
Abstract
Aerosols supply bioaccessible iron to marine biota which could affect climate through biogeochemical feedbacks. This paper review progresses in research on pyrogenic aerosol iron. Observations and laboratory experiments indicate that the iron solubility of pyrogenic aerosol can be considerably higher than lithogenic aerosol. Aerosol models highlight a significant contribution of pyrogenic aerosols (~20%) to the atmospheric supply of dissolved iron into the ocean. Some ocean models suggest a higher efficiency of pyrogenic iron in enhancing marine productivity than lithogenic sources. It is, however, challenging to quantitatively estimate its impact on the marine biogeochemical cycles under the changing air quality and climate.
Similar content being viewed by others


Introduction
Iron (Fe) is the fourth most abundant element in the Earth’s crust, but the rise of the oxygen level during the late Archean and early Paleoproterozoic periods reduced the capacity of seawater to retain dissolved Fe (DFe). In parts of the global ocean where the supply of Fe, either from the bottom (e.g., sediment and hydrothermalism) or the top (i.e., atmospheric deposition), is low, Fe becomes a micronutrient that limits the marine productivity. These oceans are often termed as the high-nutrient low-chlorophyll (HNLC) regions such as the subarctic north Pacific, the east equatorial Pacific, and the Southern Ocean1,2. It has been hypothesized that enhanced dust deposition led to an increase in the export of biogenic carbon from the surface to the deep ocean (i.e., via the biological carbon pump), substantially contributing to the lower concentration of the atmospheric carbon dioxide (CO2) during the last glacial maximum (LGM), the so-called “iron hypothesis”3.
A number of artificial Fe fertilization experiments in the open ocean confirmed that the addition of Fe in HNLC regions leads to increased phytoplankton growth4 and export of organic particles into the deep ocean5,6. Further investigations have been found in naturally Fe fertilized regions, for example, in Fe-limited regions in the Southern Ocean where the input of Fe from islands or shelf regions leads to patches of phytoplankton blooms, which can be observed in satellite images7. Furthermore, atmospheric delivery of Fe to the open ocean also stimulates nitrogen fixation and thus relieves nitrogen limitation in low latitude oceans8,9.
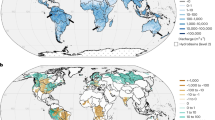
Long-range transport of desert dust to the open oceans has been well documented since the 1980s10,11. A traditional view is that mineral dust dominates the global supply of atmospheric Fe to the ocean, which is why dust aerosol was in the center of the global research in the atmospheric Fe cycle. Global aerosol models have been used to estimate atmospheric deposition fluxes of Fe in dust from arid and semi-arid regions. By assuming a solubility of Fe in the dust (i.e., the ratio of dissolved to total Fe in unit of percentage), the deposition flux of DFe could also be estimated, although a wide range of aerosol Fe solubility was reported over different oceanic regions of the world12,13. Recent atmospheric models14,15,16 estimated that the deposition flux of DFe to the ocean varied between 0.26 and 0.53 Tg Fe yr−1, which was in the higher range of estimates in earlier studies (0.05–0.54 Tg Fe yr−1)12,13 (Table 1).
In the last decade, Fe-containing aerosol from anthropogenic sources has raised attention because of its high aerosol Fe solubility17,18. The term “anthropogenic” Fe is used here as primary Fe from fossil fuel and biofuel combustion sources. Anthropogenic sources of Fe also include dust and biomass burning, which are directly or indirectly emitted due to human activities such as climate and land use/cover changes19. The emission fluxes of these sources are difficult to quantify due to the episodic nature of dust and open fire events, and thus are not explicitly disentangled in this work. Therefore, anthropogenic sources mainly contribute to pyrogenic Fe, while pyrogenic Fe from biomass burning can be partly ascribed to natural sources.
An intercomparison exercise by the Joint Group of Experts on the Scientific Aspects of Marine Environmental Protection (GESAMP) Working Group 38, “The Atmospheric Input of Chemicals to the Ocean” suggests that pyrogenic aerosols contribute to around 20% of deposited DFe to the oceans14,18. Traditionally, it is thought that pyrogenic sources are less sensitive to global environmental change due to their significantly lower Fe deposition flux compared to mineral dust, and thus have a minor effect on ocean biogeochemistry, and consequently on the carbon cycle20,21. However, high aerosol Fe solubility (>10%) is often reported at low aerosol Fe concentration over the open ocean22,23,24. The GESAMP intercomparison study suggests that this trend is attributed mainly to DFe released from pyrogenic Fe oxides18. The role of pyrogenic Fe sources in the marine Fe cycle has been explored in several ocean biogeochemical models19,20,25,26,27, which used the spatial distribution and temporary variation of Fe solubility directly supplied by the atmospheric models. Some recent model results suggested that pyrogenic Fe-containing aerosols could enhance the net primary production (NPP) more efficiently than lithogenic Fe sources19,20 and the global carbon export efficiency of pyrogenic Fe was considerably higher than lithogenic Fe by a factor of 620–919.
Atmospheric sources of Fe and its role in marine biogeochemistry have been reviewed in the last few years28,29,30,31,32. In this paper, we focus on pyrogenic sources of Fe to the ocean and track the effective fraction of Fe that is supplied by aerosols from pyrogenic sources, deposited to the sea surface, and undergoes the entire biogeochemical cycling in the ocean (Fig. 1). Therefore, we use “DFe” consistently for both the atmospheric and ocean research fields to refer to the most readily bioavailable form, bearing in mind the differences in measurements and terminologies in the different research fields32. In addition to the primary sources (as described above), this review will also discuss “secondary” DFe, which refers to the Fe solubilized during atmospheric processing of relatively insoluble Fe in aerosols (Fig. 1), in particular the acidification of Fe particles by the uptake of anthropogenic air pollutants such as sulfur dioxide (SO2), nitrogen oxides (NOx) and volatile organic compounds (VOCs).

Fe-containing particles are emitted by coal combustion, ships, metal smelting industry, and open biomass burning in addition to the emission of mineral dust particles. During the atmospheric transport, the solubility of Fe in aerosols is enhanced by photochemical processing. Dry and wet deposition unload aerosols and DFe into the ocean where aerosols participate disaggregation/aggregation and sink. DFe binds with organic ligands and enters the biological cycle, or is removed from the dissolved pool by particle adsorption (scavenging).
Figure 1 summarizes the key processes in the cycling of pyrogenic aerosol Fe: Fe-containing particles are emitted by coal combustion, ships, metal smelting industry, and open biomass burning, along with other air pollutants such as SO2, NOx, VOCs, and ammonia (NH3). Fe solubility in pyrogenic aerosols is highly dependent on the nature of the aerosol near the source regions, while it is significantly enhanced by photochemical processing during the atmospheric transport, in particular by acidification due to uptake of inorganic and organic acids. Aerosols and associated DFe enter the ocean via dry or wet deposition where the aerosol particles disaggregate, aggregate with organic particles in the ocean and sink. The deposited DFe binds with oceanic organic ligands and is taken up by phytoplankton, leading to an increase of biological production in Fe-depleted regions. A fraction of deposited DFe is lost due to particle adsorption (scavenging). Pyrogenic aerosols have an impact on climate physically by their effect on radiation balance and biogeochemically by ocean fertilization, which stimulates the marine biological carbon pump and the emission fluxes of aerosol and their precursor gases.
In the following, we will review these processes, which span three key areas (1) observational and laboratory evidences on anthropogenic signatures of atmospheric Fe aerosols; (2) global atmospheric modeling of the emission, chemistry, and deposition of pyrogenic Fe; and (3) ocean biogeochemical modeling of the impact of pyrogenic Fe deposition on marine productivity and carbon cycle.
Field observations and laboratory measurements
In this section, we will review the key observational and laboratory evidences of the direct emissions and secondary production of DFe.
Aerosol Fe emission and solubility
The estimated global total Fe emission to the atmosphere is 37–140 Tg Fe yr−1, around 95% originating from lithogenic sources, while the rest coming from pyrogenic sources including open biomass burning, coal combustion, shipping emissions, and metal smelting industry33,34,35,36,37,38. Even though mineral dust represents the largest contribution to atmospheric Fe, their solubility over low latitude source regions is low, usually <0.5%39,40,41,42. On the other hand, the pyrogenic aerosol Fe solubility varies considerably depending on the sources and can be 1–2 orders of magnitude higher than mineral dust (Table 2).
Table 2 summarizes the Fe solubilities (%) in aerosols of pyrogenic origins, including solid fuel combustion, biomass burning, and liquid fuel combustion. Solid fuel combustion includes biofuel wood and waste burning, coal combustion, and metal smelting process. The total Fe content and the estimated contribution of each source to the total Fe emissions are also reported. The total Fe emissions from solid fuel combustion and biomass burning are 0.5–1.9 Tg Fe yr−1 and 0.5–1.2 Tg Fe yr−1, respectively33,34,35,36,37,38. Minor contribution derives from liquid fuel combustion such as ship emissions, which is around 0.02%33,34,36,37,38.
The total Fe content is 0.1–12% in coal fly ash34,38,43,44,45,46,47,48, 0.02–5.5% in biomass burning aerosol45,47,49,50, and 0.1–9% in oil fly ash34,38,42,43,44,47. Emission inventories apply much higher Fe content to iron and steel smelting-related processes (26%–44%)34,38.
Schroth et al.42 found a very high aerosol Fe solubility in oil fly ash (77%–81%), where Fe is likely in the form of ferric sulfate salt (Fe2(SO4)3 · 9(H2O)). A laboratory study reported 36% aerosol Fe solubility for oil fly ash, which is significantly higher than the 0.2% aerosol Fe solubility for coal fly ash43. The dominant component of coal fly ash is the aluminosilicate glass with aggregates of Fe (oxyhydr)oxide43,46,47, which is less soluble than ferric sulfate salt. Oakes et al.51 also estimated a considerably lower aerosol Fe solubility for coal fly ash (0.06%), compared to 51–75% and 46% Fe solubilities in vehicle exhaust and biomass burning, respectively. Guieu et al.52 and Paris et al.49 reported much lower aerosol Fe solubility for biomass burning near the source (around 2%) than the 18% aerosol Fe solubility over the ocean for aerosols mainly influenced by bushfire plumes (not at source)53. Field observations and laboratory measurements suggest that Fe solubilities near the source regions vary significantly, with lower values observed near dust source regions but higher values near oil combustion and biomass burning sources (Table 2). The high Fe solubilities in aerosols from the oil and biomass combustion are attributed to the presence of Fe sulfate instead of Fe oxides or Fe-bearing silicate minerals in dust42,51. Long-range transport can alter the Fe properties and enhance Fe solubilities. It appears that current observations are inadequate to trace the aerosols from different sources and capture the variabilities in their Fe solubilities during atmospheric processing after atmospheric mixing between air masses of various origins.
Field observation on pyrogenic Fe aerosol
The most direct evidence of anthropogenic emissions of Fe-containing aerosols comes from the single-particle analysis. Anthropogenic Fe-rich particles were directly identified close to the source, (i.e., steel plants), by both single-particle mass spectrometer54 and microscopic analysis55,56. They were also identified in the marine atmosphere, for instance, over the English Channel57, Western Pacific Ocean58,59,60, and in the urban atmosphere56,61. Figure 2a shows spherical Fe-rich particles, which are likely formed under high-temperature processes, such as in steel plants.

a, b Elemental maps showing two individual sulfate particles with Fe-rich particles (as hotspots). c Ion intensity maps showing the presence of organic matter, sulfate, Fe oxide, and Fe sulfate (reproduced from Li et al.59).
Field observations have reported that magnetite is ubiquitous in anthropogenic aerosols62,63,64,65, as it crystallizes from the aluminosilicate glass during the ash formation. Since magnetite (Fe3O4) and hematite (α-Fe2O3) strongly absorb sunlight, a modified single-particle soot photometer (SP2) was employed to identify light-absorbing Fe oxide (FeOx) particles originating from anthropogenic combustion processes62,66. The field measurements over both East Asia and the Arctic reported that mass concentrations of anthropogenic FeOx were at least 20% of those of black carbon (BC)67. On the other hand, a limited number of observations suggests that magnetite is low or negligible in mineral dust from low latitude regions such as northern Africa and Asia, e.g., from not detectable to 0.1% in Saharan dust68,69 to 0.1–0.8 wt% in source regions of Asian dust70,71,72. Magnetite in high-latitude dust could be higher, for example, 1–2 wt% in Icelandic dust73. The content of magnetite in anthropogenic aerosols and their contribution to ambient bulk aerosols remain largely unknown. Therefore, more research is needed to quantify the content of magnetite in the sources and the Fe dissolution in aerosols34,74.
Several observational studies have attempted to link the aerosol Fe solubility to the aerosol chemical components. These studies suggest that the tendency of high Fe solubilities with low Fe concentrations is influenced by pyrogenic sources and/or atmospheric processing39,41,75,76,77. It is, however, difficult to quantitatively disentangle these two factors from the atmospheric observations of the elemental composition alone24,78.
Field observations on the atmospheric processing of pyrogenic Fe aerosol
Zhu et al.79 proposed that the low pH predicted in aerosol water under polluted conditions could lead to the dissolution of ferric Fe from α-Fe2O3, FeO(OH) and Fe(OH)3 minerals. Many laboratory, modeling and observation studies aimed to confirm the acid Fe dissolution hypothesis80. Laboratory experiments showed the insoluble Fe in aerosols is dissolved at low pH conditions (pH 1–3)46,47,81,82, lending some indirect evidence to this hypothesis. However, field observations have been less conclusive76,78,83,84,85,86. Recently, Li et al.59 provided convincing evidence of the acid dissolution of Fe in aerosol water. They observed Fe-containing particles in samples collected over the Yellow Sea, likely from coal combustion and steel industries, which were coated with sulfate. The single-particle analysis suggested that Fe was detected not only as “hotspots” (i.e., primary particles) but also in the sulfate coating as (water-soluble) Fe sulfate (Fig. 2b, c). Since water-soluble Fe was not detected in the freshly emitted particles, this could only be formed via the acid dissolution of the primary particles.
Laboratory experiments of Fe dissolution kinetics
Aerosol particles are subject to both physical and chemical processes during long-range transport. The chemically and photochemically based processing have been shown to have the potential to convert relatively insoluble Fe to more labile Fe forms59,76,77,87,88,89. Several studies focused on the acid and photochemical processes involved in the Fe dissolution in lithogenic and pyrogenic aerosol sources. Laboratory simulations have currently identified three principal mechanisms for the Fe dissolution: proton-promoted, ligand-promoted, and photo-reductive dissolution of Fe43,46,47,81,90,91,92,93,94,95,96,97,98,99.
The aerosol Fe solubility primarily depends on the pH of the leaching media and is enhanced as the pH decreases. At low pH, the increasing concentrations of H+ contribute to the protonation process which weakens the Fe–O bond on the particle surface favoring the detachment of Fe from the bulk oxides into solution100. Shi et al.81,98,99 investigated the effect of the proton-promoted dissolution on mineral dust during laboratory experiments to simulate acid and cloud processing, where the dust particles were subjected to multiple cycling between acidic (24 h at pH 1–2) and circumneutral pH (24 h at pH 5–6) up to 3 days. Low pH (pH 1–2), a condition relevant to fine aerosols, enhances the dissolution of metals including Fe and copper (Cu)101. Under cloud conditions (pH 5–6), the Fe dissolution is suppressed, and the formation of Fe-rich nanoparticle aggregates was observed when with no organic ligand in solution81,98,99.
Chen et al.46 simulated the acid and cloud processing of three certified coal fly ash samples, where the suspension of coal fly ash was cycled between pH 2 and pH 5 over periods of 24 h. The aerosol Fe solubility was ~20–70% after three pH cycles46, which was considerably higher than that one found in mineral dust98. Subsequently, a laboratory study90 investigated the impact of organic ligands (i.e., oxalate) on the Fe dissolution behavior at low pH of the certified coal fly ash samples in comparison with the Arizona test dust (AZTD). Chen and Grassian90 reported that at low pH the aerosol Fe solubility of the fly ash could almost double in presence of oxalic acid. The aerosol Fe solubility for the fly ash samples (~40–80%) were similar to ~60% for the AZTD after 45 h at pH 2 in presence of oxalate. Oxalate can form bidentate complexes with Fe on the particle surface, and thus it promotes the Fe dissolution process when in excess. In addition, the light-induced reduction of the structural Fe(III) to Fe(II) along with the oxidation of Fe(II)-oxalate complexes can further enhance the detachment of Fe(II) from the surface to yield dissolved Fe(II) (photo-reductive dissolution)90.
Fu et al.47 assessed the Fe dissolution kinetics of lithogenic and pyrogenic aerosol sources at pH 2. The leaching experiments were carried out in hydrochloric acid solutions in either dark conditions or the presence of light (Fig. 3). The aerosol Fe solubility after 12 h was 2.9–4.2% for coal fly ash, 74% for oil fly ash, 8.9–26.4% for biomass burning aerosols, and 4.3% for the Chinese loess. Slightly higher Fe solubilities were observed in presence of light (with no organic ligands) compared to dark conditions, due to the photochemical reduction of surface Fe(III) to Fe(II).

Total dissolved Fe concentration in a oil fly ash, b biomass burning particle, and c coal fly ash and Chinese Loess (CL) in dark conditions or under irradiation in HCl suspensions, solids loading of 1.5 g/L (reproduced from Fu et al.47). The error bars represent one standard deviation from triplicate experiments.
Borgatta et al.48 conducted laboratory experiments on coal fly ash samples collected from three distinctive locations: the USA Midwest, North-East India, and Europe. Sodium chloride (NaCl) was used to adjust the activity of proton in the acidic solutions (pH 1–2) to represent the high ionic strength in marine aerosol water. The resulting total Fe solubility at pH 2 after 24 h was 15–70%. The high variability in Fe dissolution behavior is attributed to the different physicochemical properties of the three coal fly ash samples. For example, Fe speciation, surface area, and morphology are dependent on the source region and the coal combustion process48. This is consistent with the findings in previous studies46,90. The high ionic strength influences the activity of protons and ligands in solution, hence it can affect the Fe dissolution behavior. However, currently, only a limited number of studies have considered the effect of the high ionic strength on the aerosol Fe solubility48,91,94.
Overall, oil fly ash and biomass burning aerosols showed high Fe solubilities at acidic conditions, while the coal fly ash had variable Fe solubility, similar or considerably higher than mineral dust (Fig. 3)47,93. The Fe dissolution rates determined through laboratory experiments were used to parameterize atmospheric processing and the transformation of relatively insoluble Fe into DFe according to the proton-promoted, oxalate-promoted, and photo-reductive dissolution processes in global models (Table 3). The source type is also considered, as the release rate of DFe is faster in combustion aerosols compared to mineral dust (Fig. 4)94,102.

The estimates of total dissolved Fe concentration (mg g−1) used in the IMPACT model a with no organic ligand and b with oxalate under dark conditions. The red curve was calculated for combustion aerosols102. The blue curve was calculated for mineral aerosols94. The total dissolved Fe concentration was calculated as mg of DFe per g of the solid particle.
Atmoshperhic aerosol modeling of iron
Modeling aerosol Fe emissions from lithogenic and pyrogenic sources
Traditionally, aerosol Fe emissions were associated with lithogenic sources only, assuming an average Fe content similar to the upper crustal material (3.5% for the percentage of Fe by weight)12,13. Later studies derived the Fe content in mineral dust aerosols from the geographical map of soil103,104,105. The Fe distribution in clay and silt soils was then utilized to estimate the spatial distribution of the Fe emissions94,106,107. More recently, atmospheric chemistry models implemented the emissions of pyrogenic Fe sources33,34,35,36,37,38. In practice, models need to adopt parameterizations or simplifications of the Fe emissions. The modeling approaches used to estimate the aerosol Fe solubility in pyrogenic sources at the point of emission can be classified into bulk-specific, mineral-specific, and source-specific. The bulk-specific method assumes uniform aerosol Fe solubility (i.e., 4%) for all anthropogenic combustion sources35,45, based on the limited numbers of observations available at the early research stage39. The mineral-specific approach uses a mineralogy-weighted solubility method38, based on laboratory measurements for Fe sulfate, clay minerals, and Fe oxides. The source-specific approach utilizes different Fe solubilities depending on the Fe chemical composition of the source type at the emission33,36,37, as summarized in Table 2. In the following sections, we describe a detailed analysis of lithogenic and pyrogenic Fe from the Integrated Massively Parallel Atmospheric Chemical Transport (IMPACT) model16, one of the state-of-the-art global aerosol models with detailed Fe chemistry. We also provide a brief discussion of the differences in four atmospheric Fe models, which participated in the GESAMP intercomparison study (i.e., the Community Atmosphere Model version 4 (CAM4)107, the Tracer Model 4 of the Environmental Chemical Processes Laboratory (TM4-ECPL)36 and the Goddard Earth Observing System with Chemistry (GEOS-Chem)108). IMPACT33 and TM4-ECPL36 adopted the source-specific approach for estimating aerosol Fe solubility, while CAM4 used the bulk-specific method35 and then CAM5 updated to the mineral-specific approach38. GEOS-Chem model did not include pyrogenic aerosols108.
In the IMPACT model, dust emissions are dynamically simulated using a physically-based emission scheme109,110, considering the Fe distribution in clay and silt soils94,105. On the other hand, anthropogenic emission fluxes are prescribed by the monthly estimates for each source sector34,111. The daily estimates for open biomass burning emissions are based on a terrestrial biogeochemistry model in conjunction with satellite measurements50,112. In the IMPACT model, very low aerosol Fe solubilities at the emission are considered for mineral dust (0.1%), solid fuel combustion (0%), and biomass burning aerosols (0%), while high aerosol Fe solubility is prescribed for liquid fuel combustion aerosols (58 ± 22%). Because DFe content in aerosols at emission can be highly variable, the arithmetic mean of the measurements is used for the initial Fe solubility in the oil combustion aerosol33. This reflects the lower Fe aerosol solubility for Fe-containing aluminosilicates compared to ferric sulfate and aggregated nanocrystals of magnetite observed in the laboratory measurements (Table 2)42,47. While we recognize the uncertainties, particularly for solid fuel combustion and biomass burning aerosols (Table 2), model simulations based on these prescribed solubilities are broadly consistent with field observations (see discussions later). Both the models and observations showed an increase in aerosol Fe solubility over the open ocean compared to the source region for coal fly ash, biomass burning aerosols, and mineral dust during the atmospheric transport18,40,52,53,87,113. On the other hand, sporadic high Fe solubility was reported for aerosols possibly influenced by a ship’s plume of lowest-grade fuel combustion (e.g., heavy fuel oils, residual fuel oils, crude oils, or bunker fuels) over the Arabian Sea114.
Aerosol Fe dissolution from lithogenic and pyrogenic sources
The photochemical transformation of Fe from mineral aerosols in the marine atmosphere has been initially associated with sulfuric acid originating from marine biological emissions of dimethylsulfide (DMS)115. However, these sulfuric acid concentrations are too low (much lower than those from anthropogenic sources) to induce substantial Fe dissolution from mineral dust aerosols, due to the buffering ability of alkaline minerals such as calcite45,79,80. The interaction of sub-micron aluminosilicate-rich particles with sulfuric and nitric acids during the atmospheric transport can result in very acidic solutions, particularly when sulfate is a major component116,117. The acidic conditions could lead to the substantial Fe dissolution in aerosols if the alkaline minerals were externally mixed with Fe-rich aluminosilicate minerals45,118. This is true for pyrogenic Fe-containing aerosols especially when the acidic species are substantially co-emitted (Fig. 2)59,102. Although the laboratory studies indicate that oxalate can enhance the Fe dissolution rate, only some Fe-rich particles may contain oxalate119. In fact, both the IMAPCT model results and field observations found no significant correlation between oxalate and DFe over a coastal city of China, partly because of the external mixing of oxalate-rich aerosols with Fe-rich aerosols, which can suppress the oxalate-promoted Fe dissolution near the polluted regions102,120.
The thermodynamic equilibrium modules have been used to estimate the pH in the aqueous phase of hygroscopic particles36,45,80. IMPACT model considered external mixing of size-resolved pyrogenic aerosols from alkaline mineral dust and sea-salt aerosols102, whereas TM4-ECPL36 and GEOS-Chem108 assumed internal mixing of the aerosols. The internal mixing of mineral dust with different types of aerosols led to the lower pH for the finer particles but resulted in relatively basic pH conditions over the Southern Ocean partly due to the buffering capacity of sea-salt particles36. The internal mixing of mineral dust with different size bins resulted in basic pH conditions for mineral aerosols over the Southern Ocean partly due to the buffering capacity of alkaline mineral108. CAM4107 did not include the thermodynamic equilibrium modules, and pH = 2 was prescribed when the concentration of sulfate aerosol was larger than the concentration of calcium35. The pH calculation was updated for CAM515 by lowering the prescribed aerosol pH to 1 for fine particles, compared to 2 for coarse particles.
Observations and laboratory simulations provide convincing evidence that atmospheric processing can lead to the solubilization of relatively insoluble Fe in aerosols. The modeling approach used to represent the Fe dissolution behavior during the atmospheric processing is also classified as bulk-specific, mineral-specific, and source-specific. The bulk method assumes the crystalline Fe oxides (i.e., hematite) dissolution rates for all sources35,45,80. However, the calculated Fe dissolution rates for pure hematite are much slower than those obtained from the laboratory experiments for different dust source samples97. The mineral-specific approach applies different Fe dissolution rates for each mineral15,36,106. Laboratory measurements showed that the dissolution rates of the aluminosilicate minerals such as smectite, illite, and chlorite were similar to each other after normalizing the dissolution rates to the mineral mass121. The similarity in the Fe release rates between illite and African dust samples at acidic pH values allowed us to reduce the number of mineral tracers for implementation in the models94. Thus, the source-specific method ignores the dependency of dissolution rates on the surface area of mineral particles. This simplification is supported by the laboratory experiments, which indicate the minor effects of particle size of aluminosilicate minerals on the initial Fe solubilities and dissolution rates122,123. In addition, the simulated atmospheric processing of coal fly ash samples in acid and circumneutral solutions caused the disintegration of the aluminosilicate glass into small irregular fragments, and thus produced highly dispersed Fe-bearing particles exposed to the aqueous phase, enhancing the availability of Fe-bearing species being attacked by protons unlike the crystalline aluminosilicate in mineral dust46,47,90. This supports the use of the source-specific approach94,102.
In the IMPACT model, the transformation of relatively insoluble Fe into DFe following the proton-promoted, oxalate-promoted, and photo-reductive dissolution processes is dynamically simulated according to the aerosol bin sizes and their source types. The sulfate and oxalate largely reside in the accumulation mode mainly due to cloud processing, whereas nitrate is primarily associated with aerosols in the coarse mode. Furthermore, the Fe dissolution rates in the acidic solution used for the fly ash were considerably faster than those used for the mineral dust (Fig. 4). Consequently, within the IMPACT model, the transformation of relatively insoluble Fe in fine combustion aerosols into DFe is much faster than that for coarse mineral dust aerosols during their atmospheric lifetime94,102. In fact, during the atmospheric transport, the uptake of acidic species on the surface of pyrogenic Fe-containing aerosols (Fig. 2) determines the release of DFe in the aerosol acidic deliquescent layer and enhances the aerosol Fe solubility18,59.
Below, we provide a brief overview for evaluation of aerosol Fe solubility, focusing on pyrogenic Fe. Most models12,13,36,37,108 except IMPACT do not consider Fe dissolution processing from pyrogenic aerosols explicitly. CAM4107 used the same Fe dissolution scheme for pyrogenic Fe as in lithogenic Fe in a “medium” soluble state (Table 3). CAM4 assumed lithogenic Fe to be in either a readily, medium, or slow soluble state based on previous laboratory97 and modeling106 studies.
Comparison of Fe solubility between model estimates and observational data
The comparisons of the aerosol Fe solubilities estimated by the atmospheric models with field observations are shown in Figs. 5 and 6. In addition to the compilation of measurements for the GESAMP studies18, extensive data set gathering aerosol Fe measurements across the 70°E–150 °E and 10°S–70°S sector of the Southern Hemisphere124 was used to evaluate the IMPACT model. Although mineral dust is the major source of Fe, the aerosol Fe solubility was extremely low over the Arabian sea and the eastern North Atlantic Ocean near the Saharan dust source region (Fig. 5). Much higher aerosol Fe solubility was reported in multiple field campaigns (up to 100%) far from the source regions. IMPACT simulated a wider range of aerosol Fe solubility than the other models. This may indicate that the faster dissolution rates for pyrogenic Fe contribute to improvement in simulating the larger degree of enhancement in Fe solubilities of aerosols over the open ocean that are influenced by sources of biomass burning and fossil fuel combustion, compared to that for mineral dust during atmospheric transport. The IMPACT model successfully simulated the aerosol Fe solubility in the Northern Hemisphere but did not capture the wide range of Fe solubilities over the remote ocean in the Southern Hemisphere where low aerosol Fe concentrations prevailed. TM4-ECPL and GEOS-Chem also underestimated Fe solubility in the Southern Hemisphere, whereas CAM4 overestimated this. In IMPACT, TM4-ECPL, and GEOS-Chem, Fe dissolution is suppressed mainly due to the higher pH conditions of the mineral aerosols over the Southern Ocean. In contrast, CAM4 is relatively insensitive to the acidity, as most DFe is formed via in-cloud processes. Thus, the overestimation of Fe dissolution in CAM4 was likely due to the lack of suppression mechanism for in-cloud aerosol dissolution process over the Southern Ocean. Recently, an inverse modeling technique suggested that bushfires contributed a large fraction of DFe concentrations in Fe-containing aerosols, although the substantial contribution from missing sources (e.g., coal mining activities, volcanic eruption, and secondary formation) was still inferred16. Furthermore, the external mixing of pyrogenic aerosols with mineral dust can promote acidification of the sub-micron aerosols from bushfires, which were the dominant contributor of DFe for biomass burning in contrast to mineral dust. This is reasonably consistent with field observations, which showed that higher Fe solubilities for finer aerosols influenced by biomass burning and fossil fuel combustion sources but no such trend for mineral dust-dominated aerosols76,85,86,113.

For the comparison with the observational data set (a), the daily model estimates along the cruise tracks during each sampling month were taken from each model estimate of b the IMPACT model, c TM4-ECPL, d CAM4, and e GEOS-Chem. Only the simulated data for which maximum likelihood estimates (MLEs) of total Fe concentrations fall within ±2 σ0 of the measurements were used for the comparison with field data.

For the comparison with the observational data set (a), the daily model estimates along the cruise tracks during each sampling month were taken from each model estimate of b the IMPACT model, c TM4-ECPL, d CAM4, and e GEOS-Chem. Only the simulated data for which maximum likelihood estimates (MLEs) of total Fe concentrations fall within ±2 σ0 of the measurements were used for the comparison with field data. The solid line represents a 1:1 correspondence and the dashed lines show the 10:1 and 1:10 relationships, respectively.
The IMPACT model captures enhanced Fe solubility over the Bay of Bengal, largely due to forest fires in South Asia and Southeast Asia18. Analysis of stable carbon isotopic composition of oxalic acid with Fe solubility suggests that the marine aerosols influenced by the forest fires over South Asia experience oxalate-promoted photochemistry of Fe in acidic solution over the remote Bay of Bengal125. Furthermore, a single particle analysis at a suburban coastal site in Hong Kong suggests that the oxalate−Fe photochemical processing for anthropogenic aerosols facilitates sulfur oxidation, leading to considerable sulfate formation126. This further facilitates the water uptake on aerosols and thus can explain why biomass burning particles usually undergo oxalate–Fe photolysis leading to high Fe solubility. On the other hand, the overestimates of aerosol Fe solubility over the tropical Atlantic in the Southern Hemisphere suggest that the IMPACT model overestimated the contribution of biomass burning aerosols downwind from the source regions for specific cruises (Fig. 5). The apportionment of pyrogenic Fe in mineral dust aerosols entraining into the atmosphere due to pyro-convection from vegetated lands can be sensitive to the fire severity and fuel conditions during forest fires50. Moreover, the model assumed the same Fe dissolution rate constants of coal fly ash for biomass burning aerosols, although the Fe dissolution rate was dependent on the pH, ambient temperature, and the competition for oxalate between surface Fe and DFe47,90,102. Although the proton-promoted dissolution rate used in the IMPACT model (Fig. 4) was within the range of biomass burning aerosols (Fig. 3), the oxalate-promoted Fe dissolution rate for combustion aerosols is available from only one laboratory study90. In their laboratory experiments, the acidic solution was prepared from purely oxalic acid90, while the acidity in ambient fine particles is mainly determined by sulfate with significantly lower oxalate concentration117,127. In fact, their dissolution rate for AZTD is very similar to the coal fly ash in pH 2 solutions acidified by oxalic acid90, and is much faster than that was examined for African dust samples in a more realistic aerosol solution (Fig. 4)94. The latter study set up the amount of oxalate based on the molar ratio of oxalate and sulfate in ambient fine particles127. Thus, the oxalate-promote dissolution experiments need to be examined for different types of aerosols in a more realistic aerosol solution.
The underestimates of aerosol Fe solubility over the Southern Ocean may indicate that the model underrepresented the Fe sources and the dissolution rates for Fe-containing aerosols influenced by well-mixed aerosol sources during the long-range transport (Fig. 5). A recent analysis of aerosol measurements with the IMPACT model over a coastal city of China revealed enhanced aerosol Fe solubility in fog conditions compared to other weather conditions such as haze and dust120. The median aerosol Fe solubility in foggy days was 5.8%, which was 3.3 times higher than in hazy days (1.8%), 5.2 times higher than in clear-sky days (1.1%), and 22 times higher than in dusty days (0.27%)120. The lower water content in the haze and dust particles may be responsible for the lower Fe dissolution rate in the acidic solution, and consequently, suppress the aerosol Fe solubility94. Field observations in Atlanta, southeastern USA, support that the acid dissolution driven by particle liquid water plays a dominant role and the primary emissions of pyrogenic Fe are the secondary importance128. On the other hand, the higher aerosol Fe solubility in fog cannot be reproduced by the model, possibly because the fog enhancement is not considered. For example, a laboratory study simulating cloud-water processes showed positive feedback between the SO2 uptake and the Fe dissolved on the particle surface129. Fog events are frequent in Antarctica130. The mean aerosol Fe solubilities in ice core and snow samples obtained at Dome C (10%)131 in the inland areas of Antarctica were higher than those in the coastal areas such as Berkner Island (~3%)131, Lambert Glacial Basin (~1.2%)132 and Roosevelt Island (~0.7%)133. Since anthropogenic emissions of acidic gases are substantially less in the Southern Hemisphere compared to the Northern Hemisphere, fog processing is likely to be relevant for the enhancement of aerosol Fe solubility over the Southern Ocean. The kinetic Fe dissolution experiments under fog conditions are necessary to simulate the fog enhancement.
Atmospheric deposition fluxes of lithogenic and pyrogenic DFe
At deposition to the oceans, the average aerosol Fe solubility for bulk aerosols estimated by the IMPACT model was 1.5% (Fig. 7). Biomass burning aerosols showed higher aerosol Fe solubility at deposition (38%) than anthropogenic Fe sources (12%) and mineral dust (1.2%). The model estimates of aerosol Fe solubility clearly indicate the impact of atmospheric processing on the derived aerosol Fe solubility, with higher Fe solubilities far from the source regions over the remote oceans, corresponding to lower total Fe concentrations. The relative contribution of pyrogenic versus lithogenic Fe can largely determine the bulk aerosol Fe solubility over the open oceans18,24,134. Several trace metals and their isotopes are characterized by unique biogeochemical cycling patters. Therefore, the analysis of the stable Fe isotopes may offer additional constraints on the source apportionment of aerosol Fe if the sources have distinct isotopic signatures134,135,136. However, the complexity and overlapping of the Fe isotopes in the different aerosol sources, and the kinetic isotope effect during the atmospheric processing and leaching make it difficult to provide a quantitative source apportionment for the DFe.

Each source represents a sum of all sources (1.5%), b dust (1.2%), c sum of solid fuel combustion and liquid fuel combustion (12%), and d open biomass burning (38%). The parentheses represent aerosol Fe solubility at deposition (%), which was calculated from total deposition fluxes of total Fe and DFe to the oceans.
The IMPACT model estimated an annually average deposition flux of DFe to the ocean of 271 Gg Fe yr−1 (Fig. 8). Mineral dust aerosols showed higher deposition fluxes of DFe (224 Gg Fe yr−1) than biomass burning aerosols (33 Gg Fe yr−1) and anthropogenic Fe sources (15 Gg Fe yr−1). The regions receiving the most substantial amounts of DFe from pyrogenic Fe sources were the Pacific and Southern Oceans (Fig. 8). The DFe deposition fluxes from biomass burning aerosols may be important in the past and future, particularly in the Southern Ocean19. This is already evident through the 2019–2020 bushfires in the Australian region where bushfires could contribute a large fraction of DFe concentrations in aerosols16. In the last glacial-interglacial cycle, the Fe solubilities in the North Greenland Eemian Ice Drilling (NEEM) ice core samples are higher during the warm periods than during the main cold period137. Given the significant supply of DFe from biomass burning aerosols in a warmer period when oceanic primary production may be more dependent on the nutrient input from atmospheric aerosols, the mega fires may be important to simulate the effect of Fe on ocean fertilization and need to be introduced as a potential feedback in the Earth System Models (ESMs). However, the aerosol deposition processes are highly uncertain. The atmospheric deposition flux of the radionuclide beryllium-7 (7Be) derived from its ocean inventory may provide a means to constrain the deposition fluxes of DFe138,139. This approach relies on the aerosol Fe solubility derived from the ambient aerosols sampled near the surface along the cruise tracks. However, the aerosol Fe solubility via wet deposition is often decoupled from that via dry deposition, due to different levels of atmospheric processing of aerosols at different altitudes20. Moreover, the aerosol Fe solubility for pyrogenic Fe was more than an order of magnitude higher than that of lithogenic Fe over the open ocean and thus the relative contribution of pyrogenic and lithogenic sources of DFe influences the bulk aerosol Fe solubility (Fig. 8). Therefore, models need to improve the simulation of aerosol Fe solubility in rainwater41,85,140,141,142 but more observational data are needed to validate the model estimates.

Each source represents a sum of all sources (271 Gg Fe yr−1), b dust (224 Gg Fe yr−1), c sum of solid fuel combustion and liquid fuel combustion (15 Gg Fe yr−1) and d open biomass burning (33 Gg Fe yr−1). The parentheses represent DFe deposition flux from each source to the oceans (Gg Fe yr−1).
Ocean biogeochemistry modeling of iron
The role of Fe in regulating marine productivity and carbon cycle has been widely accepted and the number of biogeochemistry models considering Fe as a micronutrient and decoupling its cycle from the cycle of carbon and macronutrients is growing. Several global models were compared within an “Fe Model Intercomparison Project” (FeMIP), and the fundamental processes in the Fe cycle considered in those models are summarized in Tagliabue et al.143. Recently, more models utilize variable aerosol Fe solubility of atmospheric input and consider the pyrogenic sources of aerosol Fe in addition to lithogenic sources. Here we focus on the processes in seawater related to the fate of the atmospheric input of DFe from pyrogenic sources and how they are parameterized in different ocean biogeochemistry models.
Atmospheric input of DFe and aerosol particles in ocean biogeochemistry models
All global ocean Fe models include an atmospheric source of DFe, whereas they differ when considering other DFe sources such as sedimentary and hydrothermal sources. Unlike ESMs which can treat dust deposition flux interactively144, ocean-only models typically prescribe the atmospheric source of dust with a spatially 2-dimensional field of deposition flux which is mostly simulated in global aerosol models14,145. Traditionally, the climatological deposition flux of mineral dust particles was used146. In most cases, the total Fe content in dust particles was obtained from the average Fe abundance in the upper crust of 3.5 wt%147. Atmospheric Fe-containing aerosol models used variable Fe content for different soils but resulted in a relatively small effect on the total Fe deposition103,148. Despite the large variability of aerosol Fe solubility over the oceans (Fig. 5), most ocean models assume a constant solubility of 1–2%. During the last decade, more model-derived deposition fluxes of DFe have become available (Table 1), encouraging ocean models20,25,26,149 to move from assuming a spatially uniform solubility towards using deposition fluxes of DFe simulated by atmospheric Fe-containing chemistry models. In the last few years, an increased number of ocean models20,26 and ESMs19,27 introduced pyrogenic sources of DFe (including solid fuel combustion, biomass burning, and liquid fuel combustion) in addition to mineral dust (Table 4). Krishnamurthy et al.25 employed combustion sources in the Biogeochemical Elemental Cycling (BEC) model. Subsequently, the long-term (multi-century) responses to changing atmospheric DFe input were investigated in equilibrium simulations with a modified BEC module in the Community Earth System Model (CESM)19 over three periods: pre-industrial, present day, and the future. For a shorter period over the last decades, Wang et al.26 examined the impact of anthropogenic source of different nutrients (N, P, and Fe) on marine productivity under realistic climate with Pelagic Interaction Scheme for Carbon and Ecosystem Studies (PISCES) coupled with the Nucleus for European Modelling of the Ocean (NEMO). In the Model for Interdisciplinary Research on Climate, Earth System version 2 for Long-term simulations (MIROC-ES2L)27, the atmospheric deposition of pyrogenic Fe-containing aerosols was dynamically simulated to study biogeochemical changes in the ocean. These studies focused on the impact of anthropogenic Fe sources on ocean biogeochemistry under the transient simulations. However, it is unclear if the large diversity in the impact of anthropogenic aerosol input on NPP change is related to the different atmospheric inputs of DFe or to the parameterization of processes in the biogeochemical cycle in different models. Thus, equilibrium sensitivity simulations were conducted to investigate a process-based understanding of the effects of atmospheric DFe input on ocean biogeochemistry20. The Ocean Ecosystem COmponent for MIROC-ES2L (OECO)27 and the Regulated Ecosystem Model version 2 (REcoM-2)150 were driven by three DFe deposition fields20. Since the same perturbations in DFe sources were applied to equilibrium simulations with the two models, more insights into differences between models in response of marine biogeochemistry were gained. REcoM-2 shows significantly higher sensitivity of ocean biogeochemistry to pyrogenic Fe source, primarily because most of the deposition of pyrogenic DFe to the open ocean occurs in Fe-limiting regions. In this review, therefore, we take REcoM-2 as a tool to analyze and illustrate the effect of the pyrogenic source of DFe on the marine productivity. We will also discuss features in different models that could result in model-dependent responses.
Atmospheric DFe is added to the uppermost model layer as DFe. A few models consider the role of lithogenic particles151,152 as well as pyrogenic particles20 in the carbon export flux and scavenging of DFe. In these models, all deposited particles are assumed to be fine particles and sink slowly on their own. The sinking of these particles is accelerated through aggregation with organic particles. All the particles provide surfaces for DFe adsorption throughout the water column and a part of adsorbed Fe is returned to DFe by desorption.
Biogeochemical cycle of Fe in the ocean
As any other tracers of solutes in models, DFe is transported with water masses and undergoes the chemical and biological cycle (Fig. 1). The chemical cycle is driven by the Fe speciation. Global ocean Fe models usually consider a simplified speciation between two size classes: DFe and particulate Fe (PFe), which represents the operational separation in measurements by a filter cut-off of 0.2 µm. Since the solubility of inorganic Fe is very low in oxygenated seawater153, the majority of DFe found in seawater exits as Fe bound with organic chelates (ligand). Therefore, in most of the models, DFe consists of free Fe (Fe′) and Fe bound with organic ligands (FeL), while few models assume that a fraction of DFe is colloidal (Fecoll)154. The concentration of FeL is calculated based on the conditional stability constant of ligand and a chemical equilibrium between Fe′ and FeL155.
Organic matter that can bind Fe contains a variety of chemical compounds. Their chemical properties, the identification and quantification of ligands in field studies have been comprehensively reviewed156,157. In global models for long-term simulations, it is cost-intensive to mathematically describe the entire pool of ligands because of the continuum of their binding stability as well as the different sources and lifetimes. Ocean modelers have made efforts to parameterize organic complexation of different complexity. The assumption of one ligand class with a uniform concentration is still used in many models. It is based on some earlier measurements of DFe concentrations with low spatial variability in the deep ocean158. With a considerable increase of measurements of DFe and ligand concentration, we know today that ligand distribution is far from uniform. Thus, this assumption should not be used anymore to reproduce observed DFe distribution, although it excludes the biological control on ligands and can still help in sensitivity studies to resolve the role of other individual processes in the Fe cycle. Introducing variable ligands into global models and simulating the spatial and temporal variability of Fe speciation may raise the computational cost, particularly because some chemical reactions require a much shorter time step than commonly used in global ocean models. This stiff problem has been numerically solved by dividing processes into “fast” and “slow” reactions and assuming that the “fast” reactions approach equilibrium within a normal model time step159. Some models linked organic complexation to other quantities simulated in models such as dissolved organic carbon (DOC), apparent oxygen utilization (AOU) and pH150,159,160. This kind of parameterizations is based on correlations found in observations or in calculations with kinetic models. It works mostly well in regions with similar conditions to the observational sites and in waters with comparable properties to the defined experiments with kinetic models. Under other conditions, the validity of these correlations needs to be carefully examined. Alternatively, sources and sinks of ligands can be described explicitly to simulate the entire cycle of ligands. It is, however, difficult to establish a proper classification of various ligands. Different criteria have been suggested, such as their sources, binding strengths, and lifetimes. Modern measurements tend to divide ligands into three classes according to the conditional stability constant156 and provide data to be compared with model results. However, modeling the cycle of several ligand classes needs to introduce a number of parameters that we know too little about. This will heavily increase the uncertainty of models. Modelers started to describe the cycle of one ligand prognostically161. More studies on mechanisms controlling the ligand cycle are needed to constrain the model parameters.
The chemical cycle of Fe in the modern ocean is largely controlled by organic complexation and scavenging (i.e., removal of DFe by particle adsorption). Particularly, DFe concentration in the deep ocean is mainly determined by the balance of these two processes. This is implemented in most models with the assumption that only Fe′, calculated as the difference between DFe and FeL, undergoes scavenging. The scavenging loss of DFe is calculated as a function of Fe′ concentration and the abundance of particles. For the latter, more and more models also treat dust particles as Fe′ scavengers. The scavenging process of Fe is very complex and there is still no consensus on a proper mechanistic description of adsorption and desorption of different Fe species in global models. Therefore, the scavenging rate is often used as one of the major tuning parameters to keep modeled DFe distribution close to observations. Both Fe′ and FeL enter the biological cycle as DFe for phytoplankton uptake in some models.
Phytoplankton groups have evolved with different Fe uptake mechanisms, for example, through membrane transporters or reduction of various Fe(III) species at the cell surface to generate Fe(II)162, and with the ability to reduce their Fe demand under Fe-limiting conditions163. This is reflected in the variety of assumptions on DFe in models. Some models assume that the entire DFe pool is equally available to all phytoplankton groups, while others consider the difference in bioavailability between Fe′ and FeL or even within FeL164. In a nutrient-phytoplankton–zooplankton-detritus (NPZD)-type ecosystem model, Fe passes from phytoplankton over zooplankton to detritus by grazing, aggregation, and mortality. A part of Fe is stored in biomass and a part is recycled and released back to the DFe pool by respiration, grazing, and remineralization. The rest of the biological form of Fe sinks deeper in the ocean with sinking organic particles and aggregates. The fraction of Fe reaching the seafloor is accumulated in sediments. Finally, some DFe is released to the water column at the bottom layer due to the degradation of organic matter as sedimentary input, which simply depends on the assumed Fe to carbon ratio for some models.
Model description and simulations to illustrate the role of pyrogenic DFe source
REcoM-2 is one of the state-of-the-art global biogeochemical models with an ecosystem compartment consisting of two phytoplankton groups: diatom (DIA) and nanophytoplankton (PHY), one zooplankton and one detritus class165. REcoM-2 considers flexible stoichiometry of carbon, nitrogen, silica for DIA only, iron and chlorophyll (C:N(:Si):Fe:Chl) and phytoplankton growth is described by multiplication of three growth limiting factors: light, temperature, and nutrients (i.e., N, Si, and Fe). Growth limitation by nutrients is calculated using the Liebig’s law of the minimum: the least available nutrient determines the current growth rate. DIA and PHY mainly differ in the model in their requirement for light and nutrients. PHY needs a higher light level for growth166. Due to the larger surface-to-volume ratio of small-sized phytoplankton, they are more efficient in nutrient uptake than large diatom cells, which is reflected in REcoM-2 by assuming a lower half-saturation concentration for dissolved inorganic nitrogen (DIN) and DFe uptake for PHY than for DIA. Therefore, PHY is less sensitive to nutrient limitation but suffers more under light limitation.
In the modeled Fe cycle, Fe′ is scavenged by both organic and lithogenic particles which undergo particle aggregation and sink to the deep ocean151. Instead of a constant ligand, a function of DOC and pH was applied to describe the dependency of ligand binding stability on biology and environmental conditions150. This parameterization essentially reduced a model-data mismatch often found in global Fe models with a constant ligand or one prognostic ligand. The latter showed largely uniform and/or too low deep DFe concentrations. Two different atmospheric deposition fields were used in two REcoM-2 runs with the surface input of DFe just from lithogenic (Rlith) and from both lithogenic and pyrogenic sources (Rlith+pyro)20, as was shown in Fig. 8. In the following, the difference between Rlith and Rlith+pyro is referred to as the pyrogenic effect to demonstrate the role of pyrogenic Fe source in regulating marine productivity.
Response of surface DFe and growth limitation of phytoplankton
Considering the pyrogenic DFe source, surface DFe (Fig. 9) mainly increases in the subtropical North Pacific, Indian Ocean, the tropical and subtropical Atlantic. It agrees well in some regions with the large deposition fluxes of DFe from pyrogenic sources (Fig. 8c, d). Regions receiving most of the pyrogenic DFe are the northwestern Pacific adjacent to the East Asia, Bay of Bengal, the eastern Indian Ocean downwind from the Southeast Asia and Australia, the tropical Atlantic and the southwestern Indian Ocean downwind from the African Continent. The differences in the pattern of the DFe deposition and surface DFe concentration could be caused by the biological consumption of Fe, which is mostly controlled by the intensity of nutrient limitation.

The surface concentration of DFe was calculated as the average over the upper 100 m. Climatological simulations were performed driven by the atmospheric input of DFe for year 2017, which was calculated with IMPACT.
REcoM-2 reproduces well as the most limiting nutrient reported so far in field studies. Most of the subtropical gyres are typically limited by the availability of macronutrients (e.g., N and P). Thus, adding more DFe from a pyrogenic source does not affect the growth of phytoplankton, which lead to an increase of surface DFe concentration when sufficient ligands exist. In other regions (e.g., the North and equatorial Pacific, in part of the South Pacific and the Southern Ocean), DFe can effectively regulate the biological productivity167. Thus, DFe from pyrogenic source is quickly consumed by phytoplankton, resulting in a strong increase in NPP dominated by PHY in most of these Fe-limited regions (Fig. 10a) and partly by DIA in the equatorial Pacific and a small area in the North Pacific (Fig. 10b). Therefore, the DFe concentration in these regions remains nearly unchanged even after the addition of pyrogenic DFe.

Climatological simulations were performed for a nanophytoplankton and b diatom driven by the atmospheric input of DFe for year 2017, which was calculated in IMPACT.
In the Pacific, phytoplankton growth is often limited by Fe in REcoM-2, which is not fully validated due to lack of observations. On the other hand, concentrations of macronutrients, such as DIN in the subtropical North Pacific Gyre, are extremely low as well. Pyrogenic DFe supports significantly higher growth of PHY in Rlith+pyro until its growth is switched from Fe-limited to N-limited. At the same time, the growth of DIA increases slightly and is switched to N-limited at the surface and to Si-limited deeper in the ocean. Because of the minimum law of nutrient limitation applied in the model, a further increase in phytoplankton growth is held back. Thus, the surface DFe concentration increases in newly established systems limited by macronutrients even though the biological uptake and production are clearly enhanced (Fig. 9).
Nanophytoplankton and diatom compete for nutrients (e.g., N and Fe) as well as light. In most of the Fe-limiting regions, the stronger response by PHY to pyrogenic Fe is explained by their more efficient Fe uptake than DIA. One exception is found in the eastern equatorial Pacific where NPP by DIA clearly increases and that by PHY slightly decreases (Fig. 10a, b). Vertically, DIA predominantly increases at the surface and declines slightly below 20–50 m, whereas PHY decreases in the entire upper 80 m. The eastern equatorial Pacific is characterized by intensive diatom blooms due to the higher silicate supply by upwelling waters. Diatom growth there is mainly limited by Fe supply. The additional DFe relieves the Fe limitation by DIA in the model and leads to a higher DIA concentration in the surface water, which results in higher absorption of light and makes the light condition crucial for PHY growth. Therefore, PHY could grow more slowly when the increase in light limitation overcomes the relief of Fe limitation. Below the surface waters, DIA also undergoes increasing light limitation, leading to a slight decrease in NPP. A similar but less intensive situation is found in the high-latitude Southern Ocean as well.
Responses of marine phytoplankton to additional pyrogenic DFe can be summarized for the HNLC regions: diatom growth is fueled in surface waters by the relief of Fe limitation; while nanophytoplankton suffers more from the intensified light limitation and can however overcome diatom where nutrients are in short supply. This could change the community composition and thus the efficiency of nutrient remineralization and of the biological carbon pump. Therefore, the competition for nutrients and light within a phytoplankton community and its representation in models are crucial to understand and predict responses of marine productivity to changing DFe input.
Effect of nutrient redistribution on primary production
The nutrient consumption in the ocean is primarily regulated by the certain elemental composition of phytoplankton, although large variations around the Redfield ratio have been found between different species168. Thus, increasing biological production in the Fe-limiting regions also consumes more macronutrients and results in more macronutrient-depleted surface waters transported to the Indian Ocean via the Indonesian Throughflow and further to the Atlantic Ocean. Consequently, NPP slightly decreases in large areas in the initially macronutrient-limited Atlantic and the Indian Ocean. A part of these waters flow southwards to the Antarctic Circumpolar Currents and causes a small decrease in NPP of PHY in the Southern Ocean.
Overall, the addition of DFe from pyrogenic source raises the global annual NPP by 6.4%, which results from an increase of 7.9% in PHY and 3.4% in DIA (Fig. 10). The global EP increases by 4.6%. Regionally, pyrogenic DFe source can have a larger contribution to the DFe pool and stimulate NPP in a more efficient way. The importance of the additional DFe source to biology is not only determined by the absolute amount of DFe input but also whether deposition fluxes coincide with a strong Fe limitation in the system before deposition. Especially, in the northeastern Pacific (Fig. 8c), the increase of NPP and EP per deposition flux of pyrogenic DFe is nearly three times the efficiency on a global scale20. In some parts of the Southern Ocean, DFe deposition flux is enhanced mainly due to wildfires and the phytoplankton community becomes more diatom-dominated, resulting in an increase of carbon export19. Since the pyrogenic aerosols from wildfires in the Southern Hemisphere deliver DFe into the HNLC regions, the interannual variability of deposition fluxes can substantially influence biogeochemical cycles in ocean models. Therefore, we estimated less NPP change, mainly because of the smaller deposition fluxes of DFe from wildfires16, compared to our previous simulations20. This unintentional ocean Fe fertilization potentially has a similar effect on phytoplankton community composition and carbon export to the artificial ocean Fe fertilizations6,169.
Factors driving different responses in models
We extend our discussion to an inter-model comparison of ocean biogeochemistry models that consider pyrogenic DFe sources (Table 4). These modeling studies demonstrated different magnitudes of global NPP change to the change in atmospheric DFe input and even the sign of NPP change differs. This is partly caused by the different experimental design and assumed different strengths of perturbations in atmospheric DFe input (up to 4-fold) in those models. Some models simulated the transient change in atmospheric DFe input from the pre-industrial time to present-day, and others calculated the equilibrium states of present-day with different atmospheric input fluxes. Other factors such as differences in processes considered in the models and their parameterization can influence the model sensitivity to perturbations as well. The obviously different results in OECO and REcoM-220 indicate the need for a better understanding of the marine Fe cycle at the process level to obtain more reliable results from global ocean biogeochemistry models.
One major factor that can influence the model sensitivity is that other external sources of DFe are less constrained than the atmospheric deposition. Thus, the input fluxes from those sources can vary over several orders of magnitudes in different models143. Indeed, REcoM-2 applied a 5–10 times lower sedimentary source than other models19,25,26 and even larger differences exist for total external DFe sources (Table 4). Thus, the contribution of pyrogenic DFe sources to the global external DFe sources in REcoM-2 is much larger, compared to other models. However, a sensitivity simulation shows that the efficiency of pyrogenic Fe to enhance NPP and EP is not strongly influenced by the strength of sedimentary source in REcoM-220, partly because it is necessary to increase the scavenging rate constant with the increased sedimentary source strengths for more realistic simulation of DFe distribution and estimates of primary production. Thus, the differences in the sensitivity could stem from different parameterizations of processes in the Fe cycle, such as organic complexation and scavenging in ocean models.
Models considering a dependence of organic complexation on biological activities (e.g., DOC- and AOU-parameterization) can have a stronger response than that assumed uniform ligand concentration due to the “knock-on” effect150: if the additional Fe supply induces phytoplankton growth, organic complexation of Fe increases with enhanced DOC production and remineralization. More deposited Fe can be kept in the dissolved form, which gives a positive feedback to the Fe supply. Models with larger external sources usually need to balance with higher scavenging rates and in many models, scavenging rate of DFe is rapidly increased at higher DFe concentrations to take into account the fast removal by “colloidal pumping”170. Therefore, even introducing the same perturbation, the speciation and residence time of added Fe could differ between models and thus induce biological production at different degrees. Furthermore, models treat elemental cycles with different complexity. For example, not all the models consider N2 fixation and denitrification in the N cycle, and the flexibility of Fe:C or Fe:N ratio varies between models, resulting in changes in phytoplankton community composition and making the inter-model comparison more difficult. The BEC model suggested that the anthropogenic atmospheric Fe deposition could have a larger effect on NPP than atmospheric N25. In contrast, in the CESM, global marine NPP and carbon cycle are relatively insensitive to changes in atmospheric deposition of DFe, while N2 fixation, denitrification, phytoplankton community structure, and carbon export are more affected19. In NEMO-PISCES, DFe plays a minor role in affecting global marine NPP in comparison to N supplied by anthropogenic aerosol26. In REcoM-2, neither the atmospheric N input nor N2 fixation and denitrification is considered.
Despite all the difficulties in the inter-model comparison, two factors strongly affecting the sensitivity of an ocean model are most likely the strength of external DFe sources and the parameterization of scavenging. Currently, these are the least constrained components in ocean models. Nevertheless, since a large area of the global ocean receiving high deposition of pyrogenic DFe is limited by Fe, it is vital to take into account this source of Fe, particularly to understand the present Fe cycle on a regional scale and to project responses of the ocean biogeochemistry to the future Fe supply.
Summary and future directions
Pyrogenic aerosol potentially contributes to ocean fertilization by supplying DFe, especially in the HNLC regions where even a small addition of Fe can trigger large phytoplankton blooms. Most previous studies, however, focused on mineral dust and neglected pyrogenic aerosol since the total Fe supply by pyrogenic aerosol is significantly lower than by other aerosol sources. In the last decade, there is increasing observational evidence on the presence of pyrogenic sources to atmospheric DFe supply in the remote marine atmosphere and the dominant role of atmospheric processing (such as acid dissolution) in producing secondary DFe. In addition, more atmospheric models calculated deposition fluxes of DFe from pyrogenic sources, and the number of ocean biogeochemistry models considering pyrogenic DFe as a source of micronutrient is growing. Our understanding on pyrogenic Fe aerosols including their emission, chemical processing, deposition, and impact on ocean biogeochemistry (Fig. 1) has improved substantially in the past few years. However, there are still major challenges that hamper our ability to more quantitatively understand the highly variable pyrogenic Fe sources, the complex aerosol and seawater chemistry, and diverse marine responses to this Fe supply, and to predict their changes under ongoing climate change. On basis of this review, we recommend that the following research should be prioritized:
-
(1)
Developing a comprehensive emission inventory of size-resolved and speciated Fe from pyrogenic sources. There are still large uncertainties in the emission of total Fe and initial Fe solubility for different sources of aerosols. Furthermore, no data is available on the size distribution of Fe species in aerosol sources. New measurements are needed to provide size-resolved emission factors of different Fe species including DFe that can be used to build emission inventories for global models, replacing simplified parameterizations in current models.
-
(2)
Determining Fe dissolution processes, involving acids and organic ligands. Current global aerosol models cannot reproduce aerosol Fe solubility over the Southern Ocean, suggesting that there is a missing source or transformation process of DFe under pristine atmospheric conditions. Furthermore, aerosol water conditions are hugely to simulate, particularly its high ionic strength and complex mixture of organics and inorganics. More research is needed to derive the ligand-promoted and photo-reductive Fe dissolution rates for different types of aerosols in realistic aerosol water conditions. Chamber studies may be needed to test whether the bulk chemistry method97 provides realistic dissolution rates. Such new studies will help to improve the parameterization of dissolution processes in aerosol Fe models.
-
(3)
Observations and source apportionment of pyrogenic Fe aerosols in remote locations. A major challenge in simulating the aerosol Fe cycle is the lack of data to validate the model results. More field observations on Fe chemistry are needed, particularly long-term observations over the more remote oceans, such as the Pacific, Indian, and the Southern Ocean. These include a comprehensive analysis of organic and inorganic species to enable a quantitative source apportionment of Fe and DFe. Such data are essential for validating the model simulations. Using the validated model simulation of deposition fluxes, it can be further examined how the temporal variability of atmospheric deposition impacts the responses of the ocean and what length of the time step in ocean models is required to predict the biological and chemical responses to the sporadic Fe input more accurately.
-
(4)
Improving atmospheric chemistry modeling of Fe. Aerosols with the same size but different compositions should be treated as external mixtures rather than internal mixtures, when calculating their pH and aerosol Fe solubility. More research is needed to quantify their mixing state. Furthermore, a more accurate estimate of the Fe solubility in rainwater is critical in the flux calculation, as the atmospheric delivery of DFe becomes the dominant input into the open ocean. The fate of labile Fe in fog, cloud, and rainwater is subject to large uncertainty, because of a lack of knowledge of specific organic ligands and their formation rates in aerosols and cloud water. While multifunctional aliphatic compounds are suggested as Fe ligands in aerosols171, strong complexation of Fe with organic compounds such as siderophores has been measured in rainwater172,173. The siderophores may be synthesized by microorganisms such as fungi and bacteria in cloud water and act as a strong complexing agent for Fe174,175. The effects of such a high molecular-weight Fe-organic complexation on Fe solubility remains an open question.
-
(5)
Improving ocean modeling of Fe biogeochemistry. Despite the expansion of our understanding of the marine Fe cycle and the improvements in model parameterizations, the observed pattern of DFe distribution is not very well reproduced in the current global Fe models, relative to the model-data agreement of macronutrients. Due to the chemical speciation and particle reactivity of Fe in seawater, the Fe cycle is more complex than cycles of macronutrients. Ocean modelers are struggling between the necessary model complexity to catch the major features of the Fe cycle, the concomitant uncertainties, and the computational costs. Simple assumptions and parameterizations are applied for many processes (e.g., input from external sources, scavenging, and cycle of organic ligands). In most global ocean models, Fe speciation is differentiated between Fe′ and FeL whereas the redox cycling and Fecoll are often implicitly treated as rapid scavenging rate of DFe at higher DFe concentration. Moreover, it is challenging to consider the variety of Fe bioavailability to different phytoplankton groups. Furthermore, only a small number of in situ nutrient limitation measurements are available for validating the modeled limitation pattern of phytoplankton growth. These simplifications and challenges are applicable for Fe from all external sources and their influences in the role of pyrogenic Fe source need to be examined thoroughly.
-
(6)
Laboratory and modeling studies on the role of organics deposited with Fe. How these organics coming with DFe into seawater alter the seawater chemistry of Fe is not well understood and, so far, ignored in ocean models. Many of these organics can form complexes with Fe which undergo interactions with oceanic ligands, and thus extend DFe residence time in the surface water176,177. The abrupt pH change at the sea surface microlayer as well as the future pH change in the ocean can alter the binding capacity with organics150,177. Therefore, more efforts need to be made to investigate the kinetics of these deposited ligands in laboratory studies, and to simulate the modification of Fe solubility by these ligands and changes in the past and future.
-
(7)
Laboratory studies on the acquisition of pyrogenic Fe by marine phytoplankton and bacteria and processing by zooplankton. Acquisition of pyrogenic Fe could be controlled by different uptake mechanisms compared to mineral dust, due to their different chemical forms and various chemical species accompanied with air pollutants. The latter may cause short- or long-term adverse effects in the aquatic environment. Laboratory experiments with phytoplankton species growing with pyrogenic Fe or mineral dust could help us to understand the bioavailability of pyrogenic Fe and further derive necessary parameterizations for models. Fe uptake by marine microbes may interact with other trace elements. For example, copper is toxic at extremely high concentrations178,179. However, its toxicity may be alleviated by Fe or increased by forming complexes with organic matter178,180,181. Developing a mechanistic understanding of the bioavailability of different trace elements found in aerosols, whether soluble or as nanoparticles98 as well as their interactions are therefore underlined. Furthermore, more laboratory and modeling works are needed to quantity the role of zooplankton, which may solubilize aerosol Fe182 due to its low pH in its gut183, in producing more bioavailable Fe for phytoplankton.
-
(8)
Systematic comparison between ocean models. Different experiment designs and parameterizations in ocean models hamper our ability to draw firm conclusions on the impact of pyrogenic Fe on marine biogeochemistry. For a proper estimation of the role of pyrogenic Fe source, simulations with different models need to be conducted using the same fields of aerosol deposition fluxes and for the same periods. Such a harmonized inter-model comparison will help us to disentangle and understand the model-dependent results.
-
(9)
Investigating the role of pyrogenic Fe source in ESMs. Pyrogenic Fe sources evolve with changes in climate and air quality in the past and future. To study the biosphere-climate feedback in ESMs, fundamental work is needed to improve our understanding of bioaccessible Fe supply due to human perturbation. To project the effects of warm climate on the high intensity and long duration of wildfires, more sophisticated fire model is needed184. To project the effects of energy policies and technologies on pyrogenic aerosol emissions, more works are needed for social-economic models185.
Data availability
The modeled and observational data set of aerosols can be obtained from https://progearthplanetsci.springeropen.com/articles/10.1186/s40645-020-00357-9 and http://advances.sciencemag.org/cgi/content/full/5/5/eaau7671/DC1.
Code availability
Code developed for the analysis of the Fe dissolution curves is available upon request from the corresponding author.
References
Boyd, P. W. et al. Mesoscale phytoplankton bloom in the polar Southern Ocean stimulated by iron fertilization. Nature 407, 695–702 (2000).
Tsuda, A. et al. A mesoscale iron enrichment in the western subarctic Pacific induces large centric diatom bloom. Science 300, 958–961 (2003).
Martin, J. H. Glacial‐interglacial CO2 change: the iron hypothesis. Paleoceanography 5, 1–13 (1990).
Boyd, P. W. et al. Mesoscale iron enrichment experiments 1993–2005: synthesis and future directions. Science 315, 612–617 (2007).
Buesseler, K. O. et al. Particle export during the Southern Ocean iron experiment (SOFeX). Limnol. Oceanogr. 50, 311–327 (2005).
Smetacek, V. et al. Deep carbon export from a Southern Ocean iron-fertilized diatom bloom. Nature 487, 313–319 (2012).
Morris, P. J. & Charette, M. A. A synthesis of upper ocean carbon and dissolved iron budgets for Southern Ocean natural iron fertilisation studies. Deep Sea Res. Part II 90, 147–157 (2013).
Capone, D. G., Zehr, J. P., Paerl, H. W., Bergman, B. & Carpenter, E. J. Trichodesmium, a globally significant marine cyanobacterium. Science 276, 1221–1229 (1997).
Falkowski, P. G. Evolution of the nitrogen cycle and its influence on the biological sequestration of CO2 in the ocean. Nature 387, 272–275 (1997).
Duce, R. A., Unni, C. K., Ray, B. J., Prospero, J. M. & Merrill, J. T. Long-range atmospheric transport of soil dust from Asia to the tropical North Pacific: temporal variability. Science 209, 1522–1524 (1980).
Uematsu, M. et al. Transport of mineral aerosol from Asia Over the North Pacific Ocean. J. Geophys. Res. 88, 5343–5352 (1983).
Fung, I. Y. et al. Iron supply and demand in the upper ocean. Glob. Biogeochem. Cycles 14, 281–295 (2000).
Jickells, T. D. et al. Global iron connections between desert dust, ocean biogeochemistry, and climate. Science 308, 67–71 (2005).
Myriokefalitakis, S. et al. The GESAMP atmospheric iron deposition model intercomparison study. Biogeosciences 15, 6659–6684 (2018).
Hamilton, D. S. et al. Improved methodologies for Earth system modelling of atmospheric soluble iron and observation comparisons using the mechanism of intermediate complexity for Modelling Iron (MIMI v.1.0). Geosci. Model Dev. 12, 3835–3862 (2019).
Ito, A. et al. Evaluation of aerosol iron solubility over Australian coastal regions based on inverse modeling: implications of bushfires on bioaccessible iron concentrations in the Southern Hemisphere. Prog. Earth Planet. Sci. 7, 42 (2020).
Mahowald, N. M. et al. Aerosol trace metal leaching and impacts on marine microorganisms. Nat. Commun. 9, 2614 (2018).
Ito, A. et al. Pyrogenic iron: the missing link to high iron solubility in aerosols. Sci. Adv. 5, eaau7671 (2019).
Hamilton, D. S. et al. Impact of changes to the atmospheric soluble iron deposition flux on ocean biogeochemical cycles in the anthropocene. Glob. Biogeochem. Cycles 34, e2019GB006448 (2020).
Ito, A., Ye, Y., Yamamoto, A., Watanabe, M. & Aita, M. N. Responses of ocean biogeochemistry to atmospheric supply of lithogenic and pyrogenic iron-containing aerosols. Geol. Mag. 157, 741–756 (2020).
Lønborg, C., Carreira, C., Jickells, T. & Álvarez-Salgado, X. A. Impacts of global change on ocean dissolved organic carbon (DOC) cycling. Front. Mar. Sci. 7, 466 (2020).
Chen, Y. & Siefert, R. L. Seasonal and spatial distributions and dry deposition fluxes of atmospheric total and labile iron over the tropical and subtropical North Atlantic Ocean. J. Geophys. Res. 109, D09305 (2004).
Baker, A. R. & Jickells, T. D. Mineral particle size as a control on aerosol iron solubility. Geophys. Res. Lett. 33, 1–4 (2006).
Sholkovitz, E. R., Sedwick, P. N., Church, T. M., Baker, A. R. & Powell, C. F. Fractional solubility of aerosol iron: synthesis of a global-scale data set. Geochim. Cosmochim. Acta 89, 173–189 (2012).
Krishnamurthy, A. et al. Impacts of increasing anthropogenic soluble iron and nitrogen deposition on ocean biogeochemistry. Glob. Biogeochem. Cycles 23, GB3016 (2009).
Wang, R. et al. Influence of anthropogenic aerosol deposition on the relationship between oceanic productivity and warming. Geophys. Res. Lett. 42, 10745–10754 (2015).
Hajima, T. et al. Description of the MIROC-ES2L Earth system model and evaluation of its climate–biogeochemical processes and feedbacks. Geosci. Model Dev. 12, 2727–2765 (2020).
Schroth, A. W. Impacts of anthropocene fossil fuel combustion on atmospheric iron supply to the ocean. Encycl. Anthropocene 1, 103–113 (2017).
Tagliabue, A. et al. The integral role of iron in ocean biogeochemistry. Nature 543, 51–59 (2017).
Kanakidou, M., Myriokefalitakis, S. & Tsigaridis, K. Aerosols in atmospheric chemistry and biogeochemical cycles of nutrients. Environ. Res. Lett. 13, 063004 (2018).
Mahowald, N. M. et al. Aerosol deposition impacts on land and ocean carbon cycles. Curr. Clim. Chang. Rep. 3, 16–31 (2017).
Meskhidze, N. et al. Perspective on identifying and characterizing the processes controlling iron speciation and residence time at the atmosphere-ocean interface. Mar. Chem. 217, 103704 (2019).
Ito, A. Global modeling study of potentially bioavailable iron input from shipboard aerosol sources to the ocean. Glob. Biogeochem. Cycles 27, 1–10 (2013).
Ito, A., Lin, G. & Penner, J. E. Radiative forcing by light-absorbing aerosols of pyrogenetic iron oxides. Sci. Rep. 8, 7347 (2018).
Luo, C. et al. Combustion iron distribution and deposition. Glob. Biogeochem. Cycles 22, GB2012 (2008).
Myriokefalitakis, S. et al. Changes in dissolved iron deposition to the oceans driven by human activity: a 3-D global modelling study. Biogeosciences 12, 3973–3992 (2015).
Wang, R. et al. Sources, transport and deposition of iron in the global atmosphere. Atmos. Chem. Phys. 15, 6247–6270 (2015).
Rathod, S. D. et al. Mineralogy-based anthropogenic combustion-iron emission inventory. J. Geophys. Res 125, e2019JD032114 (2020).
Chuang, P. Y., Duvall, R. M., Shafer, M. M. & Schauer, J. J. The origin of water soluble particulate iron in the Asian atmospheric outflow. Geophys. Res. Lett. 32, L07813 (2005).
Baker, A. R., French, M. & Linge, K. L. Trends in aerosol nutrient solubility along a west–east transect of the Saharan dust plume. Geophys. Res. Lett. 33, L07805 (2006).
Sedwick, P. N., Sholkovitz, E. R. & Church, T. M. Impact of anthropogenic combustion emissions on the fractional solubility of aerosol iron: evidence from the Sargasso Sea. Geochem. Geophys. Geosyst. 8, Q10Q06 (2007).
Schroth, A. W., Crusius, J., Sholkovitz, E. R. & Bostick, B. C. Iron solubility driven by speciation in dust sources to the ocean. Nat. Geosci. 2, 337–340 (2009).
Desboeufs, K. V., Sofikitis, A., Losno, R., Colin, J. L. & Ausset, P. Dissolution and solubility of trace metals from natural and anthropogenic aerosol particulate matter. Chemosphere 58, 195–203 (2005).
Sholkovitz, E. R., Sedwick, P. N. & Church, T. M. Influence of anthropogenic combustion emissions on the deposition of soluble aerosol iron to the ocean: empirical estimates for island sites in the North Atlantic. Geochim. Cosmochim. Acta 73, 3981–4003 (2009).
Ito, A. & Feng, Y. Role of dust alkalinity in acid mobilization of iron. Atmos. Chem. Phys. 10, 9237–9250 (2010).
Chen, H. et al. Coal fly ash as a source of iron in atmospheric dust. Environ. Sci. Technol. 46, 2112–2120 (2012).
Fu, H. et al. Solubility of iron from combustion source particles in acidic media linked to iron speciation. Environ. Sci. Technol. 46, 11119–11127 (2012).
Borgatta, J., Paskavitz, A., Kim, D. & Navea, J. G. Comparative evaluation of iron leach from different sources of fly ash under atmospherically relevant conditions. Environ. Chem. 13, 902–912 (2016).
Paris, R., Desboeufs, K. V., Formenti, P., Nava, S. & Chou, C. Chemical characterisation of iron in dust and biomass burning aerosols during AMMA-SOP0/DABEX: implication for iron solubility. Atmos. Chem. Phys. 10, 4273–4282 (2010).
Ito, A. Mega fire emissions in Siberia: potential supply of bioavailable iron from forests to the ocean. Biogeosciences 8, 1679–1697 (2011).
Oakes, M. et al. Iron solubility related to particle sulfur content in source emission and ambient fine particles. Environ. Sci. Technol. 46, 6637–6644 (2012).
Guieu, C., Bonnet, S., Wagener, T. & Loÿe‐Pilot, M. D. Biomass burning as a source of dissolved iron to the open ocean? Geophys. Res. Lett. 32, L19608 (2005).
Bowie, A. R. et al. Biogeochemical iron budgets of the Southern Ocean south of Australia: decoupling of iron and nutrient cycles in the subantarctic zone by the summertime supply. Glob. Biogeochem. Cycles 23, GB4034 (2009).
Taiwo, A. M., Beddows, D. C., Shi, Z. & Harrison, R. M. Mass and number size distributions of particulate matter components: comparison of an industrial site and an urban background site. Sci. Total Environ. 475, 29–38 (2014).
Moreno, T., Gibbons, W., Jones, T. & Richards, R. The geology of ambient aerosols: characterising urban and rural/coastal silicate PM10−2.5 and PM2.5 using high-volume cascade collection and scanning electron microscopy. Atmos. Environ. 37, 4265–4276 (2003).
Zhang, G. et al. Source and mixing state of iron-containing particles in Shanghai by individual particle analysis. Chemosphere 95, 9–16 (2014).
Choël, M., Deboudt, K., Flament, P., Aimoz, L. & Mériaux, X. Single-particle analysis of atmospheric aerosols at Cape Gris-Nez, English Channel: influence of steel works on iron apportionment. Atmos. Environ. 41, 2820–2830 (2007).
Furutani, H. et al. Single-particle chemical characterization and source apportionment of iron-containing atmospheric aerosols in Asian outflow. J. Geophys. Res. 116, D18204 (2011).
Li, W. et al. Air pollution–aerosol interactions produce more bioavailable iron for ocean ecosystems. Sci. Adv. 3, e1601749 (2017).
Moffet, R. C. et al. Iron speciation and mixing in single aerosol particles from the Asian continental outflow. J. Geophys. Res. 117, D07204 (2012).
Ault, A. P. et al. Single-particle SEM-EDX analysis of iron-containing coarse particulate matter in an urban environment: sources and distribution of iron within Cleveland, Ohio. Environ. Sci. Technol. 46, 4331–4339 (2012).
Moteki, N. et al. Anthropogenic iron oxide aerosols enhance atmospheric heating. Nat. Commun. 8, 15329 (2017).
Tavares, F. V. F. et al. Ferruginous compounds in the airborne particulate matter of the metropolitan area of Belo Horizonte, Minas Gerais, Brazil. Environ. Sci. Pollut. Res. 24, 19683–19692 (2017).
Ohata, S. et al. Abundance of light‐absorbing anthropogenic iron oxide aerosols in the urban atmosphere and their emission sources. J. Geophys. Res. 123, 8115–8134 (2018).
Yoshida, A. et al. Abundance and emission flux of the anthropogenic iron oxide aerosols from the East Asian continental outflow. J. Geophys. Res. 123, 11194–11209 (2018).
Lamb, K. D. Classification of iron oxide aerosols by a single particle soot photometer using supervised machine learning. Atmos. Meas. Tech. 12, 3885–3906 (2019).
Yoshida, A. et al. Abundances and microphysical properties of light‐absorbing iron oxide and black carbon aerosols over East Asia and the Arctic. J. Geophys. Res. 125, e2019JD032301 (2020).
Lázaro, F. J., Gutiérrez, L., Barrón, V. & Gelado, M. D. The speciation of iron in desert dust collected in Gran Canaria (Canary Islands): combined chemical, magnetic and optical analysis. Atmos. Environ. 42, 8987–8996 (2008).
Moskowitz, B. M. et al. Iron oxide minerals in dust-source sediments from the Bodélé Depression, Chad: implications for radiative properties and Fe bioavailability of dust plumes from the Sahara. Aeolian Res. 22, 93–106 (2016).
Jia, J., Wang, Y., Xia, D., Lu, H. & Gao, F. Dust sources of last glacial Chinese loess based on the iron mineralogy of fractionated source samples. Geophys. Res. Lett. 46, 2103–2110 (2019).
Maher, B. A., Mutch, T. J. & Cunningham, D. Magnetic and geochemical characteristics of Gobi Desert surface sediments: Implications for provenance of the Chinese Loess Plateau. Geology 37, 279–282 (2009).
Song, Y. et al. Distribution and composition of loess sediments in the Ili Basin, Central Asia. Quat. Int. 334–335, 61–73 (2014).
Baldo, C. et al. Distinct chemical and mineralogical composition of Icelandic dust compared to northern African and Asian dust. Atmos. Chem. Phys. 20, 13521–13539 (2020).
Hettiarachchi, E., Reynolds, R. L., Goldstein, H. L., Moskowitz, B. & Rubasinghege, G. Bioavailable iron production in airborne mineral dust: controls by chemical composition and solar flux. Atmos. Environ. 205, 90–102 (2019).
Kumar, A. & Sarin, M. M. Aerosol iron solubility in a semi-arid region: temporal trend and impact of anthropogenic sources. Tellus B 62, 125–132 (2010).
Kumar, A., Sarin, M. M. & Srinivas, B. Aerosol iron solubility over Bay of Bengal: role of anthropogenic sources and chemical processing. Mar. Chem. 121, 167–175 (2010).
Zhang, T. R., Shi, J. H., Gao, H. W., Zhang, J. & Yao, X. H. Impact of source and atmospheric processing on Fe solubility in aerosols over the Yellow Sea, China. Atmos. Environ. 75, 249–256 (2013).
McDaniel, M. F. M. et al. Relationship between atmospheric aerosol mineral surface area and iron solubility. ACS Earth Space Chem. 3, 2443–2451 (2019).
Zhu, X., Prospero, J. M., Millero, F. J., Savoie, D. L. & Brass, G. W. The solubility of ferric ion in marine mineral aerosol solutions at ambient relative humidities. Mar. Chem. 38, 91–107 (1992).
Meskhidze, N., Chameides, W. L., Nenes, A. & Chen, G. Iron mobilization in mineral dust: Can anthropogenic SO2 emissions affect ocean productivity? Geophys. Res. Lett. 30, 2085 (2003).
Shi, Z., Krom, M. D., Bonneville, S. & Benning, L. G. Atmospheric processing outside clouds increases soluble iron in mineral dust. Environ. Sci. Technol. 49, 1472–1477 (2015).
Spokes, L. J., Jickells, T. D. & Lim, B. Solubilisation of aerosol trace metals by cloud processing: a laboratory study. Geochim. Cosmochim. Acta 58, 3281–3287 (1994).
Baker, A. R., Jickells, T. D., Witt, M. & Linge, K. L. Trends in the solubility of iron, aluminium, manganese and phosphorus in aerosol collected over the Atlantic Ocean. Mar. Chem. 98, 43–58 (2006).
Buck, C. S., Landing, W. M. & Resing, J. Pacific Ocean aerosols: deposition and solubility of iron, aluminum, and other trace elements. Mar. Chem. 157, 117–130 (2013).
Buck, C. S., Landing, W. M., Resing, J. A. & Measures, C. I. The solubility and deposition of aerosol Fe and other trace elements in the North Atlantic Ocean: observations from the A16N CLIVAR/CO2 repeat hydrography section. Mar. Chem. 120, 57–70 (2010).
Trapp, J. M., Millero, F. J. & Prospero, J. M. Trends in the solubility of iron in dust‐dominated aerosols in the equatorial Atlantic trade winds: importance of iron speciation and sources. Geochem. Geophys. Geosyst. 11, Q03014 (2010).
Srinivas, B., Sarin, M. M. & Kumar, A. Impact of anthropogenic sources on aerosol iron solubility over the Bay of Bengal and the Arabian Sea. Biogeochemistry 110, 257–268 (2012).
Srinivas, B., Sarin, M. M. & Rengarajan, R. Atmospheric transport of mineral dust from the I ndo‐G angetic P lain: temporal variability, acid processing, and iron solubility. Geochem. Geophys. Geosyst. 15, 3226–3243 (2014).
Takahashi, Y., Higashi, M., Furukawa, T. & Mitsunobu, S. Change of iron species and iron solubility in Asian dust during the long-range transport from western China to Japan. Atmos. Chem. Phys. 11, 11237–11252 (2011).
Chen, H. H. & Grassian, V. H. Iron dissolution of dust source materials during simulated acidic processing: The effect of sulfuric, acetic, and oxalic acids. Environ. Sci. Technol. 47, 10312–10321 (2013).
Cwiertny, D. M. et al. Characterization and acid‐mobilization study of iron‐containing mineral dust source materials. J. Geophys. Res. 113, D05202 (2008).
Cwiertny, D. M., Young, M. A. & Grassian, V. H. Chemistry and photochemistry of mineral dust aerosol. Annu. Rev. Phys. Chem. 59, 27–51 (2008).
Fu, H., Cwiertny, D. M., Carmichael, G. R., Scherer, M. M. & Grassian, V. H. Photoreductive dissolution of Fe-containing mineral dust particles in acidic media. J. Geophys. Res. 115, D11304 (2010).
Ito, A. & Shi, Z. Delivery of anthropogenic bioavailable iron from mineral dust and combustion aerosols to the ocean. Atmos. Chem. Phys. 16, 85–99 (2016).
Paris, R. & Desboeufs, K. V. Effect of atmospheric organic complexation on iron-bearing dust solubility. Atmos. Chem. Phys. 13, 4895–4905 (2013).
Paris, R., Desboeufs, K. V. & Journet, E. Variability of dust iron solubility in atmospheric waters: Investigation of the role of oxalate organic complexation. Atmos. Environ. 45, 6510–6517 (2011).
Shi, Z. et al. Iron dissolution kinetics of mineral dust at low pH during simulated atmospheric processing. Atmos. Chem. Phys. 11, 995–1007 (2011).
Shi, Z. et al. Influence of chemical weathering and aging of iron oxides on the potential iron solubility of Saharan dust during simulated atmospheric processing. Glob. Biogeochem. Cycles 25, GB2010 (2011).
Shi, Z. et al. Formation of iron nanoparticles and increase in iron reactivity in mineral dust during simulated cloud processing. Environ. Sci. Technol. 43, 6592–6596 (2009).
Cornell, R. M. & Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses (John Wiley & Sons, 2003).
Fang, T. et al. Highly acidic ambient particles, soluble metals, and oxidative potential: a link between sulfate and aerosol toxicity. Environ. Sci. Technol. 51, 2611–2620 (2017).
Ito, A. Atmospheric processing of combustion aerosols as a source of bioavailable iron. Environ. Sci. Technol. Lett. 2, 70–75 (2015).
Hand, J. L. et al. Estimates of atmospheric processed soluble iron from observations and a global mineral aerosol model: biogeochemical implications. J. Geophys. Res. 109, D17205 (2004).
Nickovic, S., Vukovic, A., Vujadinovic, M., Djurdjevic, V. & Pejanovic, G. Technical note: high-resolution mineralogical database of dust-productive soils for atmospheric dust modeling. Atmos. Chem. Phys. 12, 845–855 (2012).
Journet, E., Balkanski, Y. & Harrison, S. P. A new data set of soil mineralogy for dust-cycle modeling. Atmos. Chem. Phys. 14, 3801–3816 (2014).
Ito, A. & Xu, L. Response of acid mobilization of iron-containing mineral dust to improvement of air quality projected in the future. Atmos. Chem. Phys. 14, 3441–3459 (2014).
Scanza, R. A. et al. Modeling dust as component minerals in the Community Atmosphere Model: development of framework and impact on radiative forcing. Atmos. Chem. Phys. 15, 537–561 (2015).
Johnson, M. S. & Meskhidze, N. Atmospheric dissolved iron deposition to the global oceans: Effects of oxalate-promoted Fe dissolution, photochemical redox cycling, and dust mineralogy. Geosci. Model Dev. 6, 1137–1155 (2013).
Kok, J. F. et al. An improved dust emission model—Part 1: model description and comparison against measurements. Atmos. Chem. Phys. 14, 13023–13041 (2014).
Ito, A. & Kok, J. F. Do dust emissions from sparsely vegetated regions dominate atmospheric iron supply to the Southern Ocean? J. Geophys. Res. 122, 3987–4002 (2017).
Hoesly, R. M. et al. Historical (1750–2014) anthropogenic emissions of reactive gases and aerosols from the Community Emission Data System (CEDS). Geosci. Model Dev. 11, 369–408 (2018).
Sato, H., Itoh, A. & Kohyama, T. SEIB-DGVM: a new dynamic global vegetation model using a spatially explicit individual-based approach. Ecol. Model. 200, 279–307 (2007).
Shelley, R. U., Landing, W. M., Ussher, S. J., Planquette, H. & Sarthou, G. Regional trends in the fractional solubility of Fe and other metals from North Atlantic aerosols (GEOTRACES cruises GA01 and GA03) following a two-stage leach. Biogeosciences 15, 2271–2288 (2018).
Siefert, R. L., Johansen, A. M. & Hoffmann, M. R. Chemical characterization of ambient aerosol collected during the southwest monsoon and intermonsoon seasons over the Arabian Sea: Labile‐Fe(II) and other trace metals. J. Geophys. Res. 104D, 3511–3526 (1999).
Zhuang, G. S., Yi, Z., Duce, R. A. & Brown, P. R. Link between iron and sulphur cycles suggested by detection of Fe(II) in remote marine aerosols. Nature 355, 537–539 (1992).
Sullivan, R. C., Guazzotti, S. A., Sodeman, D. A. & Prather, K. A. Direct observations of the atmospheric processing of Asian mineral dust. Atmos. Chem. Phys. 7, 1213–1236 (2007).
Pye, H. O. T. et al. The acidity of atmospheric particles and clouds. Atmos. Chem. Phys. 20, 4809–4888 (2020).
Kandler, K. et al. Differences and similarities of central Asian, African, and arctic dust composition from a single particle perspective. Atmos 11, 269 (2020).
Lin, Q. et al. In-cloud formation of secondary species in iron-containing particles. Atmos. Chem. Phys. 19, 1195–1206 (2019).
Shi, J. et al. High productivity of soluble iron by aerosol acidification in fog. Geophys. Res. Lett. 47, e2019GL086124 (2020).
Rozalén, M. L. et al. Experimental study of the effect of pH on the kinetics of montmorillonite dissolution at 25°C. Geochim. Cosmochim. Acta 72, 4224–4253 (2008).
Shi, Z. B. et al. Minor effect of physical size sorting on iron solubility of transported mineral dust. Atmos. Chem. Phys. 11, 8459–8469 (2011).
Marcotte, A. R., Anbar, A. D., Majestic, B. J. & Herckes, P. Mineral dust and iron solubility: effects of composition, particle size, and surface area. Atmos 11, 533 (2020).
Perron, M. M. G. et al. Origin transport and deposition of aerosol iron to Australian coastal waters. Atmos. Environ. 228, 117432 (2020).
Bikkina, S., Kawamura, K., Sarin, M. & Tachibana, E. 13C probing of ambient photo-fenton reactions involving iron and oxalic acid: Implications for oceanic biogeochemistry. ACS Earth Space Chem. 4, 964–976 (2020).
Zhou, Y. et al. Field evidence of Fe-mediated photochemical degradation of oxalate and subsequent sulfate formation observed by single particle mass spectrometry. Environ. Sci. Technol. 54, 6562–6574 (2020).
Yu, J. Z., Huang, X. F., Xu, J. & Hu, M. When aerosol sulfate goes up, so does oxalate: Implication for the formation mechanisms of oxalate. Environ. Sci. Technol. 39, 128–133 (2005).
Wong, J. P. et al. Fine particle iron in soils and road dust is modulated by coal-fired power plant sulfate. Environ. Sci. Technol. 54, 7088–7096 (2020).
Wang, Z. et al. Enhanced heterogeneous uptake of sulfate dioxide on mineral particles through modification of iron speciation during simulated cloud processing. Atmos. Chem. Phys. 19, 12569–12585 (2019).
Yang, Q., Zhang, L., Xue, Z. & Xu, C. Analyses of sea fog at Great Wall Station, Antarctica. Chin. J. Polar Res. 19, 111–120 (2007).
Conway, T. M., Wolff, E. W., Röthlisberger, R., Mulvaney, R. & Elderfield, H. E. Constraints on soluble aerosol iron flux to the Southern Ocean at the last glacial maximum. Nat. Commun. 6, 7850 (2015).
Liu, K. et al. Dissolved iron concentration in the recent snow of the Lambert Glacial Basin, Antarctica. Atmos. Environ. 196, 44–52 (2019).
Winton, V. H. L. et al. Multiple sources of soluble atmospheric iron to Antarctic waters. Glob. Biogeochem. Cycles 30, 421–437 (2016).
Conway, T. M. et al. Tracing and constraining anthropogenic aerosol iron fluxes to the North Atlantic Ocean using iron isotopes. Nat. Commun. 10, 1–10 (2019).
Kurisu, M., Takahashi, Y., Iizuka, T. & Uematsu, M. Very low isotope ratio of iron in fine aerosols related to its contribution to the surface ocean. J. Geophys. Res. 121, 11119–11136 (2016).
Majestic, B. J., Anbar, A. D. & Herckes, P. Stable isotopes as a tool to apportion atmospheric iron. Environ. Sci. Technol. 43, 4327–4333 (2009).
Xiao, C. et al. Iron in the NEEM ice core relative to Asian loess records over the last glacial−interglacial cycle. Natl Sci. Rev. nwaa144 (2020). https://doi.org/10.1093/nsr/nwaa144.
Baker, A. R. et al. Trace element and isotope deposition across the air–sea interface: progress and research needs. Philos. Trans. R. Soc. A 374, 20160190 (2016).
Kadko, D. et al. Sources, fluxes and residence times of trace elements measured during the US GEOTRACES East Pacific Zonal Transect. Mar. Chem. 222, 103781 (2020).
Baker, A. R., Adams, C., Bell, T. G., Jickells, T. D. & Ganzeveld, L. Estimation of atmospheric nutrient inputs to the Atlantic Ocean from 50°N to 50°S based on large-scale field sampling: Iron and other dust-associated elements. Glob. Biogeochem. Cycles 27, 755–767 (2013).
Theodosi, C., Markaki, Z. & Mihalopoulos, N. Iron speciation, solubility and temporal variability in wet and dry deposition in the Eastern Mediterranean. Mar. chem. 120, 100–107 (2010).
Heimburger, A., Losno, R. & Triquet, S. Solubility of iron and other trace elements in rainwater collected on the Kerguelen Islands (South Indian Ocean). Biogeosciences 10, 6617–6628 (2013).
Tagliabue, A. et al. How well do global ocean biogeochemistry models simulate dissolved iron distributions? Glob. Biogeochem. Cycles 30, 149–174 (2016).
Séférian, R. et al. Tracking improvement in simulated marine biogeochemistry between CMIP5 and CMIP6. Curr. Clim. Change Rep. 6, 95–119 (2020).
Huneeus, N. et al. Global dust model intercomparison in AeroCom phase I. Atmos. Chem. Phys. 11, 7781–7816 (2011).
Mahowald, N., Luo, C., del Corral, J. & Zender, C. S. Interannual variability in atmospheric mineral aerosols from a 22-year model simulation and observational data. J. Geophys. Res. 108, 4352 (2003).
Taylor, S. R. & McLennan, S. M. The Continental Crust: its Composition and Evolution (Blackwell Scientific, 1985).
Zhang, Y. et al. Modeling the global emission, transport and deposition of trace elements associated with mineral dust. Biogeosciences 12, 5771–5792 (2015).
Ito, T., Nenes, A., Johnson, M. S., Meskhidze, N. & Deutsch, C. Acceleration of oxygen decline in the tropical Pacific over the past decades by aerosol pollutants. Nat. Geosci. 9, 443–447 (2016).
Ye, Y., Völker, C. & Gledhill, M. Exploring the iron‐binding potential of the ocean using a combined pH and DOC parameterization. Glob. Biogeochem. Cycles 34, e2019GB006425 (2020).
Ye, Y. & Völker, C. On the role of dust-deposited lithogenic particles for iron cycling in the tropical and subtropical Atlantic. Glob. Biogeochem. Cycles 31, 1543–1558 (2017).
Pham, A. L. & Ito, T. Formation and maintenance of the GEOTRACES subsurface-dissolved iron maxima in an ocean biogeochemistry model. Glob. Biogeochem. Cycles 32, 932–953 (2018).
Liu, X. & Millero, F. J. The solubility of iron in seawater. Mar. Chem. 77, 43–54 (2002).
Aumont, O., Ethé, C., Tagliabue, A., Bopp, L. & Gehlen, M. PISCES-v2: an ocean biogeochemical model for carbon and ecosystem studies. Geosci. Model Dev. 8, 2465–2513 (2015).
Parekh, P., Follows, M. J. & Boyle, E. Modeling the global ocean iron cycle. Glob. Biogeochem. Cycles 18, GB1002 (2004).
Gledhill, M. & Buck, K. N. The organic complexation of iron in the marine environment: a review. Front. Microbiol. 3, 1–17 (2012).
Zhang, J., Kattner, G. & Koch, B. P. Interactions of trace elements and organic ligands in seawater and implications for quantifying biogeochemical dynamics: a review. Earth Sci. Rev. 192, 631–649 (2019).
Johnson, K. S., Gordon, R. M. & Coale, K. H. What controls dissolved iron concentrations in the world ocean? Mar. Chem. 57, 137–161 (1997).
Tagliabue, A. & Völker, C. Towards accounting for dissolved iron speciation in global ocean models. Biogeosciences 8, 3025–3039 (2011).
Misumi, K. et al. Humic substances may control dissolved iron distributions in the global ocean: Implications from numerical simulations. Glob. Biogeochem. Cycles 27, 450–462 (2013).
Völker, C. & Tagliabue, A. Modeling organic iron‐binding ligands in a three-dimensional biogeochemical ocean model. Mar. Chem. 173, 67–77 (2015).
Morel, F. M. M., Kustka, A. B. & Shaked, Y. The role of unchelated Fe in the iron nutrition of phytoplankton. Limnol. Oceanogr. 53, 400–404 (2008).
Strzepek, R. F. et al. Adaptive strategies by Southern Ocean phytoplankton to lessen iron limitation: Uptake of organically complexed iron and reduced cellular iron requirements. Limnol. Oceanogr. 56, 1983–2002 (2011).
Tagliabue, A., Bopp, L. & Aumont, O. Evaluating the importance of atmospheric and sedimentary iron sources to Southern Ocean biogeochemistry. Geophys. Res. Lett. 36, L13601 (2009).
Hauck, J. et al. Seasonally different carbon flux changes in the Southern Ocean in response to the Southern Annular Mode. Glob. Biogeochem. Cycles 27, 1236–1245 (2013).
Edwards, K. F., Thomas, M. K., Klausmeier, C. A. & Litchman, E. Light and growth in marine phytoplankton: allometric, taxonomic, and environmental variation. Limnol. Oceanogr. 60, 540–552 (2015).
Moore, C. M. M. et al. Processes and patterns of oceanic nutrient limitation. Nat. Geosci. 6, 701–710 (2013).
Geider, R. & La Roche, J. Redfield revisited: variability of C:N:P in marine microalgae and its biochemical basis. Eur. J. Phycol. 37, 1–17 (2002).
de Baar, H. J. W. et al. Synthesis of iron fertilization experiments: from the iron age in the age of enlightenment. J. Geophys. Res. 110, C09S16 (2005).
Honeyman, B. D. & Santschi, P. H. A Brownian-pumping model for oceanic trace metal scavenging: evidence from Th isotopes. J. Mar. Res. 47, 951–992 (1989).
Wozniak, A. S., Shelley, R. U., McElhenie, S. D., Landing, W. M. & Hatcher, P. G. Aerosol water soluble organic matter characteristics over the North Atlantic Ocean: implications for iron-binding ligands and iron solubility. Mar. Chem. 173, 162–172 (2015).
Kieber, R., Hardison, D. R., Whitehead, R. F. & Willey, J. D. Photochemical production of Fe(II) in rainwater. Environ. Sci. Technol. 37, 4610–4616 (2003).
Cheize, M. et al. Iron organic speciation determination in rainwater using cathodic stripping voltammetry. Anal. Chim. Acta 736, 45–54 (2012).
Sullivan, T. S., Ramkissoon, S., Garrison, V. H., Ramsuhlag, A. & Thies, J. E. Siderophore production of African dust microorganisms over Trinidad and Tobago. Aerobiologia 28, 391–401 (2012).
Vinatier, V. et al. Siderophores in cloud waters and potential impact on atmospheric chemistry: production by microorganisms isolated at the Puy de Dôme Station. Environ. Sci. Technol. 50, 9315–9323 (2016).
Meskhidze, N., Hurley, D., Royalty, T. M. & Johnson, M. S. Potential effect of atmospheric dissolved organic carbon on the iron solubility in seawater. Mar. Chem. 194, 124–132 (2017).
Avendaño, L., Gledhill, M., Achterberg, E. P., Rérolle, V. M. C. & Schlosser, C. Influence of ocean acidification on the organic complexation of iron and copper in Northwest European shelf seas: a combined observational and model study. Front. Mar. Sci. 3, 58 (2016).
Paytan, A. et al. Toxicity of atmospheric aerosols on marine phytoplankton. Proc. Natl Acad. Sci. USA 10, 4601–4605 (2009).
Zhang, C. et al. Fertilization of the Northwest Pacific Ocean by East Asia air pollutants. Glob. Biogeochem. Cycles 33, 690–702 (2019).
Meng, X., Chen, Y., Wang, B., Ma, Q. W. & Wang, F. J. Responses of phytoplankton community to the input of different aerosols in the East China Sea. Geophys. Res. Lett. 43, 7081–7088 (2016).
Wang, F. et al. Combined effects of iron and copper from atmospheric dry deposition on ocean productivity. Geophys. Res. Lett. 44, 2546–2555 (2017).
Schmidt, K. et al. Gut passage mobilizes lithogenic iron for ocean productivity. Curr. Biol. 26, 2667–2673 (2016).
Davis, A., Nasser, F., Lead, J. R. & Shi, Z. Development and application of a ratiometric nanosensor for measuring pH inside the gastrointestinal tract of zooplankton. Environ. Sci. Nano 7, 1652–1660 (2020).
Li, F. et al. Historical (1700–2012) global multi-model estimates of the fire emissions from the Fire Modeling Intercomparison Project (FireMIP). Atmos. Chem. Phys. 19, 12545–12567 (2019).
Nicholls, Z. R. J. et al. Reduced Complexity Model Intercomparison Project Phase 1: introduction and evaluation of global-mean temperature response. Geosci. Model Dev. 13, 5175–5190 (2020).
Acknowledgements
Support for this research was provided to A.I. by JSPS KAKENHI Grant Number 20H04329, Integrated Research Program for Advancing Climate Models (TOUGOU) Grant Number JPMXD0717935715 from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and the joint research program of the Institute for Space-Earth Environmental Research, Nagoya University. Y.Y. is funded by DFG (German Research Foundation, YE170/2-1) and PalMod (Federal Ministry of Education and Research Germany, BMBF 01LP1505C). Z.S. and C.B. are funded by UK NERC - Natural Environment Research Council (NE/S00579X/1) and NERC CENTA Ph.D. studentship grant (NE/L002493/1). We thank Mrs. Chantal Jackson at the University of Birmingham for drawing the Fig. 1.
Author information
Authors and Affiliations
Contributions
A.I. proposed the concept, hosted a workshop at Nagoya University in 2019 (A.I., Y.Y., and Z.S.), and wrote the draft of the introduction and atmospheric iron-containing chemistry models. C.B. and Z.S. wrote the draft of observations and laboratory measurements. Y.Y. wrote the draft of ocean biogeochemistry models. All authors contributed to revising and finalizing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ito, A., Ye, Y., Baldo, C. et al. Ocean fertilization by pyrogenic aerosol iron. npj Clim Atmos Sci 4, 30 (2021). https://doi.org/10.1038/s41612-021-00185-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41612-021-00185-8
This article is cited by
-
The role of biota in the Southern Ocean carbon cycle
Nature Reviews Earth & Environment (2024)
-
Climate-relevant properties of black carbon aerosols revealed by in situ measurements: a review
Progress in Earth and Planetary Science (2023)
-
Stable iron isotopic composition of atmospheric aerosols: An overview
npj Climate and Atmospheric Science (2022)
-
Size-dependent aerosol iron solubility in an urban atmosphere
npj Climate and Atmospheric Science (2022)
-
The underappreciated role of anthropogenic sources in atmospheric soluble iron flux to the Southern Ocean
npj Climate and Atmospheric Science (2022)
