
Abstract
Pre-eclampsia (PE) is a pregnancy-specific syndrome that is characterized by hypertension and proteinuria. The etiology of PE is not completely understood but is believed to involve placental insufficiency and maternal vascular damage. Growing evidence supports an important role for the apelin receptor (APJ) system in regulating cardiovascular physiology. There are two vertebrate APJ ligands, APELIN and ELABELA, both of which mediate vasodilatory functions. A recent study linked deficient ELABELA signaling and the development of PE, though the molecular mechanism remains largely unknown. In this review, we summarize the biological function of the ELABELA and APJ system in cardiovascular homeostasis and discuss the potential mechanisms by which ELABELA and APJ regulate placenta trophoblast invasion and vascular functions and participate in the development of PE.
Similar content being viewed by others

Introduction
Pre-eclampsia (PE) is a pregnancy-specific syndrome of gestational hypertension that complicates 3–5% of pregnancies worldwide [1]. PE is associated with multi-organ damage and is lethal to both gravidas and newborns if improperly addressed. The etiology of PE remains incompletely understood and is probably heterogeneous. Placental dysfunction, including inadequate cytotrophoblast migration and invasion, poor vascular remodeling of the uterine spiral arteries, and placental hypoperfusion, are believed to be central to the development of PE [2]. Placenta hypoperfusion and the associated ischemia–reperfusion (I/R) injury stimulate the placenta to release soluble antiangiogenic factors such as soluble fms-like tyrosine kinase 1 (sFlt-1) and soluble endoglin (sEng), causing widespread maternal vascular endothelial dysfunction and vasoconstriction through the nitric oxide (NO)- and endothelin-1 dependent pathways [1, 3]. In addition, early abnormal immune activity in the placenta may impact cytotrophoblast migration and invasion, and subsequent chronic systemic inflammation in the mother may also contribute to the genesis of PE [4, 5]. Therefore, PE is likely a multifactorial syndrome caused by alterations in multiple regulatory pathways at different anatomical sites. Understanding the molecular events underlying the pathology is essential for developing preventative and therapeutic approaches for PE.
The Apelin receptor APJ (also known as APLNR or AGTRL1), is a Class A G protein-coupled receptor (GPCR) [6]. APJ receptor signaling is an important regulator of blood pressure, angiogenesis, cardiovascular function, fluid homeostasis, energy metabolism, cell proliferation/migration, apoptosis, oxidative stress, and inflammation [7,8,9,10]. A recent study revealed that the APJ system also plays a critical role in the development of PE. Deletion of Elabela (also known as Apela or Toddler), a ligand for the APJ receptor, triggers PE-like symptoms such as hypertension and proteinuria in mouse, and ELABELA administration could relieve these symptoms [11]. However, the specific mechanism through which the APJ system leads to PE is unknown. In this review, we summarize the latest studies on the functions of the APJ system in cardiovascular system and discuss the potential mechanism through which the APJ system contributes to the development of PE.
Function of APELIN-APJ signaling in cardiovascular system
Activation of APELIN receptor can inhibit forskolin-stimulated cAMP production, and its activation is sensitive to pertussis toxin, indicating that APJ is coupled to inhibitory G proteins (Gi) [12]. The human APJ protein consists of 380 amino acid residues and is highly conserved among vertebrates [6]. To date, two endogenous ligands, APELIN and ELABELA have been identified; of these ligands, APELIN was the first identified and is the better studied APJ ligand [13]. APELIN is initially produced as a 77-amino-acid preproprotein. After removal of the signal peptide, the 55-residue proapelin may be further cleaved by endopeptidases at several paired basic residues (Arg-Arg and Arg-Lys) to produce a range of C-terminal fragments, including APELIN-36, −17, −13, and the post-translationally modified (Pyr1) APELIN-13. These peptides are all agonists of the APJ receptor but may present different tissue distributions and potencies [9, 14] [15]
Both APELIN and the APJ receptor are widely expressed in the heart, brain, pancreas, lung, liver, kidney and placenta [9, 16, 17], and all are highly expressed in the endothelial cells of various vessels [18,19,20]. Consistent with their prominent expression in the cardiovascular system, APELIN/APJ has well-documented vasoactive functions [21, 22]. Systemic infusions of APELIN lower blood pressure in humans and rodents [23, 24] and cause vasorelaxation of glomerular arterioles, resulting in increased diuresis [25]. Mechanistically, APELIN stimulates the expression and activity of eNOS, increases endothelial production of NO [18, 25, 26], and antagonizes the vasoconstriction activity of angiotensin II by upregulating angiotensin-converting enzyme 2 (ACE2) expression [27, 28]. In cerebral artery vascular smooth muscle cells, APELIN controls the vascular tone of the cerebral artery by inhibiting large-conductance Ca2+-activated K+ (BKCa) channel via a PI3K-dependent signaling pathway [29].
The APELIN-APJ system also possesses angiogenic potential. In vitro, APELIN treatment stimulates endothelial cells and vascular smooth muscle cells to proliferate via the PI3K-Akt, PKC, ERK and NOTCH signaling pathways [30,31,32,33]. In the postmyocardial infarction heart, APELIN enhances the homing of vascular progenitor cells, increases angiogenesis and improves cardiac repair [34], while APELIN deficiency compromises in vivo myocardial angiogenesis [35]. A recent study showed that APJ is highly enriched in tumor blood vessels and that pharmacological blockage of Apelin-APJ signaling inhibits tumor angiogenesis and growth [36].
APELIN is a potent stimulator of cardiac contractility by activating the PKCε and ERK1/2 signaling pathway [37, 38]. Systemic APELIN treatment increases coronary blood flow [23], increases cardiac contractile reserve and improves the hemodynamic profile [39], whereas mouse knockout of Apelin or APJ leads to impaired cardiac contractility under aging or stress conditions [40]. Furthermore, loss of Apelin exacerbates myocardial infarction while APELIN perfusion protects the heart from ischemia–reperfusion injury by suppressing the oxidative damage to the sarcoplasmic reticulum function [35, 41].
ELABELA is a non-redundant ligand for APJ
APJ-null, but not Apelin-null, mice display overt congenital cardiac anomalies [40, 42]. Such phenotypic inconsistency was explained by the recent discovery of ELABELA, the second endogenous ligand of APJ [43, 44]. ELABELA was initially annotated as a non-coding RNA but was later confirmed to contain a 54–amino-acid open reading frame with a predicted signal sequence [43]. The mature peptide includes ELABELA-32, ELABELA-22 and ELABELA-11 isoforms as a result of differential cleavage, with the shortest isoform 11 being highly conserved across vertebrate species [45]. ELABELA mRNA or peptide has been detected in induced pluripotent stem cells (iPSCs), human embryonic stem cells, and mouse embryonic endoderm. In the adults, ELABELA expression is detected in the kidney, prostate, placenta and plasma but not as widely as APELIN and APJ [33, 44, 46]. Like APELIN, ELABELA is also abundantly expressed in the endothelial cells of the heart and various blood vessels [47, 48].
Like APELIN, ELABELA peptide increases cardiac contractility and induces coronary vasodilation by activating ERK in the cardiac tissues, but its mechanism is independent of PKC activation [47, 48]. ELABELA also improves cardiac function by downregulating angiotensin-converting enzyme (ACE) expression in stressed hearts [49]. Injection of ELABELA increases diuresis and water intake in rats [7]. ELABELA is also essential for mouse embryonic angiogenesis [11].
ELABELA also plays a non-redundant role to APELIN. During zebrafish embryogenesis, Elabela is the earliest ligand for APJ before the onset of gastrulation, whereas apelin expression begins 5 h later, during gastrulation [44]. Loss of either Elabela or APJ impair endoderm differentiation and mesodermal cell migration and severely disrupts cardiac development, whereas the Apelin knockout has no such dramatic effect [43, 44, 50]. The Elabela null, but not Apelin null, mouse is associated with PE symptoms [11]. Moreover, ELABELA also functions as an endogenous growth factor that sustains hESC self-renewal via the PI3K/Akt pathway. Interestingly, hESCs do not express APJ, suggesting that ELABELA may signal through an alternate unknown receptor in supporting stem cell self-renewal [46].
Functions of ELABELA and APJ signaling in the pathophysiology of PE
Ho et al. reported that Elabela knockout pregnant mice exhibited PE-like symptom such as kidney glomerular endotheliosis, proteinuria, and hypertension and that these symptoms can be normalized by systemic infusion of recombinant ELABELA peptide, indicating that ELABELA signaling is necessary for regulating maternal-placental vascular homeostasis to prevent PE [11]. A lack of ELABELA causes damages to the placenta and the maternal cardiovascular system, both of which are linked to PE. In the placenta, Elabela is predominantly expressed in villous cytotrophoblasts and syncytiotrophoblasts. Loss of Elabela expression delays syncytiotrophoblast differentiation, impairs APJ signaling in the neighboring fetal endothelial cells, and disrupts placental angiogenesis. These changes result in an overall pathological change to the placenta, as evidenced by elevated hypoxia and inflammation and a thinner labyrinth. On the maternal side, Elabela knockout results in endothelial damage and higher systolic blood pressure, while ELABELA peptide infusion normalizes the blood pressure, consistent with the vasorelaxation function of ELABELA. It is currently unclear whether the development of PE-like symptoms is primarily due to placental defects or maternal vascular dysfunction or both as a result of ELABELA deficiency. It is also unclear through what mechanism does ELABELA regulate trophoblast differentiation and invasion and placenta angiogenesis. As mentioned earlier, ELABELA may have a second receptor in addition to APJ, and this second receptor might also mediate specific anti-PE functions of ELABELA in the placenta.
As discussed earlier, APJ signaling activates the PI3K/Akt pathway and NO production in endothelial cells, promoting vasodilation and angiogenesis. Competent NO signaling has protective effects on placental function and maintenance of vascular tone. In the placenta, eNOS is abundantly expressed in the syncytiotrophoblasts and endothelial cells [51]. The NO level is often decreased in the placenta and plasma of humans with PE [52, 53]; long-term NOS inhibition produces PE-like syndrome in animal models [54], while glyceryl trinitrate-induced release of NO can improve utero-placental perfusion [55], suggesting that abnormal NO signaling may be involved in the pathology of PE. As eNOS activity is regulated by PI3K/Akt signaling [56], which is downregulated in the hypoxic [57] and PE placenta [58, 59], APJ signaling may regulate placental development partly through modulating PI3K/Akt-eNOS activity in the trophoblast or fetal endothelial cells.
Increased reactive oxygen species (ROS) level or ROS-mediated damage has been observed in human PE placenta [60,61,62]. ROS can scavenge NO, forming the reactive nitrogen species ONOO−. that further induces eNOS uncoupling and compromises NO production and NO-mediated vasorelaxation [63, 64]. Thus, placental oxidative stress has been proposed as an important cause of PE [65, 66]. In cultured adipocytes, APELIN treatment can inhibit the production and release of ROS by modulating the expression of anti-oxidant and pro-oxidant enzymes in an ERK-, AMPK- and Akt-dependent manner [67]. In cultured cardiomyocytes, APELIN treatment can inhibit the generation of ROS and apoptosis following ischemic/reperfusion injury by enhancing superoxide dismutase activity and ERK and Akt signaling [68]. Furthermore, Heme oxygenase-1 (HO-1) is an important enzyme that catabolizes ROS to prevent hypertension [69]. HO-1 expression and HO-1-mediated inhibition of sEng release from the placenta are dependent on Akt [59]. Thus, APJ signaling may play an important protective role in vascular integrity by suppressing oxidative damage in the hypoperfused placenta.
The anti-apoptotic activity of Apelin has been observed in various cell types, including cardiomyocytes, vascular smooth muscle cells, endothelial cells and osteoblasts [70,71,72,73], and this effect is induced by the activation of ERK and Akt [74]. In the central nervous system, APELIN can stimulate Akt phosphorylation after hypoxic/ischemic injury, and inhibiting PI3K reverses the phosphorylation and attenuates the protective effects on apoptosis [75]. Inhibition of APJ signaling by Elabela knockout results in increased apoptosis in the placenta [11]. Thus, competent APJ signaling is required to prevent apoptosis, which is frequently observed in placentas with PE. [62, 76,77,78]
During normal pregnancy, the invasion of extravillous cytotrophoblasts into the uterine spiral arteries converts them from small, high-resistance vessels to wide caliber, low-resistance vessels. In the placenta with PE, this process is incomplete, and shallow trophoblast invasion is observed [1, 79]. The transformation of villous cytotrophoblasts into migratory and invasive extravillous cytotrophoblast at the tip of the chorionic villi requires epithelial to mesenchymal transition (EMT) [80]. Ho et al. reported that ELABELA can stimulate trophoblast-like JAR choriocarcinoma cells to acquire a more invasive phenotype in vitro, indicating that ELABELA-APJ may positively regulate trophoblast invasion, though the mechanism is unknown [11]. TGF-β is a well-known inducer of EMT during many physiological and pathological processes, such as embryonic development, cancer progression and metastasis, and post-injury organ fibrosis [81]. However, in the placenta, TGF-β inhibits rather than promotes trophoblast invasion. Its mechanism has not been fully defined but may involve inhibiting the activity and expression of multiple extracellular proteolytic enzymes [82,83,84] and suppressing vascular endothelial cadherin (VE-cadherin) expression [85]. One study provided in vitro evidence that TGF-β may inhibit trophoblast EMT by upregulating epithelial-cadherin and beta-catenin expression [86]. Increased levels of TGF-β1/3 [87, 88] and E-cadherin [87, 89, 90] and a reduced level of VE-cadherin have been detected in the trophoblasts of patients with PE [89]. APJ activation can inhibit TGF-β signaling in a number of in vivo and in vitro fibrosis models [91,92,93,94]. Therefore, APJ activity may be required in the normal placenta to counteract the inhibitory effect of TGF-β on trophoblast invasion, which may explain the impaired trophoblast invasion in the Elabela-null placenta, as reported by Ho et al (2017) [11].
Conclusion and prospects
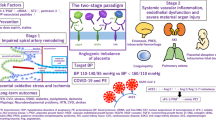
ELABELA- and APELIN-APJ signaling regulate important aspects of placental development and maternal cardiovascular homeostasis (Fig 1). ELABELA- and APELIN-APJ promote angiogenesis and cytotrophoblast invasion, support NO production to increase uterine blood flow, reduce oxidative stress, and suppress apoptosis in the placenta. Systemically, ELABELA and APELIN lower blood pressure and enhance cardiac function through their vasodilation effects. Many of these actions are executed through the activation of ERK and Akt and inhibition of TGF-β. Experiments on mouse models have demonstrated that ELABELA deficiency promotes PE and cardiovascular disease in mice. However, the clinical relevance of ELABELA, APELIN and their receptor APJ remains an open question. A recent study did not find altered expression of ELABELA in placentas from humans with PE, though the findings from 82 patients with PE need to be verified in larger cohort studies [95]. Assuming that the loss of ELABELA-APJ signaling may not account for a significant number of patients, given its widespread vasorelaxation activity, would APJ agonist treatment alleviate PE syndrome due to other causes? Wang et al (2017) found that systemic APELIN treatment significantly ameliorated the symptoms of PE in a rat model of PE induced by reduced uterine perfusion pressure [96]. Whether APELIN, or perhaps even more importantly ELABELA, has a similar therapeutic efficacy in human PE remains to be tested in the future.

The biological function of ELABELA and APJ signaling in the placental development and maternal cardiovascular homeostasis. In the placenta, ELABELA- and APELIN-APJ promote cytotrophoblast invasion/proliferation and fetal vessel angiogenesis, support NO production to increase uterine blood flow, reduce oxidative stress, and suppress apoptosis. Systemically, ELABELA and APELIN lower blood pressure and enhance cardiac function through their vasodilation effects. Loss of ELABELA or APJ signaling perturbs the placental and maternal vascular function, and contributes to PE.
References
Young BC, Levine RJ, Karumanchi SA. Pathogenesis of preeclampsia. Annu Rev Pathol. 2010;5:173–92.
Kanasaki K, Kalluri R. The biology of preeclampsia. Kidney Int. 2009;76:831–7.
Cudmore M, Ahmad S, Al-Ani B, Fujisawa T, Coxall H, Chudasama K, et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation. 2007;115:1789–97.
Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol. 2010;63:534–43.
Amaral LM, Wallace K, Owens M, LaMarca B. Pathophysiology and current clinical management of preeclampsia. Curr Hypertens Rep. 2017;19:61.
O’Dowd BF, Heiber M, Chan A, Heng HH, Tsui LC, Kennedy JL, et al. A human gene that shows identity with the gene encoding the angiotensin receptor is located on chromosome 11. Gene. 1993;136:355–60.
Deng C, Chen H, Yang N, Feng Y, Hsueh AJ. Apela regulates fluid homeostasis by binding to the APJ receptor to activate Gi signaling. J Biol Chem. 2015;290:18261–8.
Chaves-Almagro C, Castan-Laurell I, Dray C, Knauf C, Valet P, Masri B. Apelin receptors: From signaling to antidiabetic strategy. Eur J Pharmacol. 2015;763:149–59.
Chapman NA, Dupre DJ, Rainey JK. The apelin receptor: physiology, pathology, cell signalling, and ligand modulation of a peptide-activated class A GPCR. Biochem Cell Biol. 2014;92:431–40.
Bertrand C, Valet P, Castan-Laurell I. Apelin and energy metabolism. Front Physiol. 2015;6:115.
Ho L, van Dijk M, Chye STJ, Messerschmidt DM, Chng SC, Ong S, et al. ELABELA deficiency promotes preeclampsia and cardiovascular malformations in mice. Science. 2017;357:707–13.
Bai B, Tang J, Liu H, Chen J, Li Y, Song W. Apelin-13 induces ERK1/2 but not p38 MAPK activation through coupling of the human apelin receptor to the Gi2 pathway. Acta Biochim Biophys Sin. 2008;40:311–8.
Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem Biophys Res Commun. 1998;251:471–6.
Hosoya M, Kawamata Y, Fukusumi S, Fujii R, Habata Y, Hinuma S, et al. Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin. J Biol Chem. 2000;275:21061–7.
Shin K, Pandey A, Liu XQ, Anini Y, Rainey JK. Preferential apelin-13 production by the proprotein convertase PCSK3 is implicated in obesity. FEBS Open Bio. 2013;3:328–33.
Pope GR, Roberts EM, Lolait SJ, O’Carroll AM. Central and peripheral apelin receptor distribution in the mouse: species differences with rat. Peptides. 2012;33:139–48.
Medhurst AD, Jennings CA, Robbins MJ, Davis RP, Ellis C, Winborn KY, et al. Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin. J Neurochem. 2003;84:1162–72.
Tatemoto K, Takayama K, Zou MX, Kumaki I, Zhang W, Kumano K, et al. The novel peptide apelin lowers blood pressure via a nitric oxide-dependent mechanism. Regul Pept. 2001;99:87–92.
Kleinz MJ, Davenport AP. Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells. Regul Pept. 2004;118:119–25.
Kleinz MJ, Skepper JN, Davenport AP. Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells. Regul Pept. 2005;126:233–40.
Yu XH, Tang ZB, Liu LJ, Qian H, Tang SL, Zhang DW, et al. Apelin and its receptor APJ in cardiovascular diseases. Clin Chim Acta. 2014;428:1–8.
Yang P, Maguire JJ, Davenport AP. Apelin, Elabela/Toddler, and biased agonists as novel therapeutic agents in the cardiovascular system. Trends Pharmacol Sci. 2015;36:560–7.
Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, et al. Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure. Circulation. 2010;121:1818–27.
Lee DK, Saldivia VR, Nguyen T, Cheng R, George SR, O’Dowd BF. Modification of the terminal residue of apelin-13 antagonizes its hypotensive action. Endocrinology. 2005;146:231–6.
Hus-Citharel A, Bouby N, Frugiere A, Bodineau L, Gasc JM, Llorens-Cortes C. Effect of apelin on glomerular hemodynamic function in the rat kidney. Kidney Int. 2008;74:486–94.
Jia YX, Lu ZF, Zhang J, Pan CS, Yang JH, Zhao J, et al. Apelin activates L-arginine/nitric oxide synthase/nitric oxide pathway in rat aortas. Peptides. 2007;28:2023–9.
Chun HJ, Ali ZA, Kojima Y, Kundu RK, Sheikh AY, Agrawal R, et al. Apelin signaling antagonizes Ang II effects in mouse models of atherosclerosis. J Clin Invest. 2008;118:3343–54.
Sato T, Suzuki T, Watanabe H, Kadowaki A, Fukamizu A, Liu PP, et al. Apelin is a positive regulator of ACE2 in failing hearts. J Clin Invest. 2013;123:5203–11.
Modgil A, Guo L, O’Rourke ST, Sun C. Apelin-13 inhibits large-conductance Ca2+-activated K+channels in cerebral artery smooth muscle cells via a PI3-kinase dependent mechanism. PLoS ONE. 2013;8:e83051.
Masri B, Morin N, Cornu M, Knibiehler B, Audigier Y. Apelin (65-77) activates p70 S6 kinase and is mitogenic for umbilical endothelial cells. FASEB J. 2004;18:1909–11.
Liu QF, Yu HW, Sun LL, You L, Tao GZ, Qu BZ. Apelin-13 upregulates Egr-1 expression in rat vascular smooth muscle cells through the PI3K/Akt and PKC signaling pathways. Biochem Biophys Res Commun. 2015;468:617–21.
Li L, Li L, Xie F, Zhang Z, Guo Y, Tang G, et al. Jagged-1/Notch3 signaling transduction pathway is involved in apelin-13-induced vascular smooth muscle cells proliferation. Acta Biochim Biophys Sin. 2013;45:875–81.
Wang Z, Yu D, Wang M, Wang Q, Kouznetsova J, Yang R, et al. Elabela-apelin receptor signaling pathway is functional in mammalian systems. Sci Rep. 2015;5:8170.
Li L, Zeng H, Chen JX. Apelin-13 increases myocardial progenitor cells and improves repair postmyocardial infarction. Am J Physiol Heart Circ Physiol. 2012;303:H605–18.
Wang W, McKinnie SM, Patel VB, Haddad G, Wang Z, Zhabyeyev P, et al. Loss of Apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic Apelin analogues. J Am Heart Assoc. 2013;2:e000249.
Zhao H, Tian X, He L, Li Y, Pu W, Liu Q, et al. Apj(+) vessels drive tumor growth and represent a tractable therapeutic target. Cell Rep. 2018;25:1241–54.
Szokodi I, Tavi P, Foldes G, Voutilainen-Myllyla S, Ilves M, Tokola H. et al. Apelin, the novel endogenous ligand of the orphan receptor APJ, regulates cardiac contractility. Circ Res. 2002;91:434–40.
Perjes A, Skoumal R, Tenhunen O, Konyi A, Simon M, Horvath IG, et al. Apelin increases cardiac contractility via protein kinase Cepsilon- and extracellular signal-regulated kinase-dependent mechanisms. PLoS ONE. 2014;9:e93473.
Ashley EA, Powers J, Chen M, Kundu R, Finsterbach T, Caffarelli A, et al. The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc Res. 2005;65:73–82.
Kuba K, Zhang L, Imai Y, Arab S, Chen M, Maekawa Y, et al. Impaired heart contractility in Apelin gene-deficient mice associated with aging and pressure overload. Circ Res. 2007;101:e32–42.
Wang C, Liu N, Luan R, Li Y, Wang D, Zou W, et al. Apelin protects sarcoplasmic reticulum function and cardiac performance in ischaemia-reperfusion by attenuating oxidation of sarcoplasmic reticulum Ca2+-ATPase and ryanodine receptor. Cardiovasc Res. 2013;100:114–24.
Charo DN, Ho M, Fajardo G, Kawana M, Kundu RK, Sheikh AY, et al. Endogenous regulation of cardiovascular function by apelin-APJ. Am J Physiol Heart Circ Physiol. 2009;297:H1904–13.
Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S, et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343:1248636.
Chng SC, Ho L, Tian J, Reversade B. ELABELA: a hormone essential for heart development signals via the apelin receptor. Dev Cell. 2013;27:672–80.
Huang SK, Shin K, Sarker M, Rainey JK. Apela exhibits isoform- and headgroup-dependent modulation of micelle binding, peptide conformation and dynamics. Biochim Biophys Acta. 2017;1859:767–78.
Ho L, Tan SY, Wee S, Wu Y, Tan SJ, Ramakrishna NB, et al. ELABELA Is an endogenous growth factor that sustains hESC self-renewal via the PI3K/AKT pathway. Cell Stem Cell. 2015;17:435–47.
Perjes A, Kilpio T, Ulvila J, Magga J, Alakoski T, Szabo Z, et al. Characterization of apela, a novel endogenous ligand of apelin receptor, in the adult heart. Basic Res Cardiol. 2016;111:2.
Yang P, Read C, Kuc RE, Buonincontri G, Southwood M, Torella R, et al. Elabela/Toddler Is an endogenous agonist of the apelin apj receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation. 2017;135:1160–73.
Sato T, Sato C, Kadowaki A, Watanabe H, Ho L, Ishida J, et al. ELABELA-APJ axis protects from pressure overload heart failure and angiotensin II-induced cardiac damage. Cardiovasc Res. 2017;113:760–9.
Paskaradevan S, Scott IC. The Aplnr GPCR regulates myocardial progenitor development via a novel cell-non-autonomous, Galpha(i/o) protein-independent pathway. Biol Open. 2012;1:275–85.
Vatish M, Randeva HS, Grammatopoulos DK. Hormonal regulation of placental nitric oxide and pathogenesis of pre-eclampsia. Trends Mol Med. 2006;12:223–33.
Ehsanipoor RM, Fortson W, Fitzmaurice LE, Liao WX, Wing DA, Chen DB, et al. Nitric oxide and carbon monoxide production and metabolism in preeclampsia. Reprod Sci. 2013;20:542–8.
Seligman SP, Buyon JP, Clancy RM, Young BK, Abramson SB. The role of nitric oxide in the pathogenesis of preeclampsia. Am J Obstet Gynecol. 1994;171:944–8.
Podjarny E, Losonczy G, Baylis C. Animal models of preeclampsia. Semin Nephrol. 2004;24:596–606.
Luzi G, Caserta G, Iammarino G, Clerici G, Di Renzo GC. Nitric oxide donors in pregnancy: fetomaternal hemodynamic effects induced in mild pre-eclampsia and threatened preterm labor. Ultrasound Obstet Gynecol. 1999;14:101–9.
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601.
Chiang MH, Liang FY, Chen CP, Chang CW, Cheong ML, Wang LJ, et al. Mechanism of hypoxia-induced GCM1 degradation: implications for the pathogenesis of preeclampsia. J Biol Chem. 2009;284:17411–9.
Kaitu’u-Lino TJ, Hastie R, Hannan NJ, Brownfoot F, De Silva M, Cannon P, et al. Loss of Akt increases soluble endoglin release from endothelial cells but not placenta. Pregnancy Hypertens. 2016;6:95–102.
Cudmore MJ, Ahmad S, Sissaoui S, Ramma W, Ma B, Fujisawa T, et al. Loss of Akt activity increases circulating soluble endoglin release in preeclampsia: identification of inter-dependency between Akt-1 and heme oxygenase-1. Eur Heart J. 2012;33:1150–8.
Aris A, Benali S, Ouellet A, Moutquin JM, Leblanc S. Potential biomarkers of preeclampsia: inverse correlation between hydrogen peroxide and nitric oxide early in maternal circulation and at term in placenta of women with preeclampsia. Placenta. 2009;30:342–7.
Madazli R, Benian A, Aydin S, Uzun H, Tolun N. The plasma and placental levels of malondialdehyde, glutathione and superoxide dismutase in pre-eclampsia. J Obstet Gynaecol. 2002;22:477–80.
Can M, Guven B, Bektas S, Arikan I. Oxidative stress and apoptosis in preeclampsia. Tissue Cell. 2014;46:477–81.
Matsubara K, Higaki T, Matsubara Y, Nawa A. Nitric oxide and reactive oxygen species in the pathogenesis of preeclampsia. Int J Mol Sci. 2015;16:4600–14.
Dikalova AE, Gongora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2010;299:H673–9.
Seki H. Animal models of preeclampsia: an examination of usefulness and limitations based on the metabolic domino theory. Hypertens Res Pregnancy. 2017;5:52–8.
Aouache R, Biquard L, Vaiman D, Miralles F. Oxidative stress in preeclampsia and placental diseases. Int J Mol Sci. 2018;19:E1496.
Than A, Zhang X, Leow MK, Poh CL, Chong SK, Chen P. Apelin attenuates oxidative stress in human adipocytes. J Biol Chem. 2014;289:3763–74.
Zeng XJ, Zhang LK, Wang HX, Lu LQ, Ma LQ, Tang CS. Apelin protects heart against ischemia/reperfusion injury in rat. Peptides. 2009;30:1144–52.
Lu Q, Yang Y, Villar VA, Asico L, Jones JE, Yu P, et al. D5 dopamine receptor decreases NADPH oxidase, reactive oxygen species and blood pressure via heme oxygenase-1. Hypertens Res. 2013;36:684–90.
Cui RR, Mao DA, Yi L, Wang C, Zhang XX, Xie H, et al. Apelin suppresses apoptosis of human vascular smooth muscle cells via APJ/PI3-K/Akt signaling pathways. Amino Acids. 2010;39:1193–200.
Xie H, Yuan LQ, Luo XH, Huang J, Cui RR, Guo LJ, et al. Apelin suppresses apoptosis of human osteoblasts. Apoptosis. 2007;12:247–54.
Ustunel I, Acar N, Gemici B, Ozbey O, Edizer I, Soylu H, et al. The effects of water immersion and restraint stress on the expressions of apelin, apelin receptor (APJR) and apoptosis rate in the rat heart. Acta Histochem. 2014;116:675–81.
Zeng H, He X, Hou X, Li L, Chen JX. Apelin gene therapy increases myocardial vascular density and ameliorates diabetic cardiomyopathy via upregulation of sirtuin 3. Am J Physiol Heart Circ Physiol. 2014;306:H585–97.
Yang Y, Zhang X, Cui H, Zhang C, Zhu C, Li L. Apelin-13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci Lett. 2014;568:44–9.
Gu Q, Zhai L, Feng X, Chen J, Miao Z, Ren L. et al. Apelin-36, a potent peptide, protects against ischemic brain injury by activating the PI3K/Akt pathway. Neurochem Int. 2013;63:535–40.
Allaire AD, Ballenger KA, Wells SR, McMahon MJ, Lessey BA. Placental apoptosis in preeclampsia. Obstet Gynecol. 2000;96:271–6.
Tomas SZ, Prusac IK, Roje D, Tadin I. Trophoblast apoptosis in placentas from pregnancies complicated by preeclampsia. Gynecol Obstet Invest. 2011;71:250–5.
DiFederico E, Genbacev O, Fisher SJ. Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall. Am J Pathol. 1999;155:293–301.
Meekins JW, Pijnenborg R, Hanssens M, McFadyen IR, van Asshe A. A study of placental bed spiral arteries and trophoblast invasion in normal and severe pre-eclamptic pregnancies. Br J Obstet Gynaecol. 1994;101:669–74.
Pollheimer JED, Yong J, Kokkinos HE, Kalionis MI, Knofler B, Murthi M, et al. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adhes Migr. 2016;10:310–21.
Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–72.
Lash GE, Otun HA, Innes BA, Bulmer JN, Searle RF, Robson SC. Inhibition of trophoblast cell invasion by TGFB1, 2, and 3 is associated with a decrease in active proteases. Biol Reprod. 2005;73:374–81.
Meisser A, Chardonnens D, Campana A, Bischof P. Effects of tumour necrosis factor-alpha, interleukin-1 alpha, macrophage colony stimulating factor and transforming growth factor beta on trophoblastic matrix metalloproteinases. Mol Hum Reprod. 1999;5:252–60.
Graham CH. Effect of transforming growth factor-beta on the plasminogen activator system in cultured first trimester human cytotrophoblasts. Placenta. 1997;18:137–43.
Cheng JC, Chang HM, Leung PC. Transforming growth factor-beta1 inhibits trophoblast cell invasion by inducing Snail-mediated down-regulation of vascular endothelial-cadherin protein. J Biol Chem. 2013;288:33181–92.
Zhao MR, Qiu W, Li YX, Zhang ZB, Li D, Wang YL. Dual effect of transforming growth factor beta1 on cell adhesion and invasion in human placenta trophoblast cells. Reproduction. 2006;132:333–41.
Benian A, Madazli R, Aksu F, Uzun H, Aydin S. Plasma and placental levels of interleukin-10, transforming growth factor-beta1, and epithelial-cadherin in preeclampsia. Obstet Gynecol. 2002;100:327–31.
Caniggia I, Grisaru-Gravnosky S, Kuliszewsky M, Post M, Lye SJ. Inhibition of TGF-beta 3 restores the invasive capability of extravillous trophoblasts in preeclamptic pregnancies. J Clin Invest. 1999;103:1641–50.
Zhou Y, Damsky CH, Fisher SJ. Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J Clin Invest. 1997;99:2152–64.
Li HW, Cheung AN, Tsao SW, Cheung AL, Expression OWS. of e-cadherin and beta-catenin in trophoblastic tissue in normal and pathological pregnancies. Int J Gynecol Pathol. 2003;22:63–70.
Wang LY, Diao ZL, Zheng JF, Wu YR, Zhang QD, Liu WH. Apelin attenuates TGF-beta1-induced epithelial to mesenchymal transition via activation of PKC-epsilon in human renal tubular epithelial cells. Peptides. 2017;96:44–52.
Wang LY, Diao ZL, Zhang DL, Zheng JF, Zhang QD, Ding JX, et al. The regulatory peptide apelin: a novel inhibitor of renal interstitial fibrosis. Amino Acids. 2014;46:2693–704.
Pchejetski D, Foussal C, Alfarano C, Lairez O, Calise D, Guilbeau-Frugier C, et al. Apelin prevents cardiac fibroblast activation and collagen production through inhibition of sphingosine kinase 1. Eur Heart J. 2012;33:2360–9.
Chen H, Wan D, Wang L, Peng A, Xiao H, Petersen RB, et al. Apelin protects against acute renal injury by inhibiting TGF-beta1. Biochim Biophys Acta. 2015;1852:1278–87.
Pritchard N, Kaitu’u-Lino TJ, Gong S, Dopierala J, Smith GCS, Charnock-Jones DS, et al. ELABELA/APELA levels are not decreased in the maternal circulation or placenta among women with preeclampsia. Am J Pathol. 2018;188:1749–53.
Wang C, Liu X, Kong D, Qin X, Li Y, Teng X, et al. Apelin as a novel drug for treating preeclampsia. Exp Ther Med. 2017;14:5917–23.
Acknowledgements
We thank Shouyin Jiang for his assistance with the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, Y., Wang, L. & Shi, H. The biological function of ELABELA and APJ signaling in the cardiovascular system and pre-eclampsia. Hypertens Res 42, 928–934 (2019). https://doi.org/10.1038/s41440-018-0193-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-018-0193-3
Keywords
This article is cited by
-
ERK/HIF-1α/VEGF pathway: a molecular target of ELABELA (ELA) peptide for attenuating cardiac ischemia–reperfusion injury in rats by promoting angiogenesis
Molecular Biology Reports (2022)
-
Declined ELABELA plasma levels in hypertension patients with atrial fibrillation: a case control study
BMC Cardiovascular Disorders (2021)
