
Abstract
The RxPONDER and TAILORx trials demonstrated benefit from adjuvant chemotherapy in patients age ≤ 50 with node-positive breast cancer and Recurrence Score (RS) 0–26, and in node-negative disease with RS 16–25, respectively, but no benefit in older women with the same clinical features. We analyzed transcriptomic and genomic data of ER+/HER2− breast cancers with in silico RS < 26 from TCGA (n = 530), two microarray cohorts (A: n = 865; B: n = 609), the METABRIC (n = 867), and the SCAN-B (n = 1636) datasets. There was no difference in proliferation-related gene expression between age groups. Older patients had higher mutation burden and more frequent ESR1 copy number gain, but lower frequency of GATA3 mutations. Younger patients had higher rate of ESR1 copy number loss. In all datasets, younger patients had significantly lower mRNA expression of ESR1 and ER-associated genes, and higher expression of immune-related genes. The ER- and immune-related gene signatures showed negative correlation and defined three subpopulations in younger women: immune-high/ER-low, immune-intermediate/ER-intermediate, and immune-low/ER-intermediate. We hypothesize that in immune-high cancers, the cytotoxic effect of chemotherapy may drive the benefit, whereas in immune-low/ER-intermediate cancers chemotherapy induced ovarian suppression may play important role.
Similar content being viewed by others


Introduction
Most breast cancers are diagnosed in women older than 501. Age is not only a risk factor for cancer, but it also interacts with adjuvant chemotherapy benefit in hormone receptor positive/human epidermal growth factor receptor-2 negative (HR+/HER2−) breast cancers2. Three randomized trials demonstrated greater chemotherapy benefit in younger compared to older women3. The TAILORx trial showed improved invasive disease-free survival (IDFS) with chemotherapy in addition to adjuvant endocrine therapy in patients younger than 50 with lymph-node negative breast cancer and OncotypeDx 21-gene Recurrence Scores (RS) between 16 and 25, no benefit was seen in women older than 504. The RxPONDER trial randomized patients with 1–3 positive lymph nodes and RS 0–25 to either adjuvant endocrine therapy or endocrine therapy plus chemotherapy5. It also demonstrated improved IDFS with chemotherapy in premenopausal patients, or in patients 50 or younger, but no benefit was seen in older women5. In the MINDACT trial, a subset of HR+/HER2− patients with high clinical risk and low genomic risk (by the MammaPrint assay) were randomly assigned to receive adjuvant chemotherapy or not6. An exploratory analysis showed improved distant metastasis-free survival (DMFS) with chemotherapy compared to endocrine therapy alone in women younger than 50, but not in women older than 506. In all three trials, the most frequently used endocrine therapy for premenopausal women was tamoxifen.
It is unclear what explains the interaction between age and adjuvant chemotherapy benefit. Age is difficult to separate from its association with menopausal status. The mean age of onset of menopause is 51 years in Western countries and by age 55 approximately 85% of women have undergone menopause7,8. Adjuvant chemotherapy in pre-menopausal women can induce menopause in an age-dependent manner9,10. The NSABP B-47 clinical trial showed that chemotherapy induced amenorrhea in pre-menopausal women is common but it is often discordant with hormone level measurements. In this study, 85% of patients were amenorrhoeic at 12 months after starting adjuvant chemotherapy but only 28 and 22% had postmenopausal estradiol levels at 12 and 24 months11. The SOFT and TEXT trials demonstrated that in premenopausal HR+ patients ovarian suppression plus an aromatase inhibitor is more effective than tamoxifen alone to improve recurrence-free survival12,13. Chemotherapy-induced menopause can therefore contribute to adjuvant chemotherapy benefit. However, younger patients also have more chemotherapy sensitive cancers. A pooled analysis of 9000 patients enrolled in neoadjuvant chemotherapy trials showed that the pathologic complete response (pCR) rate is significantly higher in the younger HR+/HER2− patients14.
In the past 20 years, three types of molecular features emerged that predict endocrine and chemotherapy sensitivities in early stage-breast HR+/HER2− cancer; (i) expression of estrogen receptor (ER) regulated genes is a measure of endocrine sensitivity and is associated with better prognosis15, (ii) proliferation, and (iii) immune infiltration related markers are independently associated with greater chemotherapy sensitivity in neoadjuvant chemotherapy trials16,17,18.
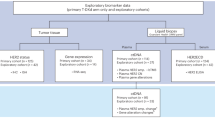
The goal of the current analysis was to compare differences in estrogen receptor (ER)-, proliferation-, and immune-related gene expressions, and somatic mutation patterns and mutation burden between younger (≤50 years of age) and older (≥55 years) patients with HR+/HER2− breast cancer that could explain the chemotherapy benefit in younger women. These age cohorts were selected because the ≤50 group is highly enriched in pre-menopausal women and represents the group where all the chemotherapy benefit accrues, whereas the ≥55 group is almost entirely composed of post-menopausal women8. We further restricted our analysis to the subset of patients who were in the lower 80% range of in silico RS distribution to mimic the RxPONDER and TAILORX populations that excluded women with RS > 25.
Results
Patient characteristics
Patient and tumor characteristics, including molecular subtype distribution, and available treatment information are presented in Table 1. The median ages of the younger and older patients ranged between 45–46 and 66–69 years across the datasets.
Differences in ER signaling, cell proliferation, and immune infiltration
ESR1 mRNA expression was significantly lower in younger women in all cohorts (P < 0.001; Fig. 1a, c, e, Supplementary Fig. 1). Lower mRNA expression in bulk RNA analysis could be due to either fewer ER-positive cancer cells, that could be reflected by lower ER percent positivity by immunohistochemistry (IHC), or to lower ER mRNA expression within ER-positive cells. To distinguish between these two possibilities, we plotted age distribution in ten IHC percent positivity brackets from 1 to 10% to >90% in increments of 10 in the TCGA data where this information was available (n = 338). We observed no statistically significant correlation between age and increasing ER IHC percent positivity (τ = 0.036, P < 0.19, Supplementary Fig. 2a). Overall, ESR1 mRNA expression increased as IHC percent positivity increased (τ = 0.27, P < 0.0001), reaching a plateau after > 40% (Supplementary Fig. 2b). ESR1 mRNA expression showed positive association with age at diagnosis (Spearman coefficient = 0.41, P < 0.0001) (Supplementary Fig. 2c). A regression model of ESR1 mRNA expression using age and IHC positivity showed contribution of both parameters but a larger effect size of age (standardized beta 0.365) than percentage of IHC positivity (standardized beta 0.215). This suggests that the overall lower ESR1 mRNA expression in younger patients is primarily driven by lower ESR1 mRNA levels in ER positive cancer cells.

a ESR1 mRNA and b MKI67 mRNA expression in TCGA cohort; c ESR1 and d MKI67 expression in Microarray Cohort-A; e ESR1 and f MKI67 expression in Microarray Cohort-B. P-values were estimated by the two-side Wilcoxon rank-sum test. Each dot represents a patient, the solid horizontal line indicates the median expression in each age group. Relationship between ERS-pos and B-Cell gene signatures in women 50 years of age or younger (g) and 55 years of age or older (h). The values from negative to positive in the X and Y axes denote increasing signature expression. Z-axis indicates case density (i.e., number of patients), color coding reflects increasing case density from dark blue to yellow.
Next, we assessed the expression of four gene signatures that are positively associated with endocrine therapy sensitivity including a 4-gene ERS19, a 7-gene ERS-Lum19, a 106-gene ERS-Pos signature15, and a 59-gene ERS-Neg signature15 which is negatively associated with ER expression and endocrine sensitivity15. Both in the TCGA and in the Metabric cohort, the ERS, ERS-Lum, and ERS-Pos signatures were all significantly lower (FDR < 0.03) while the ERS-Neg signature was higher (FDR < 0.001) in younger patients (Table 2). Similarly, in both microarray cohorts, and in the SCAN-B-cohort, the ERS-Pos signature was lower and the ERS Neg signature was higher in the younger age group (FDR < 0.002; Table 2). The two smaller signatures, ERS and ERS-Lum, showed nominally lower expressions in younger patients in cohort-A without reaching statistical significance. In cohort-B, ERS showed lower expression in young patients whereas ERS-Lum was similar between age groups (Table 2). Overall, these results indicate not only downregulation of ESR1 mRNA expression but also lower ER-associated gene expression in ER positive cancers of younger compared to older patients.
mRNA expression of the MKI67 gene, that codes for the Ki67 proliferation marker, was similar between age groups in TCGA and microarray cohort-A, but was slightly but statistically significantly higher in the younger patients in microarray cohort-B (Fig. 1b, d, f and Supplementary Fig. 1). The expression of a 12-gene mitotic kinase gene signature (MKS), that has been associated with worse prognosis in HR positive breast cancers and higher sensitivity to neoadjuvant chemotherapy14, did not differ statistically significantly between the age groups in all cohorts (Table 2). However, the most highly proliferative tumors with the highest 20% of in silico RS were not included in this analysis by design.
Next, we assessed 4 different immune cell signatures20 and a tumor inflammation signature21 that were previously shown to predict response to chemotherapy and immune checkpoint inhibitor therapy (Table 2). In the TCGA, B-cell, T-cell, Mast-cell, and TIS signatures were significantly higher, the dendritic signature only showed nominally increased expression (FDR = 0.22). In the microarray Cohort-A, B cells and mast cells were significantly higher, the T cell and TIS signatures showed a trend for higher expression. In Cohort-B, T cells, B cells, TIS, and dendritic cells signatures were significantly higher in younger patients (Table 2). We also evaluated these gene signatures in the METABRIC and SCAN-B data sets and found similar associations (Table 2). We also performed an immune cell composition analysis in the TCGA data using the ConsensusTME method22. Consistent with the gene signature results, younger patients had higher levels B cells, Cytotoxic cells, Endothelial, Fibroblasts, Plasma cells, CD4 T cell, CD8 T cells, and T regulatory cell markers (Supplementary Fig. 3).
Next, we assessed correlation between the ESR1, MKI67 expression, and the 10 gene signatures in Table 2. The MKI67 expression and MKS signature, and ESR1 expression and the ERS-Pos gene signature were each highly correlated. The correlation between ESR1 and the other ER-related gene signatures was less strong. Among the immune signatures, the T cell, B cell, and TIS signatures showed the highest co-expression. The ER-related and immune signatures showed moderate negative correlation in all 3 data sets (Pearson correlation coefficients −0.24, −0.31, −0.25) suggesting independent predictive functions (Supplementary Fig. 4). The distributions of the B cell and ERS-Pos signatures in the TCGA cohort are shown on Fig. 1g, h and illustrate that in the age ≤50 group, three patient populations are intermixed including those with immune-intermediate/ER-intermediate (largest subset), immune-low/ER-intermediate, and immune-high/ER-low (smallest subset) cancers, while in the older age group the immune-low/ER-high cancers are predominant.
Differentially expressed genes and pathways between age groups
In the TCGA, we identified 713 up- and 77 downregulated genes in younger patients (Fig. 2a and Supplementary Table 1). In microarray cohorts A and B, we found 122 and 95 upregulated and 15 and 14 downregulated genes, respectively (Fig. 2b, c, Supplementary Tables 2 & 3, and Supplementary Fig. 5). Thirty-one upregulated genes in younger patients were shared in all three analyses (Fig. 2d, e). Twenty-five and 11 of the 31 overlapped DEGs were also upregulated in young patients in SCAN-B and METABRIC cohort, respectively (Supplementary Table 4). ESR1 and CRABP2 were down-regulated in both SCAN-B and METABRIC cohorts (Supplementary Table 4). In gene set enrichment analysis, 22 biological pathways showed differential expression by age in TCGA; 7 were immune and inflammation related, the others represented estrogen, K-ras, and hedgehog signaling, epithelial mesenchymal transition, angiogenesis, and apical junction/apical surface pathways (Supplementary Table 5).

Volcano plots indicate the log2 fold change (FC) and FDR of differentially expression analysis in three cohorts, including a TCGA cohort; b microarray cohort-A; c microarray cohort-B. P-values were generated by Student’s t-test and corrected by Benjamini–Hochberg FDR. Red dots indicated genes meet criteria of fold change ≥ 1.50 or ≤ 0.67 and FDR < 0.05 (significant), and other genes were marked as gray (none, not significant). Top enriched genes with fold change ≥ 1.50 or ≤ 0.67 and FDR < 1e−05 were labeled with official gene symbols. d, e Venn diagram shows the number of upregulated (d) and downregulated (e) genes identified in the TCGA that also showed up- or downregulation in the other two data sets.
Comparison of somatic mutations and copy number variations (CNV) in younger versus older patients in TCGA
The somatic mutation burden was significantly higher in older patients (P < 0.0001; Fig. 3a), consistent with age-related accumulation of mutations23. At gene level, 13 genes had mutation frequencies ≥ 5% and only GATA3 showed a significantly higher mutation frequency in younger patients (26% versus 12%, P < 0.0001; Fig. 3b). In multivariate logistic regression analysis, luminal B tumors were associated with the enrichment of GATA3 mutations (P = 0.011, odds ratio = 2.18), younger patients also had higher rate of GATA3 mutations (P < 0.0001, odds ratio = 3.15). These results are consistent with an earlier report that showed GATA3 mutation enrichment in luminal B cancers from young women24.

a Tumor mutation burden. P-values from Wilcoxon rank-sum test. b Somatic mutation frequency. Only genes with mutation frequency > 5% in both groups are shown. ***Fisher’s exact test p value < 0.0001.
We also compared the CNV gain and loss of 705 Catalog Of Somatic Mutations In Cancer (COSMIC) genes25. We identified high rate of CNV gain of ESR1, LATS1, ARID1B, SGK1, and MYB genes (odds ratio > 8.5, FDR < 0.05) in old patients (Supplementary Table 6). Young patients have a higher rate of CNV loss of ESR1 gene (odds ratio = 0.45, FDR = 0.03, Supplementary Table 6). In addition to ESR1, we identified 19 and 29 genes have higher rate of CNV loss in young and old patients, respectively (Supplementary Table 6).
Discussion
In independent data sets including n = 4507 ER+/HER2− breast cancers, we found that cancers in patients 50 or younger have lower expression of ESR1 and ER-related genes and higher expression of immune related genes. Increasing ER expression with older age has been described in earlier studies that analyzed all breast cancer subtypes together26. A significant linear relationship between increasing age and ESR1 mRNA expression was also seen in luminal-A and -B breast cancers27. The biological reasons behind this phenomenon are unclear. In normal breast epithelium in premenopausal women, the ER expression fluctuates during the menstrual cycle, and ER expression is highest during the follicular phase28,29. Based on this observation, one would expect higher average ER expression in premenopausal women, however, we found the opposite. We hypothesize that ER expression in breast epithelial cells, and in cancers that arise from them, may increase as estrogen levels decrease with aging due to a feedback loop. Indeed, several studies showed increased ER expression in normal breast epithelium with increasing age30,31.
The clinical relevance of lower ESR1 and ER related gene expression in cancers of younger women is uncertain. However, ER-associated genes are components of all clinically validated multi-gene prognostic signatures32, and higher expression levels are associated with better prognosis with adjuvant endocrine therapy33. Higher ER-associated gene expression is also associated with longer PFS and OS in metastatic breast cancer treated with endocrine therapy34. These results suggest that lower ESR1 and ER-related gene expression in younger women may indicate lower endocrine sensitivity. Intensifying endocrine therapy could maximize benefit, which is consistent with clinical trial results that demonstrated ovarian suppression plus tamoxifen, or exemestane, is more effective than tamoxifen alone to improve recurrence-free survival in premenopausal women.
The higher immune gene expression in younger HR+/HER2− breast cancer patients compared to older patients has not previously been reported. The cause of the higher immune infiltration is unknown. Somatic mutation burden that could increase neoantigen load was lower in younger patients. The gene expression data suggests an important role for CXCL13 that was the most highly and consistently overexpressed chemokine in cancers from younger women. CXCL13 is secreted by dendritic and endothelial cells, and is a powerful B cell attractant, that can also activate helper T cells35. High expression of CXCL13 is predictive of better survival in HR+/HER2− breast cancer patients treated with adjuvant chemotherapy36, and is associated with higher pathologic complete response rate after neoadjuvant chemotherapy in HR+ breast cancers17. These observations suggest that HR+/HER2− breast cancer in younger patients may have higher chemotherapy sensitivity due to greater immune infiltration in the tumor microenvironment than cancers in older women, even if proliferation related predictive markers are similar. When we examined immune and ER related gene expression distributions jointly, we found 3 distinct sub-populations among younger women; (i) immune-high/ER-low, (ii) immune-intermediate/ER-intermediate, and (iii) immune-low/ER-intermediate cancers. The impact of adjuvant chemotherapy is likely different in these different subgroups. We hypothesize that in immune-high/intermediate and ER-low/intermediate cancers the cytotoxic effect drives the benefit, whereas in immune-low/ER-intermediate cancers chemotherapy-induced ovarian suppression plays a more important role. These observations add to the already existing literature that described general molecular differences between breast cancers in younger and older women including elevated integrin/laminin and EGFR and TGFβ signaling and numerous age-associated genes37,38,39. To increase our ability to identify differences between pre- and post-menopausal ER+ breast cancers our analysis focused on cancers from woman < 50 and >55 years of age groups and excluded the perimenopausal age group 50 to 55. We further restricted our analysis by excluding cases with the highest 20% of in silico RS. This is an important feature of our analysis that has impacted the findings, unlike all previous studies that find higher prevalence of luminal B cancers in younger women, our comparison cohorts were balanced for luminal A and B subtypes. This indicates that the higher chemotherapy benefit is not due to higher proportion of Luminal B cancers among premenopausal women with Recurrence Score <26. Finally, our purpose was to examine differences, if they exist, in carefully selected clinically validated biologic features that predict for chemotherapy and endocrine therapy sensitivity so that we could generate a hypothesis of why younger patients benefit more from chemotherapy.
This study has limitations. We were unable to assess the interaction between adjuvant treatments, molecular features and survival in the young women due to lack of patient specific treatment information in our datasets and lack of randomization. However, we describe a testable hypothesis that could be examined in future clinical trials prospectively, or retrospectively, when gene expression data becomes available from samples of the TAILORx or RxPONDER trials. We describe biological features that are highly reproducible across independent datasets and across different mRNA quantification platforms which implies that these robust gene expression features could be captured by standardized assays in the future.
Overall, our analysis suggests that both the cytotoxic and endocrine effects of adjuvant chemotherapy could contribute to the overall survival benefit seen in younger patients but the relative contributions of these effects may vary by the immune cell composition and ER expression of these cancers.
Methods
TCGA breast cancer cohort
mRNA expression, somatic mutation, and clinical data of 1085 primary breast cancer patients were obtained from TCGA (https://gdc.cancer.gov/about-data/publications/pancanatlas). The RNAseq expression matrix of Fragments per Kilobase of transcript per Million mapped reads (FPKM) was upper quantile normalized and subsequently log2 transformed. Percent ER positivity assessed by routine clinical immunohistochemistry (IHC) was available for 1037 cases40. We excluded the ER-negative (n = 238) and HER2 amplified (n = 100) cases, and cases without ER information (n = 48). We assigned HER2 status based on HER2 mRNA expression that follows a bimodal expression pattern41. We used the Bayesian information criterion to find the number of components in the Gaussian mixture model and used GaMRed (http://cellab.polsl.pl/index.php/software?id=28)42 to select the optimal threshold value (normalized FPKM equal to 15.17) to define HER2 gene overexpression. To mimick the TAILORx and RxPONDER populations we also excluded case with the top 20% in silico calculated RS score (n = 74). For final analysis, we grouped ER+/HER2− cancers (n = 530) into ≤ 50 (n = 159) or ≥ 55 years of age (n = 371) at diagnosis (Supplementary Fig. 6).
Microarray cohorts
From publicly available Affymetrix microarray datasets we identified 2007 unique, previously untreated breast cancer samples that were (i) annotated with age, (ii) had raw MAS5 data deposited, and (iii) were ER+/HER2−43 (Supplementary Fig. 6). We assembled 27 Affymetrix U133A datasets from GEO (https://www.ncbi.nlm.nih.gov/geo/) and ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) (E-TABM-158, GSE11121, GSE12276, GSE16391, GSE17907, GSE18864, GSE19615, GSE20194, GSE2034, GSE2109, GSE21653, GSE22035, GSE22513, GSE2603, GSE26971, GSE2990, GSE3494, GSE4611, GSE46184, GSE4922, GSE5327, GSE6532, GSE6532, GSE6596, GSE7390, GSE9195, MDA133) with no overlap to the RNA-Seq sample cohort from TCGA. We included only datasets with MAS5 data available (i.e., Individual sample level normalized expression data) without cohort-based normalization steps (e.g., RMA). A total of 3292 unique samples were annotated with age and had raw MAS5 data deposited. From these, we selected 2007 ER+/HER2− samples based on gene expression data as previously described43 (Supplementary Fig. 6). Supplementary Table 7 lists details for each sample including clinical information and a link to the corresponding expression data.
For the most accurate identification of differentially expressed genes, we aimed to assemble the most homogenous combined dataset with respect to technical bias and platform heterogeneity. To accomplish this, we used our previously described pipeline44 and designated this dataset as “Cohort A”. We calculated a technical comparability metric “C” which is the sum of squared normalized differences between dataset means and global means for all genes and considered datasets highly comparable if normalized C < 0.05. This resulted in 13 data sets including n = 1170 samples assigned into Cohort-A. For a second independent validation, we also combined all remaining datasets into Cohort B including n = 837 samples that correspond to data with grater technical heterogeneity (Supplementary Fig. 6).
From each cohort, we then excluded cases in the top 20% of highest in silico Recurrence score values to mimic a clinical cohort similar to that of TAILORx that included only patients with RS < 26. This resulted in n = 936 cases in Cohort A and n = 669 cases in Cohort B. For final analysis, we grouped ER+/HER2− cancers into ≤ 50 (n = 281 in cohort-A, n = 162 in cohort-B) versus ≥ 55 (n = 584 in cohort-A, n = 447 in cohort-B) years of age (Supplementary Fig. 6).
METABRIC datasets
Normalized tumor mRNA expression data and the clinical metadata of 1908 breast cancer patients45 were download from www.cbioportal.org. We excluded 723 ER-negative or HER2 amplified cases, 61 cases without ER or HER2 status, and 240 cases with the top 20% RS score. For final analysis, we grouped ER+/HER2− cancers (n = 867) into ≤ 50 (n = 157) or ≥ 55 years of age (n = 710) at diagnosis (Supplementary Fig. 6).
SCAN-B datasets
Normalized tumor mRNA expression data and the clinical metadata of 2969 breast cancer patients were downloaded from the Gene Expression Omnibus (GEO) database (GSE96058)46 (Supplementary Fig. 6). ER status assessed by immunohistochemistry was available for 2,783 patients, and HER2 status reported by situ hybridization was available for 2868 patients. We excluded the ER-negative (n = 224) and HER2 amplified (n = 378) cases, cases without ER (n = 199) or HER2 (101) status, and cases with top 20% RS score (n = 409). For final analysis, we grouped ER+/HER2− cancers (n = 1636) into ≤ 50 (n = 305) or ≥ 55 years of age (n = 1331) at diagnosis (Supplementary Fig. 6).
Calculation of in silico recurrence score
We calculated an in silico recurrence score for each sample using the oncotypedx function of the genefu R library47. These scores approximate the clinical OncotypeDX RS but are not equivalent due to different dynamic ranges of the measurements. In clinical studies, 15–20% of cases submitted for OncotypeDx testing have RS > 2548,49. In the screening phase of TAILORx, 17% of patients had RS > 25. To approximate this distribution, we excluded patients with the top 20% of the highest continuous in silico recurrence scores.
Molecular subtyping
Molecular subtype assignments of TCGA samples were obtained from Peng et al.50. To assign molecular subtypes to samples from the microarray cohorts we used the R package AIMS under R version 3.3.051.
Gene-expression signatures
To assess ER and Ki67 expression in the microarray data, we used the ESR1 probe set 205225_at, and the average of four MKI67 probe sets as previously described43. Ten mRNA expression signatures were obtained from literature including four estrogen-related signatures (e.g., ERS, ERS Luminal19, ERS Pos Symmans15, and ERS Neg Symmans15), four immune cell signatures (e.g., T Cell, B Cell, Mast Cell, Dendritic Cell20, and Tumor inflammation signature [TIS]21), and one proliferation signature (Mitosis Kinase Score, MKS19) (Supplementary Table 8). For each signature, we calculated the average normalized expression of the member genes and transformed to z-score across all cases in each cohort.
Immune-cell composition analysis
Immune cell composition was estimated using the ConsensusTME22 method that estimates the contribution of 18 immune cell types to the tissue microenvironment. We used normalized TCGA mRNA expression data as input and select ssGSEA method for immune cell signature analysis with the ConsensusTME R package22.
Differentially expressed genes
To identify differentially expressed genes (DEGs) in TCGA RNAseq data (representing 20,282 human genes), we calculated fold change and t-test p-value for each gene between younger and older cases. DEGs were defined as fold change ≥ 1.50 (i.e., upregulated) or ≤ 0.67 (i.e., downregulated) with Benjamini Hochberg corrected false discovery rate (FDR) < 0.05. To identify DEGs from Affymetrix microarray data, we applied the limma R package52. To avoid batch effects, we included the original Affymetrix source dataset as covariate. Identical fold change filters were used as for TCGA data.
Gene set enrichment analysis
Log2 transformed fold changes of all 20,282 genes of TCGA samples were used as gene rank values to perform gene set enrichment analysis using the fgsea53 package in R using the hallmark gene set (n = 50) of the Molecular Signatures Database (MSigDB)54.
Somatic mutation analysis
Somatic mutations which were available for 427 older and 183 younger TCGA breast cancer cases were obtained from the Multi-Center Mutation Calling in Multiple Cancers (MC3) dataset55. Somatic mutation burden was calculated as the total number of somatic mutations across all genes in each cancer. For comparison of gene level somatic mutation frequencies between age groups we only considered the nonsynonymous mutations, including missense, non-sense, frameshifting, in-frame shifting, or splice-site altering single-nucleotide changes or indels and statistical significance was assessed with Fisher’s exact test. A multivariate logistic regression model was used to evaluate the association of Luminal B subtype and age group with the mutation status of GATA3:
Association of ER status and age at diagnosis
We estimated the statistical significance of the trend of the ER IHC percentage categories with ESR1 mRNA expression and age at diagnosis using Jonckheere Terpstra (JT) trend analysis56. P-values were calculated using the “JonckheereTerpstraTest” function of “DescTools” R package57. Kendall’s tau (τ) coefficient was estimated to measure the increasing (positive value) or decreasing (negative value) trend for each trend analysis. We estimated the correlation between ESR1 mRNA expression and age of diagnosis using Spearman’s rank correlation analysis.
Copy number variation analysis
We obtained gene-level somatic CNV data of TCGA patients from the PanCanAtlas Aneuploidy study (https://gdc.cancer.gov/about-data/publications/pancanatlas)58. The CNVs of 25,128 genes of 513 ER+/HER2− patients were available. We focus on the 703 genes that overlapped with the COSMIC cancer gene list. The gene-level events indicate that the copy number gain/loss effect an entire chromosome arm or a specific genomic region that encodes gene. CNV was assessed with Affymetrix SNP 6.0 arrays58 and gene-level CNV values were generated by GISTIC59. A GISTIC call of +1 or +2 was considered a gain and −1 or −2 was considered a loss, and 0 as wild-type for association analysis in our study. The association of CNV gain or loss with the age group was assessed with Fisher’s exact test. Odds ratio larger than one were consider as CNVs enriched in old patients, and less than one means enriched in young patients.
Statistical analysis
The Chi-squared test was used to compare categorical variables of patient characteristics. Wilcoxon rank-sum test was used to compare the expression signatures, and somatic mutation burden. P-values were adjusted for multiple comparisons using Benjamini–Hochberg method. A regression model of ESR1 mRNA using age, ER IHC percentage categories, and their interaction was used to assess the contribution of both parameters. All analyses were performed in R version 3.6.151.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the data that support the funding in this study are public available and Web links of those datasets are available in the Methods section, additional information can be provided by the authors upon reasonable request.
Code availability
All codes for data cleaning and analysis are available at GitHub https://github.com/tao-qing/npjYoungVsOld.
References
DeSantis, C. E. et al. Breast cancer statistics, 2019. CA Cancer J. Clin. 69, 438–451 (2019).
Burstein, H. J. et al. Adjuvant endocrine therapy for women with hormone receptor-positive breast cancer: American Society of Clinical Oncology Clinical Practice Guideline update on ovarian suppression. J. Clin. Oncol. 34, 1689–1701 (2016).
Piccart, M. J. et al. Gene expression signatures for tailoring adjuvant chemotherapy of luminal breast cancer: stronger evidence, greater trust. Ann. Oncol. https://doi.org/10.1016/j.annonc.2021.05.804 (2021).
Sparano, J. A. et al. Adjuvant chemotherapy guided by a 21-gene expression assay in breast cancer. N. Engl. J. Med. 379, 111–121 (2018).
Kevin Kalinsky, W. E. B. et al. San Antonio Breast Cancer Symposium (San Antonio, 2020).
Piccart, M. et al. 70-gene signature as an aid for treatment decisions in early breast cancer: updated results of the phase 3 randomised MINDACT trial with an exploratory analysis by age. Lancet Oncol. 22, 476–488 (2021).
Broekmans, F. J., Knauff, E. A., te Velde, E. R., Macklon, N. S. & Fauser, B. C. Female reproductive ageing: Current knowledge and future trends. Trends Endocrinol. Metab. 18, 58–65 (2007).
Krailo, M. D. & Pike, M. C. Estimation of the distribution of age at natural menopause from prevalence data. Am. J. Epidemiol. 117, 356–361 (1983).
Vriens, I. J. et al. The correlation of age with chemotherapy-induced ovarian function failure in breast cancer patients. Oncotarget 8, 11372–11379 (2017).
Furlanetto, J. et al. Chemotherapy-induced ovarian failure in young women with early breast cancer: Prospective analysis of four randomised neoadjuvant/adjuvant breast cancer trials. Eur. J. Cancer 152, 193–203 (2021).
Ganz, P. A. et al. NRG Oncology/NSABP B-47 menstrual history study: Impact of adjuvant chemotherapy with and without trastuzumab. NPJ Breast Cancer 7, 55 (2021).
Francis, P. A. et al. Adjuvant ovarian suppression in premenopausal breast cancer. N. Engl. J. Med. 372, 436–446 (2015).
Regan, M. M. et al. Absolute benefit of adjuvant endocrine therapies for premenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative early breast cancer: TEXT and SOFT trials. J. Clin. Oncol. 34, 2221–2231 (2016).
Loibl, S. et al. Outcome after neoadjuvant chemotherapy in young breast cancer patients: a pooled analysis of individual patient data from eight prospectively randomized controlled trials. Breast Cancer Res. Treat. 152, 377–387 (2015).
Symmans, W. F. et al. Genomic index of sensitivity to endocrine therapy for breast cancer. J. Clin. Oncol. 28, 4111–4119 (2010).
Hatzis, C. et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 305, 1873–1881 (2011).
Denkert, C. et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J. Clin. Oncol. 28, 105–113 (2010).
Denkert, C. et al. Tumour-infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol. 19, 40–50 (2018).
Bianchini, G. et al. Proliferation and estrogen signaling can distinguish patients at risk for early versus late relapse among estrogen receptor positive breast cancers. Breast Cancer Res. 15, R86 (2013).
Danaher, P. et al. Gene expression markers of Tumor Infiltrating Leukocytes. J. Immunother. Cancer 5, 18 (2017).
Ayers, M. et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 127, 2930–2940 (2017).
Jimenez-Sanchez, A., Cast, O. & Miller, M. L. Comprehensive benchmarking and integration of tumor microenvironment cell estimation methods. Cancer Res. 79, 6238–6246 (2019).
Qing, T. et al. Germline variant burden in cancer genes correlates with age at diagnosis and somatic mutation burden. Nat. Commun. 11, 2438 (2020).
Griffith, O. L. et al. The prognostic effects of somatic mutations in ER-positive breast cancer. Nat. Commun. 9, 3476 (2018).
Tate, J. G. et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Clark, G. M., Osborne, C. K. & McGuire, W. L. Correlations between estrogen receptor, progesterone receptor, and patient characteristics in human breast cancer. J. Clin. Oncol. 2, 1102–1109 (1984).
Liedtke, C. et al. The prognostic impact of age in different molecular subtypes of breast cancer. Breast Cancer Res. Treat. 152, 667–673 (2015).
Haynes, B. P. et al. Differences in expression of proliferation-associated genes and RANKL across the menstrual cycle in estrogen receptor-positive primary breast cancer. Breast Cancer Res. Treat. 148, 327–335 (2014).
Haynes, B. P. et al. Expression of key oestrogen-regulated genes differs substantially across the menstrual cycle in oestrogen receptor-positive primary breast cancer. Breast Cancer Res. Treat. 138, 157–165 (2013).
Gulbahce, H. E., Blair, C. K., Sweeney, C. & Salama, M. E. Quantification of estrogen receptor expression in normal breast tissue in postmenopausal women with breast cancer and association with tumor subtypes. Appl Immunohistochem. Mol. Morphol. 25, 548–552 (2017).
Khan, S. A., Rogers, M. A., Khurana, K. K., Meguid, M. M. & Numann, P. J. Estrogen receptor expression in benign breast epithelium and breast cancer risk. J. Natl Cancer Inst. 90, 37–42 (1998).
Sotiriou, C. & Pusztai, L. Gene-expression signatures in breast cancer. N. Engl. J. Med. 360, 790–800 (2009).
Iwamoto, T. et al. Gene pathways associated with prognosis and chemotherapy sensitivity in molecular subtypes of breast cancer. J. Natl Cancer Inst. 103, 264–272 (2011).
Sinn, B. V. et al. SETER/PR: A robust 18-gene predictor for sensitivity to endocrine therapy for metastatic breast cancer. NPJ Breast Cancer 5, 16 (2019).
Kazanietz, M. G., Durando, M. & Cooke, M. CXCL13 and its receptor CXCR5 in cancer: Inflammation, immune response, and beyond. Front. Endocrinol. 10, 471 (2019).
Razis, E. et al. The role of CXCL13 and CXCL9 in early breast cancer. Clin. Breast Cancer 20, e36–e53 (2020).
Liao, S. et al. The molecular landscape of premenopausal breast cancer. Breast Cancer Res. 17, 104 (2015).
Rueda, O. M. et al. Dynamics of breast-cancer relapse reveal late-recurring ER-positive genomic subgroups. Nature 567, 399–404 (2019).
Osako, T. et al. Age-correlated protein and transcript expression in breast cancer and normal breast tissues is dominated by host endocrine effects. Nat. Cancer 1, 518–532 (2020).
Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Wang, J., Wen, S., Symmans, W. F., Pusztai, L. & Coombes, K. R. The bimodality index: A criterion for discovering and ranking bimodal signatures from cancer gene expression profiling data. Cancer Inf. 7, 199–216 (2009).
Marczyk, M., Jaksik, R., Polanski, A. & Polanska, J. Adaptive filtering of microarray gene expression data based on Gaussian mixture decomposition. BMC Bioinform. 14, 101 (2013).
Karn, T. et al. Data-driven derivation of cutoffs from a pool of 3,030 Affymetrix arrays to stratify distinct clinical types of breast cancer. Breast Cancer Res. Treat. 120, 567–579 (2010).
Karn, T. et al. Control of dataset bias in combined Affymetrix cohorts of triple negative breast cancer. Genom. Data 2, 354–356 (2014).
Curtis, C. et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 486, 346–352 (2012).
Brueffer, C. et al. Clinical value of RNA sequencing-based classifiers for prediction of the five conventional breast cancer biomarkers: A report from the population-based multicenter Sweden cancerome analysis network-breast initiative. JCO Precis. Oncol. https://doi.org/10.1200/PO.17.00135 (2018).
Gendoo, D. M. et al. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 32, 1097–1099 (2016).
Choi, I. S. et al. The 21-gene recurrence score assay and prediction of chemotherapy benefit: A propensity score-matched analysis of the SEER database. Cancers https://doi.org/10.3390/cancers12071829 (2020).
Stemmer, S. M. et al. Ten-year clinical outcomes in N0 ER+ breast cancer patients with recurrence score-guided therapy. NPJ Breast Cancer 5, 41 (2019).
Peng, X. et al. Molecular characterization and clinical relevance of metabolic expression subtypes in human cancers. Cell Rep. 23, 255–269 e254 (2018).
Paquet, E. R. & Hallett, M. T. Absolute assignment of breast cancer intrinsic molecular subtype. J. Natl Cancer Inst. 107, 357 (2015).
Smyth, G. K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl Genet. Mol. Biol. 3, 1–25 (2004).
Gennady Korotkevich, V. S. & Sergushichev, A. Fast gene set enrichment analysis. Preprint at bioRxiv https://doi.org/10.1101/060012 (2019).
Liberzon, A. et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 1, 417–425 (2015).
Ellrott, K. et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281 e277 (2018).
Jonckheere, A. R. A distribution-free k-sample test against ordered alternatives. Biometrika https://doi.org/10.2307/2333011 (1954).
Signorell, A. DescTools: Tools for Descriptive Statistics and Exploratory Data Analysis. https://andrisignorell.github.io (2020).
Taylor, A. M. et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell 33, 676–689 e673 (2018).
Mermel, C. H. et al. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41 (2011).
Acknowledgements
This work was supported by grants from the H.W. & J. Hector-Stiftung, Mannheim, Germany (M82) to Thomas Karn and Uwe Holtrich, and from the Susan Komen Foundation Leadership Award (SAC160076) and Breast Cancer Research Foundation Investigator Award (BCRF-21-133) to Lajos Pusztai.
Author information
Authors and Affiliations
Contributions
Concept and design: L.P.; data curation, analysis, and interpretation: T.Q., T.K., L.P., and U.H.; drafting of the manuscript: L.P., T.Q., and T.K.; critical revision of the manuscript for important: K.K., M.R., J.F., N.L.S., K.B., and F.M.B.; statistical analysis: T.Q., T.K., and M.M.; obtained funding: L.P., T.K., and U.H. T.Q. and T.K. contributed equally.
Corresponding author
Ethics declarations
Competing interests
L.P. has received consulting fees and honoraria from Pfizer, Astra Zeneca, Merck, Novartis, Bristol-Myers Squibb, Genentech, Eisai, Pieris, Immunomedics, Seattle Genetics, Clovis, Syndax, H3Bio, and Daiichi. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qing, T., Karn, T., Rozenblit, M. et al. Molecular differences between younger versus older ER-positive and HER2-negative breast cancers. npj Breast Cancer 8, 119 (2022). https://doi.org/10.1038/s41523-022-00492-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41523-022-00492-0
This article is cited by
-
Real-world use of multigene signatures in early breast cancer: differences to clinical trials
Breast Cancer Research and Treatment (2024)
