
Abstract
We describe a screen for cellular response to drugs that makes use of haploid embryonic stem cells. We generated ten libraries of mutants with piggyBac gene trap transposon integrations, totalling approximately 100,000 mutant clones. Random barcode sequences were inserted into the transposon vector to allow the number of cells bearing each insertion to be measured by amplifying and sequencing the barcodes. These barcodes were associated with their integration sites by inverse PCR. We exposed these libraries to commonly used cancer drugs and profiled changes in barcode abundance by Ion Torrent sequencing in order to identify mutations that conferred sensitivity. Drugs tested included conventional chemotherapeutics as well as targeted inhibitors of topoisomerases, poly(ADP-ribose) polymerase (PARP), Hsp90 and WEE1.
Design Type(s) | screening campaign • stimulus or stress design |
Measurement Type(s) | transposon integration • genetic mapping data |
Technology Type(s) | multiplexed sequencing library • DNA sequencing by synthesis |
Factor Type(s) | Mutation Abnormality • drug |
Sample Characteristic(s) | Mus musculus • embryonic stem cell line |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others

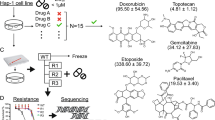

Background & Summary
Haploid mammalian cells only contain one allele of each gene. This property greatly facilitates forward genetic screens, as mutations that would be recessive in a diploid background can directly result in a phenotype. Two types of mammalian haploid cell lines have been used for genetic screens: those derived from a near-haploid human leukaemia1,2 and haploid embryonic stem cells derived from activated oocytes3,4. Both of these cell types have previously been used for forward genetic screens using insertional mutagens such as transposons and retroviruses. The screens have mainly looked for mutants with selectable phenotypes, either directly (e.g., drug or pathogen resistance1–3,5) or via a suitable reporter6, although a screen for essential genes has been recently published7. We sought to extend these screening systems to look for drug sensitivity phenotypes.
Doing so requires either creating a large array of individual mutants or a system for monitoring the abundance of each mutant in a mixed pool (Fig. 1a). The latter strategy has been used extensively to screen cells with stably-integrated short hairpin RNA (shRNA) expression vectors, by sequencing the distinct shRNA target sequences by next-generation sequencing approaches. This yields read counts for each shRNA that are proportional to the number of cells with integrations of that vector8.

(a) Screen setup. (b) Structure of mutagenic transposon, showing gene trap splice acceptor construct and random barcode regions (N25). (c) Inverse PCR strategy to associate barcodes with integration sites.
To allow us to apply a similar approach to transposon-mutagenised haploid cells, we inserted a random 25 base pair barcode at each end of the transposon, and prepared a complex pool of transposon donor plasmids with different barcodes (Fig. 1b). Since the barcodes are of identical length and average base composition, they can be quantified accurately using a PCR and sequencing approach, allowing the relative number of cells with each barcode to be inferred and compared between samples. However, unlike the hairpin sequence in shRNA vectors, the barcode sequence is of no biological relevance and does not directly identify the disrupted gene in its associated mutant. We used inverse PCR to link barcodes to the disrupted genes that they represent (Fig. 1c). The inverse PCR products generated in this way have the random barcode sequence at one end and the transposon-genome junction at the other. These PCR amplicons were sequenced using a paired end strategy on an Illumina HiSeq 2000 and used to build a database that links barcode sequences to integration sites.
Mutants were generated by transfecting purified haploid embryonic stem cells with transposon and transposase under conditions that generate mainly single copy integrations. The resulting colonies were counted and the equivalent of 1,000 colonies pooled. These 1 k pools were then mixed to generate ten pools of approximately 10,000 mutants each. We carried out screens by exposing these 10 k pools to one of 13 drugs (Table 1) at a concentration designed to kill 50% of wild type cells over the screening period (eight days). Pools treated with DMSO (the drug vehicle) were maintained in parallel to control for differences in mutant growth rates that were not linked to drug exposure. After the drug exposure period we prepared genomic DNA from surviving cells and amplified the barcode sequences. These PCR products were sequenced using the Ion Proton platform, generating read counts that represented the abundance of each barcode. These were merged with the inverse PCR data to produce read counts for each integration site under the different treatment conditions. The edgeR R package9 was used to normalise the read counts and generate fold changes and P-values for differences in barcode frequency between drug exposed and DMSO exposed cells. Analysis methods are provided to query the data for multiple insertion sites affecting the same gene that are significantly enriched or depleted.
Methods
Vector construction
The transposon vector was assembled using InFusion cloning (for sequence see Supplementary Data). Random oligonucleotides were purchased from IDT with appropriate overhangs, and introduced into the vector by InFusion cloning. The cloning reaction was used to transform E. coli cells (over 2×106 transformed colonies produced), which were grown overnight in liquid culture with ampicillin and plasmids prepared (Qiagen maxi prep). A previously described hyperactive piggyBac transposase plasmid was used10.
Embryonic stem cell culture and transfection
Haploid mouse embryonic stem cells from 129 strain mice (H129.2) were obtained from the laboratory of Anton Wutz11. Cells were cultured as previously described12, using 2i+LIF medium up until the point of mutagenesis and conventional ES cell medium with 15% serum and LIF thereafter. Prior to transfection with the transposon plasmids, cells were purified by FACS based on forward and side scatter to enrich for haploid cells13. Cells were transfected with limiting amounts of transposon plasmid to obtain mainly single-copy integrations as previously described14.
Mapping of transposon integrations by inverse PCR
Inverse PCR was based on the previously described TRIP protocol15. Genomic DNA was prepared from 1 k mutant libraries and five micrograms digested separately with BfuCI (both transposon ends) and either HhaI (PB3 end) or RsaI (PB5 end). Digested DNA was diluted to 4.5 ml in T4 ligase buffer and ligated at 16 °C overnight using 800 units of T4 DNA ligase (NEB). Ligated DNA was precipitated by adding 0.1 volume 3 M sodium acetate and two volumes ethanol and centrifuging at 4,700 r.p.m. for 45 min. The DNA pellet was washed once with 70% ethanol and resuspended in 250 μl 5 mM Tris-HCl, pH 8. Primary PCR was carried out using Q5 polymerase (NEB) in a 50 μl reaction volume using 20 μl ligated DNA. Primer sequences and cycling conditions can be found in Supplementary Data. Samples were paired-end sequenced using a HiSeq 2000 instrument, with dark cycles to ignore the low-complexity PCR primer sequences. Barcodes were annotated and the genomic insertion site sequence extracted from the paired reads (inv-hiseq.rb). Insertion sites were mapped to the mouse genome (GRCm38) using bwa16 (implemented in map.rake) and associated gene information extracted using the ruby Ensembl API (https://github.com/jandot/ruby-ensembl-api, implemented in getgene.rb). Integration sites with fewer than 10 reads/million were discarded, and sites from the same library mapping to sites within 5 bp of each other were combined.
Barcode recovery and screen analysis
Barcodes were recovered by PCR with primers tailed with barcoded Ion Torrent sequencing adapters (Supplementary Data). The primers used were based on previously published sequences for shRNA vectors8,17. PCR reactions were run as previously described8 using a total of 30 μg genomic DNA (approximately 1,000 haploid genome equivalents per mutant) template in 38 PCR reactions per sample. Samples were gel-purified at room temperature as previously described18 and sequenced on Ion Torrent P1 chips (Life Technologies), loading up to 16 samples per chip in equal amounts. Samples from the same original library were sequenced on the same chip where possible. Read counts were generated for each barcode by parsing FASTQ files (implemented in barcode-pipe.rake). Barcodes with high similarity are likely to be the result of sequencing or PCR errors (this was supported by comparison with inverse PCR data) and were grouped and counted together (group.r). Fold changes and P-values (exact test based on negative binomial distribution) were calculated using the edgeR package9 and combined with barcode mapping data (implemented in script gg_analysis.r).
Validation
A combined Cas9-sgRNA lentiviral vector was made by cloning a double stranded oligonucleotide with appropriate overhangs (Supplementary Data) into pLentiCRISPR19 (Addgene 49535). JM8A3 ES cells20 were transduced, selected in puromycin and colonies picked and screened by PCR and Western blot to identify Ewsr1 mutants. MCF7 breast cancer cells were transfected with shRNA vectors as previously described21. Cell survival after drug exposure was determined using CellTiterGlo (Promega).
Code availability
Code used to generate and analyse data is deposited on github22.
Data Records
Mappings
Inverse PCR data was processed as above to generate hashup.gene files containing mapping information (Data Citation 1).
Barcode counts
Fastq files from barcode sequencing experiments were processed as above to generate tsv files for each 10 k library screen (Data Citation 2).
Inverse PCR product sequences
Fastq files containing unmapped inverse PCR products were filtered to exclude truncated reads and reads that did not start with the expected sequence to form fastqinv files (Data Citation 3).
Technical Validation
Confirmation of mutagenic activity of barcoded transposon
In a preliminary experiment to verify that the new transposon construct was mutagenic, we transfected haploid mouse embryonic stem cells with the barcoded gene trap vector and selected cells with stable integrations using G418 to create a test library. Cells with mutations in components of the mismatch repair pathway are resistant to the modified nucleotide 6-thioguanine (6-TG). Therefore we selected this test library in 2 μM 6-TG. Eighty surviving colonies were isolated. The transposon integrations were mapped by Splinkerette PCR23. In 43 clones with mappings, a known mismatch repair gene was disrupted (Table 2 and Supplementary Table 1). Furthermore, the entire canonical mismatch repair pathway was recovered (Msh2, Msh6, Mlh1, Pms2), with multiple insertions in Msh2 and Msh6 demonstrating effective genome-scale mutagenesis.
Coverage
Analysis of the full set of integrations revealed that the libraries approached saturation with 14,132 genes in total affected by a transposon insertion (Fig. 2a). As expected, integrations were biased towards genes that are highly expressed in mouse embryonic stem cells (Fig. 2b).

(a) Cumulative number of unique genes disrupted in libraries 1–10. (b) Breakdown of genes disrupted by expression in mouse embryonic stem cells, The fraction of genes in each category with at least one integration is plotted. Single cell RNA-seq expression data and categorisation have been previously published28.
Reproducibility
Per-barcode read counts were highly correlated in the DMSO-exposed replicates for each library (mean pairwise r=0.96, Fig. 3a).

(a) Concordance of read counts in two replicate DMSO-treated samples. (b) Schematic of spiking experiment. (c) Volcano plot of spiking experiment results. P values (exact test) and fold changes were calculated using edgeR. The point corresponding to the barcode in the spiked Brca2 mutant is labelled. (d) Volcano plot of barcodes corresponding to Ewsr1 integrations in all PB3 libraries exposed to the PARP inhibitor BMN 673. (e) Validation of Ewsr1 hit. CRISPR knockout ES cells (L5.11) are more sensitive to BMN 673 than parental JM8A3 cells. Surviving fraction is measured using CellTiterGlo after five days growth in the indicated drug concentration; mean and s.d. of five replicates is plotted. (f) Western blot demonstrating knockout of Ewsr1 in CRISPR mutant. (g,h) Knockdown of the human homologue, EWSR1, in MCF7 cells using shRNA causes BMN 673 sensitivity.
Proof-of-concept
Brca2 mutant cells are highly sensitive to the PARP inhibitor olaparib (Lynparza)24. ES cells with a targeted mutation in Brca2 have previously been described25. We tagged this cell line by transfecting it with a clonal transposon plasmid with a known barcode. These tagged cells were mixed with one of the 10,000-mutant libraries (library 2) at a 1:10,000 ratio (200 cells to 2 million library cells) to form a spiked library (L2-spk, Fig. 3b). Exposure of this spiked library to olaparib as described above revealed that the tagged barcode was the most depleted in the olaparib-treated population (log2 fold change=3.6, P<1.4×10−3, exact test; Fig. 3c).
Identification of novel determinants of sensitivity to the PARP inhibitor BMN 673
We asked whether known determinants of PARP inhibitor sensitivity were hits in our screen. The most robust of these, the homologous recombination regulators Brca1 and Brca2, are known to cause a fitness defect in ES cells when fully disrupted, in contrast to the specific truncation allele in the spiked ES cells above, so may not be seen in our screen. Out of a total of 192,631 insertions mapped for the PB3 end, only five were observed in Brca1 and only one in Brca2; these genes have also been shown to be essential in a recent ES cell CRISPR screen26.
We therefore looked for novel determinants of PARP inhibitor sensitivity. We applied fold change and P-value cutoffs to the analysed barcode abundances for the potent PARP inhibitor BMN 673 (also known as talazoparib27) and DMSO exposed libraries. We linked significantly depleted barcodes to their integration sites using the inverse PCR data and observed that multiple barcodes associated with Ewsr1 integrations were depleted in the samples treated with BMN 673 (Fig. 3d). To confirm this result, we used CRISPR-Cas9 technology to knock out Ewsr1 in diploid ES cells and verified that these cells were more sensitive to BMN 673 than the parental cell line (4.2-fold lower SF50, P<0.0001, ANOVA; Fig. 3e,f). This was further validated using three different shRNA constructs to knockdown the human homologue, EWSR1, in MCF7 breast cancer cells (20-fold lower SF50, P<0.001, ANOVA; Fig. 3g,h), suggesting that EWSR1 might be a genetic determinant of PARP inhibitor sensitivity.
Usage Notes
Although 1,000 colony equivalents were pooled to form each sublibrary, there are approximately 2,000 integrations identified for each. This could be due to further transposon mobilisation events occurring in colonies after the first cell division, or a subset of cells with stable transposase expression that continue to generate integrations. Many barcodes map to multiple integration sites—this is likely due to lower than expected complexity of the barcoded plasmid population. Transposon ends PB5 and PB3 are referred to in analysis files as Sims and West respectively, corresponding to primer sequences used to prepare sequencing products8,17. Mappings were generated for all PB3 (West) libraries and PB5 (Sims) libraries L1 and L2.
Additional Information
How to cite this article: Pettitt, S. J. et al. Genome-wide barcoded transposon screen for cancer drug sensitivity in haploid mouse embryonic stem cells. Sci. Data 4:170020 doi: 10.1038/sdata.2017.20 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Carette, J. E. et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326, 1231–1235 (2009).
Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011).
Elling, U. et al. Forward and Reverse Genetics through Derivation of Haploid Mouse Embryonic Stem Cells. Cell Stem Cell 9, 563–574 (2011).
Leeb, M. & Wutz, A. Derivation of haploid embryonic stem cells from mouse embryos. Nature 479, 131–134 (2011).
Reiling, J. H. et al. A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin. Proc. Natl. Acad. Sci. USA 108, 11756–11765 (2011).
Leeb, M., Dietmann, S., Paramor, M., Niwa, H. & Smith, A. Genetic exploration of the exit from self-renewal using haploid embryonic stem cells. Cell Stem Cell 14, 385–393 (2014).
Blomen, V. A. et al. Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 (2015).
Sims, D. et al. High-throughput RNA interference screening using pooled shRNA libraries and next generation sequencing. Genome Biol. 12, R104 (2011).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Yusa, K., Zhou, L., Li, M. A., Bradley, A. & Craig, N. L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 108, 1531–1536 (2011).
Leeb, M. et al. Germline potential of parthenogenetic haploid mouse embryonic stem cells. Development 139, 3301–3305 (2012).
Pettitt, S. J. et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS ONE 8, e61520 (2013).
Pettitt, S. J., Tan, E. P., Yusa, K. in Methods in Molecular Biology Vol. 1239 Chromosomal Mutagenesis (ed. Shondra, M. & Pruett-Miller ) Ch. 2, 15–28 (Springer, New York, 2015).
Wang, W., Bradley, A. & Huang, Y. A piggyBac transposon-based genome-wide library of insertionally mutated Blm-deficient murine ES cells. Genome Res. 19, 667–673 (2009).
Akhtar, W. et al. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154, 914–927 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Westerman, B. A. et al. A genome-wide RNAi screen in mouse embryonic stem cells identifies Mp1 as a key mediator of differentiation. J. Exp. Med. 208, 2675–2689 (2011).
Quail, M. A. et al. A large genome center's improvements to the Illumina sequencing system. Nat. Methods 5, 1005–1010 (2008).
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014).
Pettitt, S. J. et al. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 6, 493–495 (2009).
Bajrami, I. et al. Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 74, 287–297 (2014).
Pettitt, S. J. Source code for: Genome-wide barcoded transposon screen for cancer drug sensitivity in haploid mouse embryonic stem cells. Github https://github.com/GeneFunctionTeam/pooled-transposon (2016).
Li, M. A., Pettitt, S. J., Yusa, K. & Bradley, A. in Methods in Enzymology Vol. 477, 217–242 (Elsevier, 2010).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005).
Tutt, A. et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. EMBO J. 20, 4704–4716 (2001).
Koike-Yusa, H., Li, Y., Tan, E. P., Velasco-Herrera, M. D. C. & Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 32, 267–273 (2013).
Shen, Y. et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin. Cancer Res. 19, 5003–5015 (2013).
Tang, F. et al. Tracing the derivation of embryonic stem cells from the inner cell mass by single-cell RNA-Seq analysis. Cell Stem Cell 6, 468–478 (2010).
Data Citations
Pettitt, S. J. Figshare https://doi.org/10.6084/m9.figshare.3469157.v1 (2016)
Pettitt, S. J. Figshare https://doi.org/10.6084/m9.figshare.3469112.v1 (2016)
Pettitt, S. J. Figshare https://doi.org/10.6084/m9.figshare.4213170.v3 (2016)
Acknowledgements
We thank Martin Leeb and Anton Wutz for providing H129.2 haploid cells. This work was funded by Cancer Research UK, Breast Cancer Now, as part of their funding to the Breast Cancer Now Research Centre, and the Breast Cancer Research Foundation. We acknowledge NHS funding to the NIHR Royal Marsden Hospital Biomedical Research Centre.
Author information
Authors and Affiliations
Contributions
S.J.P., A.A., C.J.L.: Study conception and design, data generation, data analysis, manuscript writing. D.B.K., H.N.P., Y.F., J.F., F.L.R., R.B., F.S., I.B., R.R., F.W., I.K., K.F., J.A.G., A.S.: Data generation. D.B.K., H.N.P., A.G., J.C.: Data analysis. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0 Metadata associated with this Data Descriptor is available at http://www.nature.com/sdata/ and is released under the CC0 waiver to maximize reuse.
About this article

Cite this article
Pettitt, S., Krastev, D., Pemberton, H. et al. Genome-wide barcoded transposon screen for cancer drug sensitivity in haploid mouse embryonic stem cells. Sci Data 4, 170020 (2017). https://doi.org/10.1038/sdata.2017.20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.20
This article is cited by
-
Identification of genes that promote PI3K pathway activation and prostate tumour formation
Oncogene (2024)
-
The ubiquitin-dependent ATPase p97 removes cytotoxic trapped PARP1 from chromatin
Nature Cell Biology (2022)
-
Mammalian haploid stem cells: establishment, engineering and applications
Cellular and Molecular Life Sciences (2019)
-
Knocking down the obstacles to functional genomics data sharing
Scientific Data (2017)
-
Genome-wide barcoded transposon screen for cancer drug sensitivity in haploid mouse embryonic stem cells
Scientific Data (2017)
