS fluorescence observed in the presence of the freshly dissolved Aβ1-40 sample (red) is only slightly blue-shifted from that of the spectrum of bis-ANS alone (black), indicating limited binding of bis-ANS to monomeric Aβ1-40 and a significant difference in the hydrophobic exposure compared to the freshly dissolved Aβ1-40 or the Aβ1-40 fibrils.
In addition to the bis-ANS measurements, dynamic light scattering (DLS) studies revealed distinct size distributions of these disordered Aβ oligomers when compared to the freshly dissolved Aβ1-40 sample (Fig. 1d and Supplementary Fig. 2). DLS measured a RH of ~5.1 nm with a polydispersity of 32.2% for the oligomers isolated by spin-X filtration, whereas the RH of the freshly dissolved Aβ1-40 sample was ~1.4 nm with a polydispersity of 19.0%. To complement our optical spectroscopy data, representative electron micrographs and AFM images of the amyloid preparations showed oligomeric and fibrillar forms present in the aggregated sample of Aβ1-40 (Fig. 1e,f). All AFM images showed the disordered Aβ oligomers exhibit an amorphous, spherical morphology (Fig. 1e). The oligomers were found present among a dense fibrillar network whose bundled, twisted features were apparent in all electron micrographs as observed in Fig. 1f. Taken together, all biophysical measurements point to the existence of a disordered and soluble Aβ1-40 oligomer coexisting as a minority population amongst Aβ1-40 fibrils. This finding is very intriguing as it indicates Aβ1-40 is simultaneously undergoing β-sheet and non-β-sheet aggregation pathways. For this reason, we aimed to find distinct structural features that contributed to the formation of such an Aβ oligomer.
The RFDR-based 2D 1H/1H chemical shift correlation provides site-specific information on a minority population of disordered Aβ1-40 oligomers in a fiber-containing sample
To obtain an atomic-level picture of these disordered oligomers, we use a combination of solution- and solid-state NMR experiments. RFDR is different from the NOESY experiment in that the incorporation of rotor-synchronized π-pulses during the mixing period reintroduces coherent homonuclear dipole-dipole interactions (Supplementary Fig. 3). We first performed RFDR-based 2D 1H/1H experiments on two types of samples: the unfiltered, aggregated Aβ1-40 sample used in the CD experiments and a control sample of freshly dissolved Aβ1-40 without fibers made primarily of monomeric and low MW species. An overlay of the two 2D RFDR spectra is shown in Fig. 2. Only a few cross-peaks are observed in the 2D 1H/1H spectrum of the freshly dissolved Aβ1-40 sample (red in Fig. 2), probably due to the rapid tumbling of monomeric and/or low molecular weight Aβ1-40. The few cross-peaks that do appear in the spectrum of freshly dissolved Aβ1-40 likely arise from early aggregates and the presence of peaks appearing near 0 ppm in the 1D 1H spectrum suggests that this is indeed the case (Supplementary Fig. 4). Such peaks (near 0 ppm) are commonly observed in the spectra of amyloidogenic peptides and have been shown to occur due to the presence of an oligomeric species in which aliphatic protons are solvent protected and thus shifted to the high field region of the 1D 1H spectrum39,40. However, with the exception of these few cross-peaks, the RFDR-based 2D 1H/1H spectra of unaggregated Aβ1-40 is very sparse, consistent with weak (to negligible) 1H-1H dipolar couplings in low MW samples. The RFDR-based 2D 1H/1H experiment therefore acts as an efficient spectral filter for intermediate sized oligomers; low MW exhibit few cross-peaks because of weak (to negligible) 1H-1H dipolar couplings while the linewidth from very high MW species like amyloid fibers is too large (due to very strong dipolar couplings) to generate a detectable signal under the slow MAS speeds used in this study.

RFDR-based 2D 1H/1H chemical shift correlation spectra of freshly dissolved (red) and aggregated (blue) forms of Aβ1-40.
(a) Side-chain to Hα, (b) side-chain and (c) Hβ-Hα and Hα -Hα regions of the overlaid 2D spectra were recorded under 2.7 kHz MAS. The dotted circle highlights the Ser and Gly fingerprints of the aggregated Aβ1-40 sample. Peak assignments are given for the mixed Aβ1-40 sample. The spectra were acquired with a 50 ms mixing time at 600 MHz in 100% D2O, 10 mM sodium phosphate buffer, pH 7.4 and 37 °C. Total Aβ1-40 concentrations for both samples were 462 μM; the estimated oligomer concentration in the aggregated sample is 35 ± 12 μM. The acquisition time was 4 days.
In contrast to the sparse spectra obtained from unaggregated Aβ1-40, the RFDR-based 2D 1H/1H spectra of the aggregated sample showed multiple cross-peaks consistent with the presence of the disordered oligomer suggested by the CD, fluorescence and AFM experiments (Fig.1). The dominant feature of the RFDR-based 2D 1H/1H spectra is a very strong upfield shift of Hα resonances compared to both the expected random coil values and the unaggregated, primarily monomeric sample. Upfield shifts typically derive from two primary sources: the formation of helical secondary structure and the shielding of the residue from solvent. Unfortunately, the spectra in Fig.2 are not sufficient to distinguish between these two sources.
However, this spectrum was taken under rather extreme conditions of low concentration (~35 μM) and high heterogeneity (the oligomers only constitute 7–10% of the entire sample) to test the limits of the technique to samples not traditionally considered amenable for NMR. To see if additional structural details could be resolved in a more conformationally pure sample, we performed the RFDR-based 2D 1H/1H experiment on purified oligomers by removing the fibers through Spin-X filtration using 10% D2O instead of 100% D2O to resolve the amide resonances. We first tested the influence of the MAS rate by increasing it up to 15 kHz (Supplementary Fig. 5). The fact that the resolution of 1D 1H spectra does not improve with the increasing MAS rate suggests that oligomers are not sedimenting out of solution at the speeds used in the experiment, although the selective sedimentation of fibers may play a role in enhancing the resolution of the mixed fiber/oligomer sample30.
Under 10 kHz MAS, we were able to obtain a well-resolved RFDR-based 2D 1H/1H spectrum of the pure oligomer sample (Fig. 3). A majority of peaks assigned in the RFDR-based 2D 1H/1H spectrum of the unfiltered sample (Fig.2) are identified in the spectrum of the filtered oligomer sample as well (Fig. 3), suggesting peaks appearing in both samples arise from conformationally similar species. Under these conditions, it was possible to perform a partial assignment of the resonances using TOCSY experiments under MAS (Fig. 3, red spectrum). The lack of complete connectivity hampered the complete assignment of peaks in RFDR-based 2D 1H/1H spectra. Accordingly, the assignments of a partially folded structure of Aβ1-40 guided unambiguous assignment of 2D 1H/1H NMR spectra14. While the lack of connectivity is unavoidable given these constraints, the results suggest a spectrum that, while unusual for either a well-folded or unstructured protein, is consistent with results obtained from other biophysical experiments shown in Fig. 1. At the atomic level, the RFDR-based 2D 1H/1H spectrum contains inter-residue cross-peaks between the aliphatic and alpha protons of K28-G29, S26-N27, H13-G38 and S8-E11. These inter-residue contacts are observed for both 20 and 50 ms RFDR mixing times (Supplementary Fig. 6), indicating the interaction of these residues are prominent features for the oligomer’s structure. Moreover, the Ser and Gly fingerprints are observed only for the disordered oligomer species (circled cross-peaks in Fig. 2), suggesting the involvement of these residues in the oligomer formation and stabilization. More generally, though there are very strong up-field shifts for the resonances that usually suggest helix formation, no medium range αN (i, i + 3/i + 4) connectivity was found indicative of an α- or 310- helical conformation. Rather, the strong upfield shift appears to be the result of the oligomerization of peptide and the consequent shielding of the residues from solvent. Taken together, this finding suggests a dynamic and disordered structure with a high degree of turns and twists but without a well-defined secondary structure.

MAS spectra of the filtered disordered Aβ1-40 oligomer.
An overlay of the assigned Hα regions from RFDR-based 2D 1H/1H (blue) and 2D 1H/1H TOCSY (red) spectra acquired at 298 K with 10 kHz MAS. The filtered oligomer sample was lyophilized and re-hydrated to double its initial concentration, making for a total Aβ1-40 of ~35 μM. The RFDR and TOCSY based 2D 1H/1H spectra were acquired with mixing times of 50 and 70 ms, respectively, at 600 MHz in 100% D2O, 10 mM sodium phosphate buffer, pH 7.4. RFDR spectra were acquired at 25 °C under 10 kHz MAS. Assignments are given for the RFDR-based 2D 1H/1H spectra. The non-uniform sampling based data acquisition time was 4 hours.
We do not rule out the possibility that highly mobile regions of the fibril may contribute in part to the RFDR spectrum recorded on the mixed fibril-oligomer sample. The serine and glycine fingerprints highlighted in Fig. 2 are not found in the RFDR spectra of the filtered oligomer nor are they found in the freshly dissolved samples of Aβ1-40. Nevertheless, a majority of the cross-peaks assigned in the RFDR spectra of the filtered oligomer are the same as those assigned in the unfiltered Aβ1-40 sample. Given that any highly mobile residue from the Aβ fibril would largely maintain the fibril’s correlation time, it is more than likely that cross-peaks observed in the RFDR spectrum of the unfiltered sample derive from the disordered Aβ1-40 oligomer as MAS experiments performed on the filtered oligomer (Fig. 3 and Supplemental Fig. 5). Isolation of the oligomer, although helpful in improving the resolution (compare Fig. 2 and Fig. 3), is not strictly necessary.
The disordered Aβ1-40 oligomer is conformationally stable and grows in size
In light of our ability to purify the disordered Aβ oligomer, we were interested in whether the oligomers would aggregate into a fibrillar state. CD and DLS measurements confirm that the disordered nature of the oligomer is maintained while increasing in size over the course of 19 days (Fig. 4a,b). The CD spectrum of the spin-X-isolated oligomer at 19 days (Fig. 4b) shows a minimum intensity ca. 198 nm, indicative of random coil structure and the DLS experiments indicate an increase in hydrodynamic radius of the oligomer from ~5.1 nm to ~8.6 nm (Fig. 4a).

The disordered Aβ1-40 oligomer grows in size while maintaining its morphology.
(a) DLS experiments at 19 days demonstrate that the disordered oligomer not only remains disordered, but also grows in size as well with a distribution of hydrodynamic radii at 8.6 nm and 65.3 nm and polydispersity of 14.9% and 37.8%, respectively. (b) The strong minimum at ~200 nm in the CD spectrum of the disordered oligomer after 19 days reveals that the oligomer does not progress to a fibrillar state. Two-dimensional spectra of the fingerprint region of (c) TOCSY (70 ms mixing) and (d) NOESY (250 ms mixing) of the disordered Aβ1–40 oligomers recorded at 4 days (blue) and 19 days (red). The filtered oligomer sample was lyophilized and re-hydrated to double its initial concentration, making for a total Aβ1-40 of ~35 μM. After 19 days (red), almost all peaks are broadened beyond detection in both TOCSY and NOESY spectra.
Solution and MAS NMR experiments further demonstrate that the spin-X-isolated Aβ oligomers are conformationally diverse and grow over time, yet maintain their disordered nature. The 1D profiles of 1H MAS spectra do not change significantly over a period of 16 days, indicating the general fold of the oligomer is preserved. Between 4 and 9 days, the two sharp peaks at 1.20 and 1.18 ppm broaden beyond detection, while at the same time the oligomer peak at 0.16 ppm undergoes similar line-broadening (Supplementary Fig. 7). We attribute this line-broadening to the increasing size of disordered Aβ oligomers. Even after 4 days of aggregation, cross-peaks were not observed in 2D NOESY spectra under MAS conditions (Supplementary Fig. 8).
RFDR reveals details that solution NMR cannot for intermediate size oligomers
Since the RFDR pulse sequence (Supplementary Fig. 3) utilizes the transfer of proton magnetization via coherent 1H-1H dipolar couplings and an incoherent cross-relaxation from the NOE to generate cross-peaks34, we would expect the RFDR pulse sequence to be more sensitive to larger oligomers than the traditional NOESY experiment utilizing only NOE cross-relaxation. Accordingly, cross-peaks were not observed in 2D NOESY spectra obtained under MAS conditions and very few peaks were observed under static conditions (Supplementary Fig. 8), indicating the dipolar interaction among protons dominates the transfer of magnetization between nuclei of the disordered Aβ1-40 oligomer as evidenced by the RFDR spectrum in Fig. 3 (blue).
We further tested this observation by comparing the results from the RFDR experiment to solution NMR experiments using the time dependent growth of the oligomer from 4 to 19 days (Fig. 4c,d). Static TOCSY spectra (Fig. 4c) over this timeframe showed a severe decrease in the number of cross-peaks observed, while NOESY experiments under static conditions yielded little to very few cross-peaks at 4 days (Fig. 4d) and no cross-peaks were observed for the NOESY experiment at 19 days (data not shown). Similarly, 2D 1H/15N heteronuclear single-quantum correlation (HSQC) experiments performed on the spin-X-isolated oligomer of Aβ1-40 after 4 days (Fig. 5) exhibit drastically different HSQC spectra than that observed for a freshly dissolved Aβ1-40 sample41,42. Only four peaks are observed in the HSQC spectrum of the isolated, disordered Aβ1-40 oligomer after 4 days of aggregations; these likely coming from highly mobile residues and/or mobile side-chains. These results then suggest that molecular motions do not average out the 1H-1H dipolar interaction of the oligomer and therefore the peaks are broadened beyond detection in solution NMR experiments. We therefore conclude that while the oligomer is observable by traditional solution NMR experiments, only limited information can be acquired, in contrast to the high-resolution information obtained from the RFDR-based 2D 1H/1H MAS solid-state NMR experiment.

Comparison of 1H/15N HSQC spectra of the freshly dissolved (red) and the spin-X-isolated disordered oligomer (blue) of Aβ1-40 (after 4 days) recorded from a 900 MHz spectrometer.
Both experiments were performed in 10 mM phosphate buffer, pH 7.4 and 10% D2O at 25 °C.
Conclusion
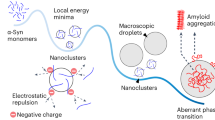
Using the 1H/1H RFDR technique, we were able to reveal the dynamic and disordered structure comprised of turns and twists for the intermediate size (5–10 nm) oligomers. By implementing both solution- and solid-state NMR experiments, particularly through the 1H-1H dipolar couplings recoupled by RFDR, we have characterized high-resolution structural properties of a dynamic and disordered Aβ1-40 oligomer and the development of early amyloid aggregates. Disordered and/or micelle-like structures have been observed for other amyloid-forming proteins and peptides as well43,44,45,46. In terms of a stable conformation, the oligomers studied here resemble, at least biophysically, conformers generated by small molecule amyloid inhibitors (such as polyphenols like EGCG41,47 or resveratrol48); i.e., inert Aβ species that are large and predominantly unstructured. However, the stable oligomer studied here occurs without small molecule perturbations or chemical modifications in the Aβ peptide sequence. Furthermore, this disordered oligomer forms simultaneously with the highly ordered and well- structured β-sheet fibrils, indicating that a single aggregation pathway is not necessarily prevalent for a given preparation of Aβ (Fig. 6). Whether the disordered Aβ oligomer studied here is cytotoxic remains to be determined; however, the fact that this disordered conformation persists to an end-state that is not fibrillar is unexpected in light of the concept of nucleated conformational conversion and Aβ1-40 aggregation pathways in general49,50.

Simultaneously occurring aggregation pathways of Aβ1-40.
Early aggregates maintain structural similarity to the stable, disordered Aβ1-40 oligomers observed at late aggregation stages. The early aggregates either (i) solely nucleate the disordered oligomers or (ii) act as a single nucleating seed from which the two distinct aggregation pathways bifurcate.
In a more general sense, our results demonstrate the value of the RFDR-based 2D 1H/1H experiment in obtaining high-resolution information on supramolecular assemblies not easily amenable to analysis by other biophysical techniques, including solution NMR and other solid-state NMR experiments. Specifically, most of the high resolution data so far on amyloid oligomers and fibers has come from solid-state NMR experiments. These experiments have been invaluable in advancing our understanding of amyloid and other supramolecular assemblies. However, solid-state NMR experiments are inherently insensitive and require frozen or lyophilized samples that must also be isotopically labeled, often in a site-specific fashion that requires chemical synthesis rather than recombinant expression. Importantly, the 1H/1H RFDR experiment can be run on aggregated solution samples using proton detection. For this reason, it is more sensitive than most other solid-state NMR experiments but is able to access a size range inaccessible to solution NMR experiments. Since it is run under solution conditions that allow dynamic averaging and very large oligomeric species are spectrally filtered out by the procedure, it is also very tolerant to both conformational heterogeneity and heterogeneous oligomer size distributions. As demonstrated in this study, the signal from the oligomer can be resolved without purification even though the oligomer comprises only 5% of the total sample. Finally, the experiment does not require isotopic labeling. All of these characteristics make it ideal for medically relevant samples that have been difficult to characterize, such as amyloid oligomers directly derived from the brain, which in some cases have shown intriguingly different properties than the corresponding recombinant or synthetic Aβ peptide13,51,52,53,54.
Methods
Peptide Synthesis
Aβ1-40 was synthesized manually by solid-phase Fmoc-based chemistry using the dimethoxybenzyl-protected (dmn) dipeptide, Fmoc-Val-(Dmb)-Gly-OH at positions 36 and 37 for the purpose of preventing aggregation during synthesis. The peptide was cleaved from the resin using 92.5% trifluoroacetic acid (TFA), 2.5% H2O, 2.5% ethanedithiol and 2.5% anisole. The crude peptide was dissolved in 20% acetic acid (v/v) and purified by reverse-phase HPLC using a Waters semipreparative C18 column equilibrate in 0.1% TFA. The peptide was eluted with a linear gradient of 0–80% acetonitrile at a flow rate of 10 ml/min. Proper synthesis and purification were validated using matrix-assisted laser desorption ionization mass spectrometry, which gave a value corresponding to the correct mass 4329.9 Da.
Sample Preparation
To remove preformed aggregates, the purified peptide was dissolved in 1% ammonium hydroxide (v/v) at a concentration of 1 mg/ml followed by removal of the solvent by lyophilization for 24 hours in aliquots of either 0.1 or 0.3 mg. The aliqouted peptide was then stored at −20 °C and only used once.
Preparation of Aggregated Aβ1-40 Sample
For preparation of Aβ1-40 aggregated sample containing a mixture of fibers and disordered oligomers, 0.1 or 0.3 mg of the lyophilized peptide was solubilized in a 10 mM sodium phosphate buffer (pH 7.4) solution at a concentration of 1 mg/ml (231 μM) and incubated for 48 hours at 37 °C under agitation at 1000 rpm. This aggregated Aβ1-40 sample was then used to seed 5% of the total concentration of freshly dissolved Aβ1-40 in the same buffer conditions at a total Aβ1-40 concentration of 1 mg/ml. The seeded Aβ1-40 sample was incubated for 48 hours at 37 °C under agitation at 1000 rpm to form a sample containing a minority of Aβ1-40 oligomers (17.0 ± 6.0 μM or ~10% of the total peptide concentration) amongst a much larger population of Aβ1-40 fibrils. Concentrations were determined by the Thermo-Scientific BCA protein assay kit from 3 independent samples.
Isolation of Disordered Oligomers
Upon completion of preparing the seeded Aβ1-40 aggregates (i.e. at the end of the 4 day incubation period), disordered oligomers were isolated using a Spin-X microcentrifuge spin column (Corning Inc.), containing a 0.22 μm cellulose acetate filter. The filtrate contained the isolated Aβ1-40 disordered oligomer (the 4-day old oligomer) and the Aβ1-40 fibrils were retained in the retentate. Due to the concentrations of the disordered oligomers being very low for NMR measurements, these samples could be lyophilized and rehydrated at double their concentration (quantified above). This had no effect on the solution characteristics of the sample, namely its size, morphology, or secondary structure as verified by CD and DLS.
Monomer preparation (i.e., Freshly Dissolved Aβ1-40)
Preparation of Aβ1-40 monomer sample was performed as described previously14. Briefly, 0.1 mg of the lyophilized peptide was first dissolved in 10 μl of 1 mM NaOH and sonicated until the peptide was solubilized. The peptide solution was then hydrated in H2O (or 100% D2O for MAS NMR measurements), buffered in 10 mM sodium phosphate (pH 7.4) and diluted to 76 μM (0.1 mg in 300 μl).
NMR Spectroscopy
All NMR data was processed using TopSpin 2.1 (Bruker). 1D data were analyzed using TopSpin 2.1 and 2D data were analyzed using SPARKY. In all NMR spectra, the 1H peak from H2O was used as a chemical shift reference by setting its frequency at 4.7 ppm.
MAS NMR Spectroscopy
MAS NMR experiments were performed at 298 K or 310 K on an Agilent/Varian VNMRS 600 MHz solid-state NMR spectrometer using a 4 mm 1H/X double-resonance Nanoprobe. The spectrometer was operated with a deuterium field lock and a MAS spinning speed of 2.7 kHz. The proton carrier frequency was set to the resonance frequency of water for all experiments and 1H2O signal was suppressed using a 10 Hz saturation RF pulse for 1 s at the beginning of NOESY or RFDR-based 2D 1H/1H experiments. The radio frequency field strength used for the 90° and 180° pulses was 61 kHz. The NOESY and RFDR-based 2D 1H/1H spectra were recorded using 1100 scans, 200 t1 increments, 6252 t2 complex points and a spectral width of 11 ppm in both frequency dimensions. The experimental data sets were zero-filled in both t1 and t2 dimensions to form a 2048 × 4096 data matrix. Phase shifted sine bell multiplication was applied to both dimensions prior to Fourier transformation. Since the maximum MAS rate possible with the Agilent Nanprobe is 2.7 kHz, to achieve higher MAS rates while maintaining the utility of deuterium locking and pulse-field gradients, we used a Bruker complete multi-phase (CMP) probe on a Bruker 600 MHz Avance III NMR spectrometer. 1D 1H NMR spectra were acquired for MAS speeds from 5 to 15 kHz (Supplementary Figure 5). RFDR-based 2D 1H/1H experiments were performed under 10 kHz MAS with mixings times of 20 and 50 ms and using non-uniformly sampled (NUS), interleaved datasets. NUS datasets, where each data is split into its 2 component dataets, was done using the Split program and processed separately with the standard ‘xfb’ command, regenerating the missing points and transforms the datasets.
Assignment of proton resonances was done using SPARKY with published assignments for Aβ1-40 as a guide. 1D 1H MAS experiments were recorded with 20000 scans, 16 dummy scans, a spectral width of 12 ppm and an acquisition time of 0.5 s. The proton carrier frequency was set at water resonance for all experiments and 1H2O resonance was suppressed using a 50 Hz RF pulse for 1.5 s.
Solution NMR Spectroscopy
Solution NMR spectroscopy was performed on the filtered oligomer. Data were acquired on a 900 MHz Bruker NMR Spectrometer equipped with a cryogenic triple-resonance pulse-field gradient probe. 1D and 2D NMR spectra were collected at either 298 K or 310 K. NOESY spectra were acquired with a spectral width of 12 ppm in both dimensions, with 400 (ω1) and 2048 (ω2) complex points using a 1.5 s recycle delay. The NOESY experiments were acquired with two different mixing times: 250 and 600 ms. Solvent suppression was done using gradient pulses centered at 1H resonance frequency of water. The same parameters were used for TOCSY experiments; however, mixing times of 70 and 100 ms were used. The experimental data sets were zero-filled to form a 2048 × 4096 data matrix and a phase-shifted sine bell multiplication was applied to both dimensions prior to Fourier transformation. 15N-labeled Aβ1-40 was purchased from rPeptide (Athens, GA, U.S.A.) and used for HSQC experiments. The exact same sample preparation protocols detailed above were used for 15N-labeled Aβ1-40 samples. Each spectrum was obtained from 128 t1 experiments, 92 scans (for the spin-X-isolated Aβ1-40 oligomer) and 1 s recycle delay.
Circular Dichroism (CD)
CD measurements were performed on JASCO J-715 Spectropolarimeter using a 0.1 cm path length cell. Isolated oligomer and fibril samples of Aβ1-40 were prepared as described above. Molar CD per residue values were calculated using ε = θobsd/(3298lcn), where θobsd is the observed ellipticity measured in millidegrees, c is the molar concentration, l is the cell path length in centimeters and n is the number of residues in the peptide.
bis-ANS and Thioflavin T Fluorescence Assays
Aβ1-40 fibril formation was measured by increased fluorescence emission upon binding of amyloid fibers to the commonly used amyloid-specific dye, thioflavin T (ThT). Aβ1-40 oligomer and fibril formation were also measured by fluorescence emission spectra of the less specific dye, 4,4′-Dianilino-1,1′-Binaphthyl-5,5′-Disulfonic Acid (bis-ANS); purchased from Santa Cruz Biotechnology, Inc. bis-ANS exhibits limited fluorescence in water; however, becomes considerably fluorescent upon binding hydrophobic surfaces. Isolated oligomeric and fibrillar Aβ1-40 species were prepared as described above. The retentate containing Aβ1-40 fibrils was dissolved in 200 μL of 10 mM sodium phosphate buffer, pH 7.4, of which 90 μL was then aliquoted into microcentrifuge tubes. Similarly, the filtrate containing the Aβ1-40 oligomers was aliquoted into 90 μL quantities as well. Either ThT or bis-ANS fluorescent dye was then added to a peptide aliquot at a concentration of 10 μM and fluorescence emission was measured on a Horiba FluoroMax 4 spectrofluorometer. An excitation wavelength of 446 nm and 350 nm was used for ThT and bis-ANS, respectively.
Atomic Force Microscopy (AFM)
An aggregated Aβ1-40 sample prepared as described above was deposited onto freshly cleaved mica and incubated for twenty minutes at room temperature. The surfaces was rinsed with nanopure water and dried under nitrogen flow. Dry imaging was carried out in tapping mode using a Nanoscope III atomic force microscopy (AFM) and JZ Scanner (Veeco) with VistaProbes T300R (NanoScience, AZ; nominal radius 10 nm, force constant 40 N/m, resonance frequency 300 kHz). The AFM was calibrated with a 100 nm x 100 nm standard (2D-100, NANOSENSORS, Switzerland). After calibration, the percent error was −0.6%. Random locations on the sample were selected for imaging. Particles were detected and height measurements were made using SPIP 6.0.13 software (NanoScience Instruments).
Dynamic Light Scattering (DLS)
Light scattering experiments were performed on Aβ1-40 samples prepared as described using a DynaPro Nanostar instrument from Wyatt Technology (Santa Barbara, CA). Light scattering was measured at 90°. The intensity correlation function and the distribution of the hydrodynamic radii (RH) of the particles contributing to the scattering were determined using DYNAMICS software (Wyatt Technology).
Transmission Electron Microscopy (TEM)
Samples for negative stain TEM analysis were deposited on continuous carbon films on copper rhodium 100 mesh grids (Electron Microscopy Sciences, EMS Hatfield PA.). Prior to adding samples, the grids were charged using a glow discharger for 15 s at 30 mA negative discharge. Fibrillar and oligomer sample solutions at 1 mg/ml were adsorbed to the grids for 2 minutes prior to rinsing with two 10 μL drops of water for 10 s. Samples were blotted using No. 2 Whatman filter paper. Samples for TEM were then stained with a 10 μL drop of freshly filtered 2% uranyl acetate (EMS) for 15 s before blotting excess stain. Samples were analysed using a Philips CM-100 microscope operating at 80 kV.
Additional Information
How to cite this article: Kotler, S. A. et al. High-resolution NMR characterization of low abundance oligomers of amyloid-ß without purification. Sci. Rep. 5, 11811; doi: 10.1038/srep11811 (2015).
References
Alzheimer’s disease facts and figures. Alzheimer’s Dement. 11, 332–384 (2015).
Sakono, M. & Zako, T. Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 277, 1348–58 (2010).
Benilova, I., Karran, E. & De Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–57 (2012).
Selkoe, D. J. The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498 (1991).
Lin, H., Bhatia, R. & Lal, R. Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J 15, 2433–2444 (2001).
Butterfield, S. M. & Lashuel, H. a. Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 49, 5628–54 (2010).
Bernstein, S. L. et al. Amyloid β-protein: monomer structure and early aggregation states of Aβ42 and its Pro19 alloform. J. Am. Chem. Soc. 127, 2075–84 (2005).
Ball, K. A., Phillips, A. H., Wemmer, D. E. & Head-Gordon, T. Differences in β-strand populations of monomeric Aβ40 and Aβ42. Biophys. J. 104, 2714–24 (2013).
Baumketner, A. et al. Amyloid β-protein monomer structure: a computational and experimental study. Protein Sci. 15, 420–8 (2006).
Tycko, R. Solid-state N. M. R. studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 62, 279–99 (2011).
Schütz, A. K. et al. Atomic-Resolution Three-Dimensional Structure of Amyloid β Fibrils Bearing the Osaka Mutation. Angew. Chem. Int. Ed. Engl. (2014). 10.1002/anie.201408598
Petkova, A. T. et al. Self-propagating, molecular-level polymorphism in Alzheimer’s beta-amyloid fibrils. Science 307, 262–265 (2005).
Lu, J.-X. et al. Molecular structure of β-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154, 1257–68 (2013).
Vivekanandan, S., Brender, J. R., Lee, S. Y. & Ramamoorthy, A. A partially folded structure of amyloid-beta(1-40) in an aqueous environment. Biochem. Biophys. Res. Commun. 411, 312–6 (2011).
Chimon, S. et al. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol. 14, 1157–64 (2007).
Ahmed, M. et al. Structural conversion of neurotoxic amyloid-beta(1-42) oligomers to fibrils. Nat. Struct. Mol. Biol. 17, 561–567 (2010).
Sarkar, B. et al. Significant structural differences between transient amyloid-β oligomers and less-toxic fibrils in regions known to harbor familial Alzheimer’s mutations. Angew. Chem. Int. Ed. Engl. 53, 6888–92 (2014).
Scheidt, H. A., Morgado, I. & Huster, D. Solid-state NMR reveals a close structural relationship between amyloid-β protofibrils and oligomers. J. Biol. Chem. 287, 22822–6 (2012).
Tay, W. M., Huang, D., Rosenberry, T. L. & Paravastu, A. K. The Alzheimer’s amyloid-β(1-42) peptide forms off-pathway oligomers and fibrils that are distinguished structurally by intermolecular organization. J. Mol. Biol. 425, 2494–508 (2013).
Lendel, C. et al. A Hexameric Peptide Barrel as Building Block of Amyloid-β Protofibrils. Angew. Chem. Int. Ed. Engl. 53, 12756–60 (2014).
Yu, L. et al. Structural characterization of a soluble amyloid beta-peptide oligomer. Biochemistry 48, 1870–7 (2009).
Ladiwala, A. R. A. et al. Conformational differences between two amyloid β oligomers of similar size and dissimilar toxicity. J. Biol. Chem. 287, 24765–73 (2012).
Ono, K., Condron, M. M. & Teplow, D. B. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. U. S. A. 106, 14745–50 (2009).
Luo, J., Wärmländer, S. K. T. S., Gräslund, A. & Abrahams, J. P. Alzheimer peptides aggregate into transient nanoglobules that nucleate fibrils. Biochemistry 53, 6302–8 (2014).
Serra-Vidal, B. et al. Hydrogen/Deuterium Exchange-Protected Oligomers Populated during Aβ Fibril Formation Correlate with Neuronal Cell Death. ACS Chem. Biol. 9, 2678–2685 (2014).
Stroud, J. C., Liu, C., Teng, P. K. & Eisenberg, D. Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. U. S. A. 109, 7717–22 (2012).
Jehle, S. et al. Solid-state NMR and SAXS studies provide a structural basis for the activation of alphaB-crystallin oligomers. Nat. Struct. Mol. Biol. 17, 1037–42 (2010).
Corsale, C. et al. Entrapment of Aβ(1-40) peptide in unstructured aggregates. J. Phys. Condens. Matter 24, 244103 (2012).
Suzuki, Y. et al. Resolution of oligomeric species during the aggregation of Aβ1-40 using (19)F NMR. Biochemistry 52, 1903–12 (2013).
Bertini, I. et al. Formation kinetics and structural features of Beta-amyloid aggregates by sedimented solute NMR. Chembiochem 14, 1891–7 (2013).
Bennett, A. E., Griffin, R. G., Ok, J. H. & Vega, S. Chemical shift correlation spectroscopy in rotating solids: Radio frequency-driven dipolar recoupling and longitudinal exchange. J. Chem. Phys. 96, 8624 (1992).
Raya, J. et al. Proton Dipolar Recoupling in Resin-Bound Peptides under High-Resolution Magic Angle Spinning. J. Magn. Reson. 157, 43–51 (2002).
Pandey, M. K. et al. Proton-detected 2D radio frequency driven recoupling solid-state NMR studies on micelle-associated cytochrome-b(5). J. Magn. Reson. 242, 169–79 (2014).
Ramamoorthy, A. & Xu, J. 2D 1H/1H RFDR and NOESY NMR experiments on a membrane-bound antimicrobial peptide under magic angle spinning. J. Phys. Chem. B 117, 6693–700 (2013).
Aucoin, D. et al. High-resolution 1H MAS RFDR NMR of biological membranes. J. Magn. Reson. 197, 77–86 (2009).
Kotler, S. A. et al. Gangliosides Mediate a Two-Step Mechanism of Membrane Disruption by Beta-Amyloid: Initial Pore Formation Followed by Membrane Fragmentation. Biophys. J. 104, 217A–217A (2013).
Bolognesi, B. et al. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 5, 735–40 (2010).
Guerrero-Muñoz, M. J., Castillo-Carranza, D. L., Sengupta, U., White, M. A. & Kayed, R. Design of metastable β-sheet oligomers from natively unstructured peptide. ACS Chem. Neurosci. 4, 1520–3 (2013).
Narayanan, S. & Reif, B. Characterization of chemical exchange between soluble and aggregated states of beta-amyloid by solution-state NMR upon variation of salt conditions. Biochemistry 44, 1444–52 (2005).
Huang, R. et al. NMR characterization of monomeric and oligomeric conformations of human calcitonin and its interaction with EGCG. J. Mol. Biol. 416, 108–20 (2012).
Hyung, S.-J. et al. Insights into antiamyloidogenic properties of the green tea extract (-)-epigallocatechin-3-gallate toward metal-associated amyloid-β species. Proc. Natl. Acad. Sci. U. S. A. 110, 3743–8 (2013).
Fawzi, N. L., Ying, J., Ghirlando, R., Torchia, D. A. & Clore, G. M. Atomic-resolution dynamics on the surface of amyloid-β protofibrils probed by solution NMR. Nature 480, 268–272 (2011).
Brender, J. R. et al. Probing the Sources of the Apparent Irreproducibility of Amyloid Formation: Drastic Changes in Kinetics and a Switch in Mechanism Due to Micellelike Oligomer Formation at Critical Concentrations of IAPP. J. Phys. Chem. B 119, 2886–2896 (2015).
Walsh, P., Neudecker, P. & Sharpe, S. Structural properties and dynamic behavior of nonfibrillar oligomers formed by PrP(106-126). J. Am. Chem. Soc. 132, 7684–95 (2010).
Rhoades, E. & Gafni, A. Micelle formation by a fragment of human islet amyloid polypeptide. Biophys. J. 84, 3480–7 (2003).
Carulla, N. et al. Experimental characterization of disordered and ordered aggregates populated during the process of amyloid fibril formation. Proc. Natl. Acad. Sci. U. S. A. 106, 7828–33 (2009).
Lopez del Amo, J. M. et al. Structural properties of EGCG-induced, nontoxic Alzheimer’s disease Aβ oligomers. J. Mol. Biol. 421, 517–24 (2012).
Ladiwala, A. R. A. et al. Resveratrol selectively remodels soluble oligomers and fibrils of amyloid Abeta into off-pathway conformers. J. Biol. Chem. 285, 24228–37 (2010).
Lee, J., Culyba, E. K., Powers, E. T. & Kelly, J. W. Amyloid-β forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 7, 602–609 (2011).
Serio, T. R. Nucleated Conformational Conversion and the Replication of Conformational Information by a Prion Determinant. Science 289, 1317–1321 (2000).
Moore, B. D., Rangachari, V., Tay, W. M., Milkovic, N. M. & Rosenberry, T. L. Biophysical analyses of synthetic amyloid-beta(1-42) aggregates before and after covalent cross-linking. Implications for deducing the structure of endogenous amyloid-beta oligomers. Biochemistry 48, 11796–806 (2009).
Hartley, D. M. et al. Transglutaminase induces protofibril-like amyloid beta-protein assemblies that are protease-resistant and inhibit long-term potentiation. J. Biol. Chem. 283, 16790–800 (2008).
Townsend, M., Shankar, G. M., Mehta, T., Walsh, D. M. & Selkoe, D. J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. J. Physiol. 572, 477–92 (2006).
Reed, M. N. et al. Cognitive effects of cell-derived and synthetically derived Aβ oligomers. Neurobiol. Aging 32, 1784–94 (2011).
Acknowledgements
This study was supported by research funds from NIH and the Protein Folding Disease Center at the University of Michigan (to A.R.). S.K was partly supported by the Molecular Biophysics Training Grant from NIH to the University of Michigan. We would also like to thank Dr. Ari Gafni and Dr. Duncan Steel for providing access to their lab equipment for optical spectroscopy measurements.
Author information
Authors and Affiliations
Contributions
S.A.K., J.R.B., S.V., K.Y., J.K., M.M. and A.R. performed NMR experiments and analyzed NMR results, S.A.K., Y.S., E.N.G.M. synthesized the peptide, M.C. and M.M.B.H. performed AFM experiments, S.A.K., P.W. and J.R.B. performed all other biophysical experiments, S.A.K., J.R.B. and A.R. analyzed all the results and wrote the paper. S.A.K., J.R.B. and A.R. designed the project and A.R. directed the research. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Kotler, S., Brender, J., Vivekanandan, S. et al. High-resolution NMR characterization of low abundance oligomers of amyloid-β without purification. Sci Rep 5, 11811 (2015). https://doi.org/10.1038/srep11811
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11811
This article is cited by
-
Fluorescence-based techniques for the detection of the oligomeric status of proteins: implication in amyloidogenic diseases
European Biophysics Journal (2021)
-
Epitope Mapping by NMR of a Novel Anti-Aβ Antibody (STAB-MAb)
Scientific Reports (2019)
-
Transthyretin Aggregation Pathway toward the Formation of Distinct Cytotoxic Oligomers
Scientific Reports (2019)
-
His6, His13, and His14 residues in Aβ 1–40 peptide significantly and specifically affect oligomeric equilibria
Scientific Reports (2019)
-
Unpacking the aggregation-oligomerization-fibrillization process of naturally-occurring hIAPP amyloid oligomers isolated directly from sera of children with obesity or diabetes mellitus
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
