
Abstract
The lifespan of an acute brain slice is approximately 6–12 hours, limiting potential experimentation time. We have designed a new recovery incubation system capable of extending their lifespan to more than 36 hours. This system controls the temperature of the incubated artificial cerebral spinal fluid (aCSF) while continuously passing the fluid through a UVC filtration system and simultaneously monitoring temperature and pH. The combination of controlled temperature and UVC filtering maintains bacteria levels in the lag phase and leads to the dramatic extension of the brain slice lifespan. Brain slice viability was validated through electrophysiological recordings as well as live/dead cell assays. This system benefits researchers by monitoring incubation conditions and standardizing this artificial environment. It further provides viable tissue for two experimental days, reducing the time spent preparing brain slices and the number of animals required for research.
Similar content being viewed by others

Introduction
Following the pioneering work of Henry McIlwain in the early 50's and 60's1,2,3, the in vitro brain slice preparation has become an accepted and powerful experimental approach in the field of neuroscience. Indeed much of our understanding of neuronal function at the cellular and synaptic level is derived from this approach. The brain slice preparation remains one of the most commonly used experimental preparations in neuroscience. It is employed to investigate questions across the neuroscience spectrum, including immunohistochemistry, brain anatomy, bio-molecular and pharmacological studies to investigate channelopathies and electrophysiological studies to characterize properties of individual neurons and synapses, along with glial and neuronal networks4,5,6,7.
In vitro brain slice preparations provides a means of examining metabolic parameters and electrophysiological properties without contamination from anaesthetics, muscle relaxants or intrinsic regulatory substances. The rapid preparation time further avoids the prolonged use of anaesthetics. The fact that brain slices maintain their structural integrity, unlike cultures or cell homogenates, allows the study of specific circuits and brain networks in isolation4,8, such as the thalamocortical pathway9,10. The stability of electrophysiological recording in acute slices is superior to in vivo recordings as the heartbeat and respiration of the experimental animal are eliminated, which also allow longer periods of cellular recording. Moreover, direct visualization of the slice enables researchers to locate, identify and easily access the cells being studied11 and also allows for local drug application, which is otherwise blocked by the blood brain barrier.
In recent years, the use of brain slices has greatly increased our knowledge about the mammalian central nervous system and is predicted to remain a valuable experimental method into the future5. However, this experimental method has a number of limitations that constrain its use. One of the major limitations is the lifespan of a brain slice, as this limits the time available to study the neuronal properties of the slice. Normally, the lifespan of a brain slice, from either a rat or a mouse, is limited to 6–12 hours. Moreover, it has been shown that the vast majority of the cells in hippocampal and neocortical thin slices can only be maintained in a healthy state for about 4 hours12. The main reasons for this short life span can be divided into two main categories: external properties and internal properties.
The external properties that may decrease cell viability in the slice may include changes in pH, temperature, oxygen and glucose levels13,14,15. Furthermore, as the slice is typically maintained in a non-sterile environment, acute slices are environmentally defenceless and vulnerable to increased bacteria numbers that release endotoxins such as lipopolysaccharide, leading to neurodegeneration and impacting cell survival16,17. Antibiotics can be used to reduce bacteria levels, however the addition of antibiotics poses a problem since many antibiotics have been shown to activate neurons18, hence impacting on cellular physiology and potentially biasing results.
Bacterial numbers in the recovery chamber increase with time, which is mainly due to the fact that the ideal conditions for maintaining tissues are also ideal for bacterial growth. Bacteria display a characteristic four-phase pattern of growth in liquid media (reviewed by Zwietering and colleagues19). The initial Lag Phase is a period of slow growth during which the bacteria are adapting to the conditions in the fresh medium. This is followed by a Log Phase during which bacterial growth is exponential, doubling every replication cycle. The Stationary Phase occurs when the supply of nutrients becomes a limiting factor and the rate of multiplication equals the rate of death. Finally, the Logarithmic Decline Phase occurs when bacteria die faster than they are replicated. Usually, recordings from brain slices are constrained to the Lag phase, in which the amount of bacteria is low and not affecting cell viability.
Bacteria stimulate glial cells to produce Nitric Oxide (NO) as part of the antimicrobial immune response to different toxins such as lipopolysaccharides, lipopeptides and other cytokines20. Previous studies showed that NO levels produced in glial cells increased dramatically over time due the release of bacterial lipopeptides and lipopolysaccharides, accumulating to a massive level of 40 mMol/μg protein after 12 hrs20,21,22. These studies imply that under regular artificial cerebrospinal fluid (aCSF) incubation conditions, bacterial levels increase over time, reaching the Log Phase after 6–12 hrs and results in accelerated cell death.
Internal properties include tissue deterioration as a secondary injury process that follows damage caused by the slicing procedure itself. Toxicants such as excitatory amino acids (EAA) released from the damaged and dead cells during the slicing procedure leads to excitotoxicity and neuronal vulnerability23.
Potentially, the key to maintaining a healthy slice over a long period is the condition of the solution in which the slices are incubated. The medium in which the slice is maintained is under the full control of the investigator. Unfortunately, there is no “gold standard” recovery incubation chamber that encapsulates the control of temperature, pH, NO and bacteria numbers. The lack of such a chamber may be a factor in any discrepancy in results reported across different labs.
Attempts have been made previously to enhance brain slice viability by improving the metabolism of the slices through the addition of adenylate precursors, amino acids or vitamins (reviewed by Reid et al., 198814). Although neuronal extracellular recordings following 20–24 hours were occasionally reported, their success rate was very low13,24,41,42,43. These efforts to extend the lifespan of brain slices were not successful, as the slices became increasingly fragile with time, lost their structural integrity and dissociated into monolayers of cells after prolonged incubation14,25.
We hypothesised that the lifespan of acute brain slices could be dramatically extended by slowing bacterial growth using hypothermia and reducing bacteria numbers by irradiating the aCSF with UV light. As the intrinsic membrane conductances and voltage gated channel kinetics in neurons are also dependent on temperature26, we further hypothesised that reducing the temperature in the recovery incubation chamber would also slow the deterioration of neurons.
Results
Under our experimental conditions, two types of gram-negative bacteria were identified: Pseudomonas species and stenotrophomonas maltophilia. These types of bacteria are mesophilics, hence their optimal growth conditions are 28°C for Pseudomonas27 and 35°C for stenotrophomonas maltophilia28. Also, as both bacteria types are neutrophils, their optimal growth is under a neutral pH of 6.5–8.529 making brain slice recovery chambers a perfect breeding habitat. In order to assess the impact of UVC light and low temperature on bacterial growth, we compared two experimental regimes. In the Braincubator (Figure 1), slices were kept at 15–16°C and the aCSF was constantly filtered through a UVC chamber, while in control conditions, slices were kept at room temperature (~22°C) and without UVC filtration. As seen in figure 2, under control conditions, the exponential bacterial growth (Log phase) in both slices and solution started between 12–16 hrs, reaching a maximal growth after 36 hrs. Although bacterial growth in the Braincubator was comparable to control conditions until 6 hrs, it showed a clear divergence from 12 hrs onwards (p < 0.0001; Mixed two-way ANOVA), hence extending the growth Lag Phase.

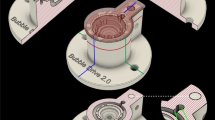
Schematic diagram of the UVC (a) and Main (b) chambers of the Braincubator.

Bacterial growth over time.
(a) IR-DIC image of brain slice surface after 30 hours under control conditions indicating bacterial colonies (white arrows), which later lead to necrotic tissue. See also supplementary video 1. (b) Graph depicting the bacterial numbers over time following incubation with either control (n = 4) or Braincubator (n = 4) conditions. Exponential square root fit was 0.9 for both control measurements and 0.97 for Braincubator data. (c) Expansion of the area marked in B depicting the significant changes started after 12 hrs. Data displayed as Mean ± S.E.M (** P < 0.01; ***P < 0.001, mixed two-way ANOVA).
Brain slice viability in the Braincubator
To evaluate brain slice viability, we co-incubated the slices with the dead cell marker propidium Iodide (PI)30,31 and the DNA specific fluorescent probe DAPI. Slice viability was assessed as the ratio of live cells (PI negative) out of the total visible cells (DAPI positive + PI positive) in the field of view. To avoid discrepancy between different areas in the slice, we confined our measurement to the cerebral cortex (Fig. 2). We have imaged 54 slices under Braincubator and 54 slices under control conditions. Acute brain slices were imaged at time points of 1, 6, 12, 16, 20, 24, 28, 32 and 36 hours post slicing. Under Braincubator conditions, slice deterioration was reduced significantly compared to slices incubated in the control recovery chamber (compare supplementary video 1 and supplementary video 2). After one hour, the average cell viability ratio in the Braincubator was 73 ± 4%; (n = 9) and no changes were observed between control and Braincubator slices (Fig. 3). However, during later incubation periods (>6 hrs), a significant difference of the live/total cell ratio was detected (11.3%; p ≤ 0.01; Two-way ANOVA), reaching a maximal difference of 28% after 24 hours (45 ± 3% vs 17 ± 3%, p < 0.0001, two-way ANOVA). Moreover, after 24 hours under control conditions, some of the dead cells morphology was changed, depicting extensive fragility, probably due to membrane deterioration (Fig. 3C).

Slices viability over time.
Confocal microscopic serial images (×40) of acute brain slices following different incubation time at control (a) and Braincubator (b) conditions. All sections were sliced at the same time. The combination of DAPI (blue) and PI (red) allows the simultaneous visualisation of all cells in the slice. Scale bar = 50 μm. b) Graph depicting the slice viability as assessed from the cell death/total assay. For each time point n = 6, data displayed as Mean ± S.E.M (c) Expansion of the marked areas in A depicting the morphology of dead cells following 24 hours in control (Top) and Braincubator (Bottom).
Electrophysiological properties of layer 5 pyramidal neurons in the Braincubator
In order to evaluate individual cell viability following different incubation times in the Braincubator, we monitored both passive (Vm; Rin; Tau) and active (action potential properties) membrane properties of layer V pyramidal neurons. All cells had the morphology of pyramidal neurons and responded to a depolarizing current stimulus with tonic, adapting patterns of action potentials, which categorize them as regular spiking (RS) neurons32,33,34 (Fig. 4). Only neurons with resting membrane potentials greater than −60 mV and action potentials that overshot 0 mV were included in the analysis. The recorded neurons were divided into four groups according to the time since slicing (Table 1). As no significant differences were detected between neurons from slices that were incubated in the Braincubator for less than 3 hours to neurons incubated under control conditions for the same period of time (two tailed t-test; Table 1), we referred to the first group (1–3 hours) as a baseline and compared these basal cellular characteristics to the other groups (using one way ANOVA). In general, all passive and active membrane properties were similar to previous reports of layer V pyramids32,35,36 and remained constant between different incubation time groups (Table 1). We also compared their membrane resonance frequency, which is a property determined by the interplay between neuronal active (voltage gated currents) and passive (capacitance; leak currents) properties and describes the ability of neurons to respond selectively to inputs at preferred frequencies37. In cortical neurons, the resonance frequency is dependent on the interplay between two currents, a slowly activating and non-inactivating K+ current and a fast persistent Na+ current38. The average resonance frequency ranged between 1 and 3 Hz (average 1.7 ± 0.1 Hz; n = 38), as reported previously35 and did not differ between groups (p < 0.84, one way ANOVA).

Active and passive membrane properties of layer V pyramidal neurons.
(a) I-V traces. Increasing step currents of 500 ms were injected into the soma through the recording electrode to reveal the input resistance and firing properties. (b) The resonance frequency was measured by injecting a chirp stimulation of 60 pA (peak to peak). (c) Sample traces of spontaneous synaptic activity recorded from a neuron after 31 hrs in the braincubator.
Miniature postsynaptic currents (mPSC's) are generated as a consequence of spontaneous vesicular release, hence reflecting the functional synapses onto layer V pyramidal neurons. We therefore assessed mEPSC's characteristics as a means of assessing the spontaneous activity of individual neurons in the slice. These characteristics were similar between neurons incubated in the Braincubator and under control conditions for periods of less than 3 hours (two tailed student t test), as well as to previous studies39,40 and did not show any significant changes between different time groups (one-way ANOVA), implying that the majority of excitatory synaptic inputs onto layer V neurons were intact and functional even 36 hrs after the slicing procedure (Table 1).
Discussion
We have studied the impact of a controlled environment on the long-term viability of brain slices, to enable the isolation and recording from neurons in the slices for up to 36 hours. Our results indicate that a significant extension of the viability of brain slices can be achieved through the use of a specialised recovery chamber, termed the Braincubator, that allowed a) the reduction of the temperature of the extracellular aCSF in the recovery chamber and b) reducing bacterial growth in the aCSF by UVC irradiation.
We have shown that over a period of 24–36 hours there is a substantial increase in the numbers of bacteria in brain slices prepared using conventional slicing and incubation techniques and that this bacterial growth is correlated with the decline in slice viability over that period. The two types of bacteria found in the slices in this study were gram-negative aerobic bacteria that, along with gram positive bacteria, can induce neuronal injury that is necrotic and apoptotic44. The mechanisms by which bacteria cause neuronal damage and cell deterioration are not fully understood, however, several reports have identified the involvement of bacterial secretion of toxins such as TNF-α, H2O2 and pneumolysin44,45 serving as apoptosis inducing factors (AIF) by leading to Ca2+ translocation and mitochondrial damage44,46.
To impede the growth of bacteria in the brain slices we used two approaches: reduced temperature and UVC irradiation of the solution in which the brain slice was placed. Temperature is the cardinal factor controlling the rate of growth, where other factors such as nutrients and water are not limited47. Each microbial species requires a temperature growth range that is determined by the heat sensitivity of its particular enzymes, membranes, ribosomes and other components. The species of bacteria found in our slices are mesophilic and therefore have an optimum growth temperature at around 37°C, with a minimum temperature of 10–15°C48. The temperature of the solution in our incubation chamber was lowered to 16°C to restrict the bacterial growth to the Lag phase. We did not investigate the effect of temperature alone on the growth of bacteria in the brain slice, however there is a substantial amount of work on the protective role of hypothermia on neuronal survival49, possibly via reduction of reactive oxygen species production.
The low temperature was combined with UVC irradiation of the solution in the incubation system. UVC irradiation leads to bonding of adjacent thymine bases, thus create a dimmer that prevents DNA replication50. Hence, UV disinfection is a physical process rather than a chemical disinfectant, which eliminates the need to introduce toxic and hazardous chemicals that might damage the tissue.
Our assessment of the viability of the brain slices following treatment in the Braincubator involved electrophysiological recording from cortical neurons within the slice. We observed that the resting membrane potential of individual neurons remained constant, illustrating that the electrochemical balance of the main contributing ions, Na+, K+ and chloride over different incubation times imply that the extracellular environment remains constant, as also shown by Mass-spectrometry (Supplementary Fig. 1). We recorded from layer 5 pyramidal neurons that are projection neurons from the cerebral cortex to subcortical targets51. These neurons are part of local networks termed cortical modules that along with other interconnected neurons in the module form recurrent neural networks. An early and sensitive indication of slice damage may be the loss of recurrent synaptic activation15 in neurons within a cortical module, as such we assessed the level of spontaneous synaptic activity within neurons to gauge the state of the synaptic connectivity of neurons in the brain slices. The frequency of spontaneous miniature excitatory postsynaptic currents (mEPSC's) is correlated with the extent of viable synaptic connections in the cortical network52. Our results indicate that neurons in brain slices incubated in the Braincubator retained their functional excitatory synaptic connections, as evidenced by the lack of significant changes in both frequency and amplitude of mEPSC's.
Although the overall slices viability after 20 hrs in control conditions was comparable to slices incubated for 36 hrs in the Braincubator (Figure 3b), we could rarely see viable cells under IR microscopy (−20–70 μm), hence it was extremely difficult to patch these cells. This is probably due the fact that most cells proximal to the slice surface were susceptible to the toxins released by the bacteria (that accumulated on the surface, see figure 2a) and the viable cells were located deep in the slice. Extending the useful period over which data can be gathered from neurons within a brain slice provides the opportunity to ask questions about neuron function not possible with conventional preparation systems and has the potential to reduce the number of animals that need to be scarified.
Methods
Animals
For this study we used 2–5 week old Wister rats. All animals were healthy and handled with standard conditions of temperature, humidity, twelve hours light/dark cycle, free access to food and water and without any intended stress stimuli. All experiments were approved and performed in accordance with the University of Western Sydney committee for animal use and care guidelines (Animal Research Authority #A9452).
Slice preparation and recording
Wister rats were deeply anesthetized by inhalation of isoflurane (5%), decapitated and their brains were quickly removed and placed into ice-cold physiological solution (artificial CSF) containing (in mM): 125 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 2 CaCl2, 25 NaHCO3, 25 dextrose and saturated with carbogen (95% O2 -5% CO2 mixture; pH 7.4). Parasagittal brain slices (300 μm thick) were cut with a vibrating microtome (Camden Instruments, UK) and transferred to a holding chamber containing carbogenated aCSF for 30 min at 35°C. Sequentially, slices were either allowed to cool to room temperature (~22°C) in the same recovery chamber (Control, built as reported by11) or transferred into a custom-made incubation system that closely monitored and controlled pH levels, carbogen flow and temperature as well as irradiating bacteria through a separate UV chamber (Fig. 1, the Braincubator™). Slices were kept in the incubation system (either control or in the Braincubator) for at least 30 min before any measurement.
Evaluating slice viability
Acute slices were incubated for 15 min with the selective dead cell fluorescent marker propidium iodide (PI, 1 μg/ml) as reported previously30. For total cell counts, slices were co-incubated with the nuclear marker DAPI (1 μg/ml). Following incubation, slices were washed with fresh aCSF for 10 min. Images were acquired using a Zeiss LSM-510 Meta confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 40×-oil immersion objective in the inverted configuration. Z plane optical sections (4 μm) were taken at −20 to −70 μm depth from the surface of the cerebral cortex to produce an image stack. The DAPI signal was obtained using Argon laser excitation at 488 nm; PI was excited with 543-nm HeNe laser. Images were visualized using ZEN software and processed using ImageJ. Slice viability was assessed as the ratio between dead/total cells in the visual field.
Electrophysiological recording and stimulation
The recording chamber was mounted on an Olympus BX-51 microscope equipped with IR/DIC optics. Following the incubation period in the Braincubator at 16°C, slices were mounted in the recording chamber for a minimum of 15 min to allow warming up to room temperature (~22°C) and were constantly perfused (2–3 ml/min) with oxygenated solution as reported previously7. Whole cell recordings were performed from the soma of layer 5 pyramidal neurons in the somatosensory cortex with patch pipettes (5–7 MΩ) containing (in mM) 130 K-Methansulfate, 10 HEPES, 0.05 EGTA, 7 KCl, 0.5 Na2GTP, 2 Na2ATP, 2 MgATP, 7 phosphocreatine, 0.1 Alexa Fluor-488 (Molecular Probes) and titrated with KOH to pH 7.2 (~285 mOsm). Stimulation protocols were designed using the pClamp 10 software suit (Molecular devices, Sunnyvale, CA) and stimulation currents were injected through the recording electrodes. Voltages were recorded in current clamp mode using a multiclamp 700B dual patch-clamp amplifier (Axon instruments, Foster city, CA), digitally sampled at 30–50 kHz, filtered at 10 kHz and analysed off-line using pClamp software. The access resistance was corrected on-line and recordings were included in the analysis if the access resistance was <30MΩ. Cells were considered stable and suitable for analysis if the access resistance, input resistance and resting membrane potential did not change by more than 20% from their initial value during recording. At the termination of each experiment, the location and morphology of neurons were examined by fluorescence microscopy and digitally recorded (ROLERA-XR, Q-Imaging).
Determining the resonance frequency
In order to reveal the resonance frequency of the cells, we used the impedance analysis described by Gutfreund and colleagues38. In short, a 20 second subthreshold sinusoidal current with a linear increase in frequency from 0.1–20 Hz (chirp stimulation) was applied through the recording electrode. The impedance amplitude profile (ZAP) was generated by transforming the input current (I) and the voltage response (V) into the frequency domain using a fast Fourier transform (FFT) and then dividing the voltage transformation FFT(V) by the current stimulus transformation FFT(I). The stimulus file was generated by Python based software, imported into pClamp and applied as described above. The resonance frequency (fR) was determined as the peak of the ZAP profile.
Detecting spontaneous activity
To evaluate the functional activity of the slice, we have recorded the spontaneous miniature excitatory postsynaptic currents (mEPSC's) in layer 5 pyramidal neurons. mEPSC's were recorded in the whole-cell voltage-clamp configuration at a holding potential of −70 mV to avoid NMDA conductance and in the presence of 1 μM tetrodotoxin (TTX) and 50 μM picrotoxin, to block GABAergic receptors. mEPSC's were analyzed off-line using pClamp 10 software. The frequency and amplitude of events were calculated over 5 min periods.
Bacteria culturing and detection
aCSF and acute brain slice samples were collected from either the Braincubator or control recovery chamber at set time points (1, 6, 12, 16, 20, 24, 30 & 36 hrs) after slicing. For aCSF samples, 1 mL of solution was aspirated by sterile syringe, diluted and cultured on agar plates for 24 hrs at 37°C. For brain slices, 1 mL of solution together with the brain slice was aspirated from each group at set time point by sterile syringe, smashed into a thick suspension and centrifuged (1000 rpm for 5 min). Supernatant was collected, diluted and cultured on agar plates for 24 hrs at 37°C. Following incubation, colony-forming units (CFU) were counted and inspected qualitatively for common colonies. Sequentially, colonies were smeared (in triplicate) with a sterile toothpick onto a matrix-assisted laser desorption/ionization (MALDI) stainless steel target plate and allowed to dry at room temperature in a fume hood. 1 μL of 70% formic acid was overlaid onto the dried sample and allowed to dry. Automated spectrum acquisition was performed using a Daltonik MALDI Biotyper (Bruker, Germany) and analysed with Flex Analysis MALDI Biotyper software (Bruker, Germany).
The Braincubator – a recovery incubation system
To closely monitor and control the environment of the brain slices, we have built a closed loop incubation system consisting of two chambers (Fig. 1). Slices were placed in the main chamber, which contained probes for pH and temperature. The main chamber was covered with polystyrene foam for thermal insulation and UVC blockade. The second chamber (UVC chamber) was isolated from the main chamber and exposed to 1.1 W UVC light (254 nm, 5W/2P Philips Ultra Violet sterilizer UV lamp) in order to eradiate bacteria floating in the solution. UVC light timing was controlled via a programmable timer using a random feature which turns ON at times varying between 15 and 26 min every 15 to 30 min. The UVC chamber volume was set to 120 ml and the flow rate was set to 12 ml/min. Application of UVC was intermittent and randomised to avoid excessive heating of the aCSF temperature and to prevent the increase of bacterial UVC resistance. The UVC chamber was covered with aluminium foil to prevent UVC illumination outside the chamber, which can damage neuronal tissue. A peristaltic pump circulated the solution through the two chambers and a Peltier thermoelectric cold plate cooler (TE technology, Traverse city, MI) enabled the solution to be either cooled or heated to temperature in the range of 0–50°C.
Statistical analysis
Data is reported as mean ± S.E.M. Statistical comparisons were done using two tailed unpaired student t test; one-way ANOVA and two-way ANOVA according to the experimental design.
References
McIlwain, H., Buchel, L. & Cheshire, J. D. The inorganic phosphate and phosphocreatine of Brain especially during metabolism in vitro. Biochem. J. 48, 12–20 (1951).
Yamamoto, C. & McIlwain, H. Electrical activities in thin sections from the mammalian brain maintained in chemically-defined media in vitro. J. Neurochem. 13, 1333–43 (1966).
Collingridge, G. L. The brain slice preparation: a tribute to the pioneer Henry McIlwain. J. Neurosci. Methods 59, 5–9 (1995).
Colbert, C. M. Preparation of cortical brain slices for electrophysiological recording. Methods Mol. Biol. 337, 117–25 (2006).
Khurana, S. & Li, W.-K. Baptisms of fire or death knells for acute-slice physiology in the age of “omics” and light. Rev. Neurosci. 24, 527–536 (2013).
Buskila, Y. et al. Enhanced Astrocytic Nitric Oxide Production and Neuronal Modifications in the Neocortex of a NOS2 Mutant Mouse. PLoS One 2, 9 (2007).
Buskila, Y. & Amitai, Y. Astrocytic iNOS-dependent enhancement of synaptic release in mouse neocortex. J. Neurophysiol. 103, 1322–1328 (2010).
Luhmann, H. J. & Kilb, W. Isolated Central Nervous System Circuits. Neuromethods 73, 301–314 (2012).
Agmon, A. & Connors, B. Thalamocortical responses of mouse somatosensory (barrel) cortexin vitro. Neuroscience 41, 365–379 (1991).
Agmon, A. & Connors, B. W. Correlation between Intrinsic Firing Patterns and Thalamocortical Synaptic Responses of Neurons in Mouse Barrel Cortex. J. Neurosci. 12, 319–329 (1992).
Gibb, A. & Edwards, F. Patch clamp recording from cells in sliced tissues. Microelectrode Tech. 255–274 (1994).
Fukuda, A., Czurk, A., Hidal, H., Muramatsu, K. & Nishino, H. Appearance of deteriorated neurons on regionally different time tables in rat brain thin slices maintained in physiological condition. Neurosci. Lett. 184, 13–16 (1995).
Schurr, A., Reid, K. & Tseng, M. A Dual chamber for comparative studies using the brain slice preperation. Comp. Biochem. physiol 82, 701–704 (1985).
Reid, K. H., Edmonds, H. L., Schurr, a., Tseng, M. T. & West, C. a. Pitfalls in the use of brain slices. Prog. Neurobiol. 31, 1–18 (1988).
Dingledine, R., Dodd, J. & Kelly, J. The in vitro brain slice as a useful neurophysiological preperation for intracellular recording. J. Neurosci. Methods 2, 323–362 (1980).
Qin, L., Wu, X., Block, M., Liu, Y. & Breese, G. Systemic LPS Causes Chronic Neuroinflammation and Progressive Neurodegeneration. Glia 55, 453–462 (2007).
Ahmed, S.-H. et al. Effects of Lipopolysaccharide Priming on Acute Ischemic Brain Injury Editorial Comment. Stroke 31, 193–199 (2000).
Grøndahl, T. O. & Langmoen, I. A. Epileptogenic effect of antibiotic drugs. J. Neurosurg. 78, 938–43 (1993).
Zwietering, M. Modeling of the Bacterial Growth Curve. Appl. Enviromental Microbiol. 56, 1875–1881 (1990).
Terenzi, F. et al. Bacterial Lipopeptides Induce Nitric Oxide Synthase and Promote Apoptosis through Nitric Oxide-independent Pathways in Rat Macrophages. J. Biol. Chem. 270, 6017–6021 (1995).
Knowles, R. G., Merrett, M., Salter, M. & Moncada, S. Differential induction of brain, lung and liver nitric oxide synthase by endotoxin in the rat. Biochem. J. 270, 833–6 (1990).
Moncada, S. & Higgs, E. molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 9, 1319–1330 (1995).
Schurr, A., Payne, R., Heine, M. & Rigor, B. Hypoxia, excitotoxicity and neuroprotection in hippocampal slice preperation. J. Neurosci. Methods 59, 129–138 (1995).
Schurr, A., Reid, K. H., Tseng, M. T. & Edmonds, H. L. The stability of the hippocampal slice preparation " an electrophysiological and ultrastructural analysis. Brain Research 297, 357–362 (1984).
Gähwiler, B. Slice cultures of cerebellar, hippocampal and hypothalamic tissue. Experientia 40, 235–243 (1984).
Thompson, S. M., Masukawa, L. M. & Prince, D. a. Temperature dependence of intrinsic membrane properties and synaptic potentials in hippocampal CA1 neurons in vitro. J. Neurosci. 5, 817–24 (1985).
Jaouen, T. & Dé, E. Pore size dependence on growth temperature is a common characteristic of the major outer membrane protein OprF in psychrotrophic and mesophilic Pseudomonas species. Appl. Enviroumental Microbiol. 70, 6665–6669 (2004).
Denton, M. & Kerr, K. Microbiological and Clinical Aspects of Infection Associated with Stenotrophomonas maltophilia. Clin. Microbiol. Rev. 11, 57–80 (1998).
Thomas, K. L., Lloyd, D. & Boddy, L. Effects of oxygen, pH and nitrate concentration on denitrification by Pseudomonas species. FEMS Microbiol. Lett. 118, 181–6 (1994).
Buskila, Y., Farkash, S., Hershfinkel, M. & Amitai, Y. Rapid and reactive nitric oxide production by astrocytes in mouse neocortical slices. Glia 52, 169–176 (2005).
Sasaki, D. T., Dumas, S. E. & Engleman, E. G. Discrimination of viable and non-viable cells using propidium iodide in two color immunofluorescence. Cytometry 8, 413–20 (1987).
Chagnac-Amitai, Y., Luhmann, H. J. & Prince, D. a. Burst generating and regular spiking layer 5 pyramidal neurons of rat neocortex have different morphological features. J. Comp. Neurol. 296, 598–613 (1990).
Zhu, J. J. & Connors, B. W. Intrinsic firing patterns and whisker-evoked synaptic responses of neurons in the rat barrel cortex. J. Neurophysiol. 81, 1171–83 (1999).
Schubert, D. et al. Layer-specific intracolumnar and transcolumnar functional connectivity of layer V pyramidal cells in rat barrel cortex. J. Neurosci. 21, 3580–92 (2001).
Buskila, Y., Morley, J. W., Tapson, J. & van Schaik, A. The adaptation of spike backpropagation delays in cortical neurons. Front. Cell. Neurosci. 7, (2013).
Gil, Z., Connors, B. W. & Amitai, Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron 19, 679–86 (1997).
Hutcheon, B. & Yarom, Y. Resonance, oscillation and the intrinsic frequency preferences of neurons. Trends Neurosci. 23, 216–222 (2000).
Gutfreund, Y., Yarom, Y. & Segev, I. Subthreshold oscillations and resonant frequency in guinea-pig cortical neurons: physiology and modelling. J. Physiol. 483, 621–40 (1995).
Fortin, D. a. & Levine, E. S. Differential effects of endocannabinoids on glutamatergic and GABAergic inputs to layer 5 pyramidal neurons. Cereb. Cortex 17, 163–74 (2007).
Marek, G. J. & Aghajanian, G. K. 5-HT 2A receptor or a 1 -adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur. J. Pharmacol. 197–206 (1999).
Satinoff, E. et al. Do the suprachiasmatic nuclei oscillate in old rats as they do in young ones? Am. J. Physiol. 265, R1216–R1222 (1993).
Palovcik, R. a. & Phillips, M. I. A constant perfusion slice chamber for stable recording during the addition of drugs. J. Neurosci. Methods 17, 129–39 (1986).
Tcheng, T. & Gillette, M. A novel carbon fiber bundle microelectrode and modified brain slice chamber for recording long-term multiunit activity from brain slices. J. Neurosci. Methods 69, 163–169 (1996).
Braun, J. S. et al. Pneumococcal pneumolysin and H 2 O 2 mediate brain cell apoptosis during meningitis. J. Clin. Invest. 109, 19–27 (2002).
Zychlinsky, a. & Sansonetti, P. Perspectives series: host/pathogen interactions. Apoptosis in bacterial pathogenesis. J. Clin. Invest. 100, 493–5 (1997).
Nau, R. & Brück, W. Neuronal injury in bacterial meningitis: mechanisms and implications for therapy. Trends Neurosci. 25, 38–45 (2002).
Ratkowsky, D. & Olley, J. Relationship between temperature and growth rate of bacterial cultures. J. Bacteriol. 149, 1–5 (1982).
Todar, K. Online textbook of bacteriology. 2012 www.textbookofbacteriology.net (13/05/2014).
Kil, H. Y., Zhang, J. & Piantadosi, C. A. Brain temperature alters hydroxyl radical production during cerebral ischemia/reperfusion in rats. J. Cereb. blood flow Metab. 16, 100–6 (1996).
Cadet, J., Sage, E. & Douki, T. Ultraviolet radiation-mediated damage to cellular DNA. Mutat. Res. 571, 3–17 (2005).
Markram, H. A network of tufted layer 5 pyramidal neurons. Cereb. Cortex 7, 523–33 (1997).
Segal, M. Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur. J. Neurosci. 31, 2178–84 (2010).
Acknowledgements
We thank Sindy Kueh, James Wright and David Harman (the UWS Mass Spectrometry Facility) for technical assistance. This work was supported by seed funding grant (UWS) to Y.B. and the innovation office at UWS.
Author information
Authors and Affiliations
Contributions
Y.B., P.P.B., J.W.M., J.T. and A.v.S. designed the project. Y.B. and P.P.B. built the Braincubator. Y.B. performed and analysed the electrophysiological recordings. M.B. performed and analysed the bacterial growth and confocal imaging. All authors wrote and approved the paper.
Ethics declarations
Competing interests
Yes there is potential Competing Interest. Dr Buskila and Dr Breen have applied for a patent on the Braincubator™. Mr Barton, Prof. van Schaik, Prof. Tapson and Prof. Morley declare no potential conflict of interest.
Electronic supplementary material
Supplementary Information
Supplementary Information
Supplementary Information
Supplementary video 1
Supplementary Information
Supplementary video 2
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Buskila, Y., Breen, P., Tapson, J. et al. Extending the viability of acute brain slices. Sci Rep 4, 5309 (2014). https://doi.org/10.1038/srep05309
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep05309
This article is cited by
-
Chronic neuroinflammation during aging leads to cholinergic neurodegeneration in the mouse medial septum
Journal of Neuroinflammation (2023)
-
Three dimensional microelectrodes enable high signal and spatial resolution for neural seizure recordings in brain slices and freely behaving animals
Scientific Reports (2021)
-
Metabolic Changes in Brain Slices over Time: a Multiplatform Metabolomics Approach
Molecular Neurobiology (2021)
-
Roles Played by the Na+/Ca2+ Exchanger and Hypothermia in the Prevention of Ischemia-Induced Carrier-Mediated Efflux of Catecholamines into the Extracellular Space: Implications for Stroke Therapy
Neurochemical Research (2020)
-
Increasing cellular lifespan with a flow system in organotypic culture of the Laterodorsal Tegmentum (LDT)
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
