
Abstract
Purpose
Infantile Caffey disease is a rare disorder characterized by acute inflammation with subperiosteal new bone formation, associated with fever, pain, and swelling of the overlying soft tissue. Symptoms arise within the first weeks after birth and spontaneously resolve before the age of two years. Many, but not all, affected individuals carry the heterozygous pathogenic COL1A1 variant (c.3040C>T, p.(Arg1014Cys)).
Methods
We sequenced COL1A1 in 28 families with a suspicion of Caffey disease and performed ultrastructural, immunocytochemical, and biochemical collagen studies on patient skin biopsies.
Results
We identified the p.(Arg1014Cys) variant in 23 families and discovered a novel heterozygous pathogenic COL1A1 variant (c.2752C>T, p.(Arg918Cys)) in five. Both arginine to cysteine substitutions are located in the triple helical domain of the proα1(I) procollagen chain. Dermal fibroblasts (one patient with p.(Arg1014Cys) and one with p.(Arg918Cys)) produced molecules with disulfide-linked proα1(I) chains, which were secreted only with p.(Arg1014Cys). No intracellular accumulation of type I procollagen was detected. The dermis revealed mild ultrastructural abnormalities in collagen fibril diameter and packing.
Conclusion
The discovery of this novel pathogenic variant expands the limited spectrum of arginine to cysteine substitutions in type I procollagen. Furthermore, it confirms allelic heterogeneity in Caffey disease and impacts its molecular confirmation.
Similar content being viewed by others

INTRODUCTION
Caffey disease or infantile cortical hyperostosis (OMIM 114000) is a rare heritable disorder, characterized by subperiosteal new bone formation leading to cortical thickening (hyperostosis) of the underlying bone [1]. It is accompanied by inflammation and swelling of the overlying soft tissues, pain, hyperirritability, and fever. Laboratory findings usually show increased levels of alkaline phosphatase and signs of systemic inflammation, with increased C-reactive protein and immunoglobulin levels, elevated erythrocyte sedimentation rates, and sometimes anemia [2]. Symptoms typically arise within the first five months of life and spontaneously resolve by the age of two years, leaving no trace of the hyperostosis on radiographs [3]. The most frequently involved parts of the skeleton are the diaphyses of the long bones, the mandible, and clavicle. Despite the self-limiting nature of the disease, patients are treated with corticosteroids or nonsteroidal anti-inflammatory drugs such as indomethacin, to reduce pain and inflammation [3, 4]. Recurrences have occasionally been reported in childhood and rarely in adulthood [5]. Sequelae are rare, but may include synostosis, scoliosis, short stature, and persistent bone deformity [5]. Due to the sometimes subtle phenotype and the spontaneous resolution, the exact prevalence of the disorder remains unknown.
Caffey disease can be either sporadic or familial [2], the latter showing an autosomal dominant (AD) inheritance pattern with incomplete penetrance and variable expression [3]. Gensure and colleagues identified a pathogenic heterozygous variant (c.3040C>T, p.(Arg1014Cys)) in COL1A1 in three unrelated families from different origins [3]. COL1A1 encodes the proα1(I) chain of the fibrillar type I procollagen, which when processed to collagen is the most abundant protein in bone, ligaments, dermis, and blood vessel walls. Two proα1(I) chains and one proα2(I) chain (encoded by COL1A2) form a type I procollagen molecule that consists of a triple helical domain, flanked by globular amino- and carboxy-terminal propeptides. Each triple helical domain consists of uninterrupted Gly-Xaa-Yaa triplet repeats that extend over 1,014 residues, where the small glycine (Gly) residue is crucial to ensure efficient winding into a triple helix. After cleavage of the propeptides, the collagen molecules form collagen fibrils and fibers in the extracellular matrix (ECM).
The identified COL1A1 c.3040C>T variant gives rise to a substitution of arginine by cysteine (Arg-to-Cys) p.(Arg1014Cys), which is located in the Xaa position of the Gly-Xaa-Yaa triplet repeat of the proα1(I) chain. In normal circumstances, no cysteine residues are present in the triple helical domain of type I procollagen, and the introduction of a cysteine residue allows the formation of disulfide-bonded proα1(I) dimers when two altered chains are in the same molecule. These are detected as an additional band on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of collagen extracted from patient skin fibroblasts [3]. Since its initial identification, the COL1A1 c.3040C>T variant has been reported in literature in over 60 Caffey disease patients. However, the absence of the variant in some familial cases with Caffey disease argued for allelic and/or locus heterogeneity [3, 4, 6]. The latter was confirmed recently by the identification of a homozygous nonsense variant in the AHSG gene in an infant with typical signs of Caffey disease [7].
In addition to the classical form of Caffey disease, which is self-limiting and relatively benign, a lethal prenatal form has also been described. It becomes evident during ultrasound studies before 35 weeks of gestation and is characterized by polyhydramnios, severe bone lesions, and perinatal lethality, mainly caused by respiratory problems due to prematurity or lung hypoplasia [8]. To date, the COL1A1 c.3040C>T variant was reported in only one case with prenatal cortical hyperostosis [3, 4], and no other genes have been identified for this condition.
Here we report 28 novel families with Caffey disease. In 23 of these families we detected the previously described COL1A1 c.3040C>T variant. In five other families, we discovered a novel COL1A1 variant, c.2752C>T, (p.(Arg918Cys)) within the proα1(I) triple helical domain. Both variants lead to the formation of procollagen molecules that, when they contain two altered proα1(I) chains, can form disulfide bonds within the triple helical domain, and ultrastructural collagen fibril abnormalities.
MATERIALS AND METHODS
Patient materials
Blood samples were collected to extract genomic DNA. Skin biopsies were used to grow primary dermal fibroblast cultures under standard conditions or for performing transmission electron microscopic studies. Controls are age- and sex-matched.
Molecular analysis
For families 1–23, the coding sequence of exon 41 of COL1A1 (RefSeq NM_000088.4) was polymerase chain reaction (PCR) amplified. Amplicons were sequenced by bidirectional Sanger sequencing using the BigDye Terminator Cycle Sequencing kit and ABI 3730XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA). For families 24–26, all exons and flanking intronic sequences of COL1A1 and AHSG (RefSeq NM_001622.4) were PCR amplified and analyzed according to Illumina’s sequencing-by-synthesis technology using the MiSeq desktop automatic sequencer (Illumina, San Diego, CA, USA), followed by Sanger sequencing for confirmation. For families 27 and 28, coding sequences of exons 39 and 41 of COL1A1 were amplified and sequenced. The predicted pathogenic effect of the variants was determined with the Alamut Visual software (v2.5, Interactive Biosoftware, Rouen, France). Variants were classified according to the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines. Human Genome Variation Society (HGVS) guidelines (http://www.hgvs.org/mutnomen/) were followed for variant nomenclature.
SDS-PAGE
(Pro)Collagen extraction and labeling for SDS-PAGE analysis was performed as described previously [9].
Immunocytochemistry
Cultured fibroblasts were seeded and stained as described previously [10]. Primary antibodies against type I (pro)collagen (1/50, Ab758, Merck, Darmstadt, Germany), PDI (protein disulfide isomerase) (1/300, Ab3672, Abcam, Cambridge, UK) or GOLPH4 (Golgi-localized phosphoprotein 4) (1/400, Ab28049, Abcam) and secondary antibodies conjugated with Alexafluor 488 and Alexafluor 594 (1/750, Life Technologies, Eugene, OR, USA) were used. Samples were visualized using the Axio Observer.Z1 fluorescence microscope (Carl Zeiss Microscopy, Thornwood, NY, USA). The Zen pro software was used for image capturing, analysis, and processing. Quantitative analysis of intracellular type I procollagen was performed as described previously [11].
Immunoblotting
Fibroblasts were seeded at 1 × 105 cells/ml in culture medium. Cells were lysed after three days using RIPA lysis buffer (Sigma-Aldrich, Saint-Louis, MO, USA) supplemented with protease inhibitor cocktail (Roche, Basel, Switzerland) and phosphatase inhibitor cocktail 2 and 3 (Sigma-Aldrich). SDS-PAGE was performed on 4–12% NuPage BT gels (ThermoScientific, Life Technologies Europe, Ghent, Belgium), followed by transfer to nitrocellulose membranes using the iBlot System (Life Technologies Europe). The membranes were blocked with 2% membrane blocking agent (ThermoScientific) for 2 hours and incubated overnight with primary antibodies against BiP (1/500, 3183, Cell Signaling Technologies [CST], Bioké, Leiden, The Netherlands), eIF-2α (1/1000, 9722, CST), phospho-eIF-2α (1/750, 9721 S, CST), ATF6 (1/500, 65880, CST), PERK (1/1,000, 3192, CST), phospho-PERK (1/200, sc32577, Santa Cruz Biotechnology, Dallas, TX, USA), JNK (1/1,000, 9252, CST) or phospho-JNK (1/1,000, 4688, CST). HRP-coupled secondary antibody (CST) was added for 2 hours. Blots were developed using SuperSignal West Dura Extended Duration Substrate (ThermoScientific) and scanned with ChemiDoc-It 500 Imaging System (UVP, Upland, CA, USA). Membranes were stripped with Restore PLUS Western blot stripping buffer (ThermoScientific) and reprobed with antibodies directed against β-tubulin (1/1,000, Ab6046, Abcam) for loading control. For P26 and a matched control, semiquantitative analysis was performed on triplicates as previously described [10].
Transmission electron microscopy
Skin biopsies for transmission electron microscopy (TEM) analysis were obtained from the inside of the upper arm, fixed in glutaraldehyde and conventionally processed and imaged in the Department of Dermatopathology, University Hospital of Sart-Tilman (Liège, Belgium) for patient 1 and in the Institute of Pathology, Heidelberg University Hospital (Heidelberg, Germany) for patient 26 as described previously [12, 13].
Statistical analysis
Data are expressed as mean ± standard error (SEM). P values < 0.05 were considered significant. Statistical calculations were performed using GraphPad Prism 5 (La Jolla, CA, USA).
RESULTS
Clinical findings
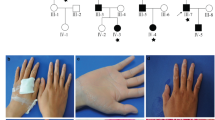
Of 28 probands who received a clinical diagnosis of Caffey disease, 18 were male, 9 were female, and for one proband, gender was not reported. The age at diagnosis ranged between 4 days up to 9 months after birth. Affected bones included the mandible, clavicle, humerus, radius, ulna, femur, and tibia, the latter being most frequently affected (9/28) (Fig. 1a, b). In two single cases, the parietal skull and scapula showed signs of hyperostosis. Other signs of connective tissue fragility were rare. One patient displayed pronounced tibial bowing at the age of 2.5 years, which slowly resolved by the age of 12 years (Fig. 1b). In two independent probands, a femur fracture and pedes plani were respectively reported. In 11 families, the clinical family history was positive for signs of Caffey disease, such as hyperostosis or painful swelling during early childhood. An extensive clinical report of family 1 is available as Supplemental information.

(a) Radiographic images showing hyperostosis of the mandible (upper figure) and clavicle, humerus (distal part), and ulna (lower figure) in proband 19 at age 7 weeks. (b) Radiographic images showing mild hyperostosis and pronounced bowing of the tibia in the proband of family 28 at age 2.5 years. The bowing was improved at age 12 years. (c) Pedigrees of the families in whom DNA of two or more members was analyzed. Full symbol: symptomatic individual. Dotted symbol: asymptomatic individual carrying variant. NA not analyzed. (d) Sanger sequencing identified the previously reported heterozygous variant COL1A1 c.3040C>T, p.(Arg1014Cys), located just outside MLBR-2 in families 1–23, and a novel heterozygous variant COL1A1 c.2752C>T, p.(Arg918Cys), located within MLBR-2 in family 24–28. Ligands binding to procollagen, the proα1(I) chain or the proα2(I) chain are indicated with a full line, a dashed line above the sequence or a dashed line below the sequence, respectively. Scissors: MMP-1 cleavage site. APP amyloid precursor protein, COMP cartilage oligomeric matrix protein, DDR2 discoidin domain receptor 2, HSP47 heat shock protein 47, MLBR-2 major ligand binding region, MMP matrix metalloproteinase, SPARC secreted protein acidic and rich in cysteine binding site, THP/α1 α2β1 triple helical peptide binds integrin receptors, THP/FA+ triple helical peptide promotes fibroblast adhesion. Figure adapted from Sweeney et al. [14].
Genetic testing was requested for confirmation of the clinical diagnosis of Caffey disease in 27 of the 28 probands and in none was abuse or osteogenesis imperfecta (OI) suspected.
Molecular analysis
Genomic DNA was used to perform molecular analyses. In probands 1 to 23, the previously reported heterozygous COL1A1 variant c.3040C>T (p.(Arg1014Cys)) in exon 41 was identified. Sequencing analysis was performed in 17 at-risk relatives from 9 families. Among these, only one did not carry the variant. Of the 16 relatives with the variant, 4 were clinically affected and 12 reported no symptoms.
In the five probands without the c.3040C>T variant (probands 24–28), molecular analysis revealed a novel heterozygous nucleotide change c.2752C>T (p.(Arg918Cys)) located in exon 39 of COL1A1. In four of the five families, ten additional family members were tested, of whom three carried the variant; only one had displayed clinical symptoms, the other two were asymptomatic. The variant was shown to be de novo in the proband of family 25 (Fig. 1c). The c.2752C>T variant was absent from the general population database gnomAD, whereas the c.3040C>T variant was reported in 2 of 250,334 alleles (https://gnomad.broadinstitute.org/ version 2.1.1, consulted on 28 September 2020). Other noncysteine substitutions of the same two residues were reported in low frequencies. Molecular analysis of AHSG in probands 24–26 did not show any sequence alterations.
Both variants cause an Arg-to-Cys substitution in the Xaa position of the Gly-Xaa-Yaa triplet repeat of the proα1(I) chain and are 96 amino acids apart in the triple helical region of the molecule. The previously reported p.(Arg1014Cys) resides in the gap between two regions referred to as Major Ligand Binding Region (MLBR) 2 and 3 in a region that appears to form a binding site for interleukin-2 (IL-2) [14, 15]. The newly identified p.(Arg918Cys) substitution is located in the MLBR-2 (Fig. 1d) in a region that appears to form a binding site for the α1β1 integrin [14].
Biochemical (pro)collagen analysis
SDS-PAGE of procollagens extracted from the cell layer fraction of cultured skin fibroblasts and treated with pepsin from proband 1 with the p.(Arg1014Cys) variant showed the presence of an additional band that represented disulfide-bonded α1(I) dimers (Fig. 2). These were derived from type I procollagen molecules that had two proα1(I) chains with the substitution. This band was present in the medium fraction as well, albeit to a lesser extent. An extra band was also present in the cell layer fraction of fibroblasts from proband 26 carrying the novel p.(Arg918Cys) substitution, but the dimer was not detected in the secreted collagen fraction. The band migrated higher compared to the extra band observed in proband 1.

Detection of disulfide-linked α1(I) dimers with SDS-PAGE from extracted (pro)collagen of dermal fibroblasts. In P1 (p.(Arg1014Cys)), an α1(I) dimer, indicated by an arrow, is detected in the intracellular and secreted fraction. In P26 (p.(Arg918Cys)), an α1(I) dimer is detected intracellularly, but not in the medium.
Effect on collagen organization
To examine the effect of the variants on the organization of type I collagen, we performed immunocytochemistry with antibodies against type I collagen, PDI and GOLPH4 on dermal fibroblasts of proband 1 (p.(Arg1014Cys() and proband 26 (p.(Arg918Cys)) and age-matched controls (Fig. 3). In the ECM, the architecture and quantity of type I collagen appeared normal, forming dense networks parallel to the long axis of the fibroblasts. TEM analysis of the probands’ skin biopsies however revealed slightly abnormal collagen fibril morphology (Fig. 4), with variable fibril diameters and irregular interfibrillar spaces, confirming that introduction of an Arg-to-Cys substitution in the Xaa position interferes with collagen fibril organization in the matrix.

Images from cultured skin fibroblasts from the proband of family 1 (p.(Arg1014Cys)), family 26 (p.(Arg918Cys)) and age- and sex-matched controls, showing extracellular type I collagen (scale bar: 100 µm), co-staining of intracellular type I procollagen (green) with ER marker PDI (magenta) and co-staining of intracellular type I procollagen (green) with Golgi marker GOLPH4 (magenta) (scale bar: 50 µm).

Affected collagen ultrastructure in TEM images of skin biopsies showing variable fibril diameters and increased interfibrillar space in probands 1 and 26, compared to control collagen (scale bar: 500 nm).
We did not observe intracellular accumulation of type I procollagen on immunocytochemistry (Fig. 3 and Supplemental Fig. S1). However, immunoblotting for ER stress markers in cell lysates of dermal fibroblasts of proband 26 (p.(Arg918Cys)) and a matched control revealed a significant upregulation of BiP, uncleaved ATF6 and phosphorylation of PERK and a trend toward more phosphorylation of JNK, but not of (phospho-)eIF2α (Supplemental Fig. S2). Together these data suggest that the presence of mutant chains may activate the unfolded protein response, although no overt intracellular accumulation of type I procollagen is observed.
DISCUSSION
Since its discovery in 2005, the p.(Arg1014Cys) substitution in the proα1(I) chain of type I procollagen has been the only known molecular case for AD Caffey disease [3]. We now report a second heterozygous COL1A1 variant (c.2752C>T, p.(Arg918Cys)), which results in a substitution of a different arginine by cysteine 96 residues amino-terminal to the known residue in the triple helical domain of the proα1(I) chain. The latter sequence alteration causes a phenotype clinically indistinguishable from that caused by the c.3040C>T variant. Both for the c.3040C>T and the c.2752C>T variants, inter- and intrafamilial variability were noted in severity and localization of bone lesions. In nine families with the previously reported variant and in three families with the novel variant, sequence analysis confirmed AD inheritance; however, multiple family members who carried one of the variants were asymptomatic. Some did not recall symptoms in childhood while others were not certain. These observations reinforce the striking developmental features of the disorder and suggest that there may be incomplete penetrance or considerable variability in clinical expression. In one family, the c.2752C>T variant was de novo. In 14 families with the c.3040C>T variant and in one family with the c.2752C>T variant we could not complete studies of additional family members. In accord with previous studies [16, 17], distinct abnormalities of the connective tissues, such as overt bone fragility, blue sclerae, generalized joint hypermobility, skin hyperextensibility or vascular fragility, which are observed for other COL1A1 pathogenic variants, were absent.
Recently, a patient with typical signs of infantile Caffey disease, clinically indistinguishable from those with a heterozygous COL1A1 variant, was reported to harbor a homozygous pathogenic variant in AHSG [7] which encodes fetuin-A or α2-HS-glycoprotein. Only a single unresolved case with infantile Caffey disease is reported to date in whom sequencing of COL1A1 revealed no variants [6]. However, variants in AHSG were not excluded as it was not then known to be associated to the disorder. Together, these findings unequivocally confirm allelic and locus heterogeneity for infantile Caffey disease, with autosomal dominant as well as autosomal recessive inheritance patterns [8]. This has important implications for the diagnostic strategy for confirmation of Caffey disease. First, molecular analysis should include at least the entire COL1A1 and AHSG genes. Second, they show the importance of identifying the underlying molecular defect for accurate genetic counseling, in view of the different modes of inheritance, and thus the different recurrence risks. Third, sequence analysis should include both parents of children with COL1A1 alterations because of the variable expression and lost medical history.
The finding of COL1A1 variants in an inflammatory bone disease was quite unexpected, since variants in this gene classically give rise to OI, characterized by bone fragility, with multiple fractures and bone deformities [18]. The bulk of the known missense variants in both COL1A1 and COL1A2 result in substitutions for glycine, disrupting the triple helix [18]. Substitutions of nonglycine residues are rare, and when present, they usually involve Arg-to-Cys substitutions in the Xaa or the Yaa position of the triple helical domain [12, 19,20,21,22]. The identification of the p.(Arg918Cys) variant brings the number of Arg-to-Cys substitutions in the proα1(I) chain to eight (Table 1) [12, 19,20,21,22]. Besides the two variants associated with Caffey disease, a p.(Arg312Cys) variant was identified in a number of patients with a phenotype of classical Ehlers–Danlos syndrome (EDS), characterized by skin hyperextensibility and fragility, atrophic scarring, easy bruisability, and joint hypermobility, in association with vascular ruptures [12, 19, 23, 24]. In addition, p.(Arg574Cys) and p.(Arg1093Cys) were each found in one patient with isolated vascular ruptures [12], p.(Arg958Cys) was found in a patient with mild OI [20], and p.(Arg1036Cys) and p.(Arg1066Cys) were identified in families associated with an EDS/OI overlap phenotype [21, 22]. None of these patients were reported to have Caffey-like episodes of temporary hyperostosis with systemic inflammation [25]. Intracellular proα1(I) dimer formation within procollagen molecules with or without partial secretion, along with aberrations in the collagen ultrastructure, was demonstrated in each (Table 1).
For all of these substitutions, it is expected that incorporation of the proα1(I) chains into type I procollagen trimers is not disturbed so that one quarter of the molecules would have two aberrant chains and half will have one chain with the cysteine in the triple helical domain. For the approximately 80 positions within the triple helical domain in which there are substitutions of glycine for cysteine in individuals with OI [26], the side chains most likely point to the interior of the molecule, leading to more robust intramolecular dimer formation. In contrast, the Xaa and Yaa position have the side chains pointing outward and so disulfide bond formation within the trimer is less efficient [21]. Thus, the disulfide bonds may be formed between procollagen molecules (instead of within) during trafficking through the Golgi apparatus when the molecules align in register into prefibrils [27]. This could disrupt the staggered alignment in mature fibrils and affect their integrity, causing ultrastructural abnormalities. Aberrations in the periodic gap-overlap pattern due to misalignment might in addition impact intrafibrillar mineralization, which is normally mediated by fetuin-A [28]. However, since collagen fibril abnormalities are also observed in patient samples where molecules with dimers are not detected in the secreted fraction [12, 19], it is possible that collagen molecules containing a single aberrant chain can disrupt proper fibril integrity due to impaired interactions with other ECM molecules. Alternatively, the absence of the band representing the dimer in the secreted fraction possibly indicates a technical inability to detect (very) low levels of the disulfide-linked molecules.
We and others have noticed that the electrophoretic mobility of the proα1(I) dimers varies with position of the altered residue in the triple helical domain, regardless of whether the substitution is for a glycine or an Xaa or Yaa position residue. The migration of the dimers into the gel decreases as the location nears the center of the triple helical domain and are farthest with locations near the ends. This appears to reflect a change in protein volume that is largest with the cruciform structure with the disulfide bond in the center and smallest with it at the ends which allows significant rotation and linear orientation.
Ultrastructural studies reveal collagen fibrils that are more widely spaced and more variable in diameter in the dermis of all patients with Arg-to-Cys substitutions in the triple helical domain of proα1(I) chains that were tested [3, 12, 19, 21]. In contrast, immunocytochemistry on patients fibroblasts with either the p.(Arg1014Cys) or the p.(Arg918Cys) variant uncovered no defects in the architecture of type I collagen in the ECM. Possible explanations are the lower magnification/resolution and/or the more elaborate processing of skin biopsies used in immunocytochemistry compared to TEM. The absence of intracellular accumulation of type I procollagen suggests that the nonsecreted molecules that contain dimers are degraded before excessive intracellular buildup occurs. Yet, moderate signs of ER stress, such as a (mildly) dilated ER in p.(Arg312Cys) and in p.(Arg958Cys) fibroblasts, and an upregulation of ER stress markers in p.(Arg918Cys) and p.(Arg958Cys) have been exposed in this and other studies [19, 20].
To date it is unclear why the proα1(I) p.(Arg918Cys) and p.(Arg1014Cys) substitutions lead to an early-onset, self-limiting disorder with localized subperiosteal new bone formation and local and systemic inflammation, and absence of overt signs of connective tissue fragility. We suspect that the phenotype is caused by alterations in cell signaling due to the spatiotemporal disruption of specific interactions with type I (pro)collagen molecules containing either one or two aberrant (pro)α1(I) chains, rather than due to the structural abnormalities of the connective tissue. This could explain the variable phenotypes associated with Arg-to-Cys substitutions. Analogous to what is observed in type I procollagen, Arg-to-Cys substitutions in type II procollagen result in a phenotype that is distinct from the chondrodysplasia spectrum normally caused by glycine substitutions [29].
Currently, the pathogenic mechanism behind Caffey disease remains elusive, but multiple hypotheses are proposed. The two identified COL1A1 variants most likely affect (a) common signaling pathway(s). Candidate molecules that are suspected to play a role in the pathogenesis are prostaglandin E (PGE) and transforming growth factor beta (TGF-β). Both can induce cortical hyperostosis when upregulated [25, 30, 31]. Long-term PGE administration in children with ductus-dependent cyanotic congenital heart disease is associated with new bone formation [30]. In Ghosal syndrome, the characteristic increased bone density with predominant diaphyseal involvement is believed to be the result of elevated levels of PGE caused by homozygous missense variants in TBXAS1, encoding the enzyme thromboxane synthase, which converts prostaglandins to thromboxane A2 [32]. Elevated PGE levels are reported in patients with Caffey disease and treatment with indomethacin, a prostaglandin synthetase inhibitor, is shown to decrease the symptoms [33].
TGF-β1 is constitutively active in Camurati–Engelmann disease, due to pathogenic variants in the latency-associated peptide of TGF-β1, and results in cortical hyperostosis [31]. TGF-β cytokines are antagonized by fetuin-A, which was recently reported to be associated with Caffey disease and which has homology to the TGF-β receptor type II in a cysteine-flanked 18–19 amino acid sequence known to form a disulfide-linked loop in fetuin-A [7, 34, 35]. Fetuin-A deficient mice display enhanced bone formation presumed to be due to failure to block TGF-β signaling in osteoblasts [36]. TGF-β levels have not been reported yet for Caffey disease, but the variants in type I procollagen may disrupt interactions, which in turn can lead to TGF-β activation.
The two Arg-to-Cys substitutions causing Caffey disease are both located close to the region interacting amongst others with α2β1 integrin, fibronectin and IL-2. α2β1 integrin signaling promotes osteoblast differentiation by activating Runx2 and downregulating the TGF-β receptors expression in osteoblastic cells, thus reducing the inhibition of terminal osteoblast differentiation by TGF-β [37, 38]. Furthermore, fibronectin incorporates latent TGF-β binding proteins (LTBP) into the ECM, which regulate the storage and availability of TGF-β. Abrogation of fibronectin results in elevated free TGF-β [39]. Therefore, the disrupted interactions of mutant type I collagen with both α2β1 integrin and fibronectin (and possibly other partners) and consequential aberrations in TGF-β signaling could be at the basis of the observed increased bone formation [25].
The interaction of IL-2 with type I collagen in this region offers a hypothesis for the temporary inflammatory symptoms in Caffey disease [2, 3, 25]. IL-2 is an important cytokine involved in neonatal adaptive immunity [40], so disturbed interactions due to variants in this region of type I collagen possibly induce temporary inflammation in young children, which decreases with the maturation of their immune system. Although the interaction of IL-2 with type I collagen is only suggested to occur in the region of p.Arg1014, a role for the p.Arg918 residue is not excluded [14, 15]. In the patient with the homozygous AHSG variants, the inflammatory signs are presumably the result of the loss of the anti-inflammatory capacity of fetuin-A [34]. Apart from the maturity of the immune system, the self-limiting character of the symptoms associated with Caffey disease may be the result of differences in bone (re)modeling between young children and adults due to distinct developmental demands.
Future studies focusing on obtaining mechanistic insights should focus on bone tissue, ideally collected during an episode of hyperostosis. In addition, the generation of animal models harboring these specific COL1A1 variants might shed more light tpdelon the disease mechanisms and will allow to study relevant tissues.
In summary, we report a novel Arg-to-Cys substitution in the proα1(I) chain of type I procollagen that is associated with infantile Caffey disease. This discovery expands the molecular spectrum of the disease, confirming allelic heterogeneity, and revises the diagnostic strategy. The two Caffey-associated Arg-to-Cys substitutions that are now described add to the limited group of Arg-to-Cys substitutions in the proα1(I) chain of type I procollagen. This group contains different connective tissue disorders, where Caffey disease is the only one associated with subperiosteal hyperostosis and inflammation of the overlying soft tissue. The reason for the unique and temporary nature of Caffey disease remains elusive. Further studies of the pathological mechanisms behind this spatiotemporally limited bone disorder in a relevant animal model and/or the affected tissues of a patient can enhance our understanding of bone formation in normal and diseased states.
Data availability statement
All data will be made available upon request.
References
Caffey J. On some late skeletal changes in chronic infantile cortical hyperostosis. Radiology. 1952;59:651–657.
Kamoun-Goldrat A, le Merrer M. Infantile cortical hyperostosis (Caffey disease): a review. J Oral Maxillofac Surg. 2008;66:2145–2150.
Gensure RC, et al. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J Clin Invest. 2005;115:1250–1257.
Kamoun-Goldrat A, et al. Prenatal cortical hyperostosis with COL1A1 gene mutation. Am J Med Genet A. 2008;146A:1820–1824.
Navarre P, Pehlivanov I, Morin B. Recurrence of infantile cortical hyperostosis: a case report and review of the literature. J Pediatr Orthop. 2013;33:e10–17.
Kitaoka T, et al. Two Japanese familial cases of Caffey disease with and without the common COL1A1 mutation and normal bone density, and review of the literature. Eur J Pediatr. 2014;173:799–804.
Merdler-Rabinowicz R, et al. Fetuin-A deficiency is associated with infantile cortical hyperostosis (Caffey disease). Pediatr Res. 2019;86:603–607.
Schweiger S, Chaoui R, Tennstedt C, Lehmann K, Mundlos S, Tinschert S. Antenatal onset of cortical hyperostosis (Caffey disease): case report and review. Am J Med Genet A. 2003;120A:547–552.
Syx D, et al. Bi-allelic AEBP1 mutations in two patients with Ehlers–Danlos syndrome. Hum Mol Genet. 2019;28:1853–1864.
Delbaere S, et al. Novel defects in collagen XII and VI expand the mixed myopathy/Ehlers–Danlos syndrome spectrum and lead to variant-specific alterations in the extracellular matrix. Genet Med. 2020;22:112–123.
Van Damme T, et al. Biallelic B3GALT6 mutations cause spondylodysplastic Ehlers–Danlos syndrome. Human molecular genetics. 2018;27:3475–3487.
Malfait F, et al. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers–Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28:387–395.
Zimmer AD, et al. Sixteen novel mutations in PNPLA1 in patients with autosomal recessive congenital ichthyosis reveal the importance of an extended patatin domain in PNPLA1 that is essential for proper human skin barrier function. Br J Dermatol. 2017;177:445–455.
Sweeney SM, et al. Candidate cell and matrix interaction domains on the collagen fibril, the predominant protein of vertebrates. J Biol Chem. 2008;283:21187–21197.
Somasundaram R, et al. Collagens serve as an extracellular store of bioactive interleukin 2. J Biol Chem. 2000;275:38170–38175.
Cerruti-Mainardi P, et al. Infantile cortical hyperostosis and COL1A1 mutation in four generations. Eur J Pediatr. 2011;170:1385–1390.
Cho TJ, et al. The c.3040C>T mutation in COL1A1 is recurrent in Korean patients with infantile cortical hyperostosis (Caffey disease). J Hum Genet. 2008;53:947–949.
Forlino A, Marini JC. Osteogenesis imperfecta. Lancet. 2016;387:1657–1671.
Nuytinck L, Freund M, Lagae L, Pierard GE, Hermanns-Le T, De Paepe A. Classical Ehlers–Danlos syndrome caused by a mutation in type I collagen. Am J Hum Genet. 2000;66:1398–1402.
Makareeva E, et al. Substitutions for arginine at position 780 in triple helical domain of the alpha1(I) chain alter folding of the type I procollagen molecule and cause osteogenesis imperfecta. PLoS One. 2018;13:e0200264.
Cabral WA, et al. Y-position cysteine substitution in type I collagen (alpha1(I) R888C/p.R1066C) is associated with osteogenesis imperfecta/Ehlers–Danlos syndrome phenotype. Hum Mutat. 2007;28:396–405.
Lund A, Joensen F, Christensen E, Duno M, Skovby F, Schwartz M. A novel arginine-to-cysteine substitution in the triple helical region of the alpha1(I) collagen chain in a family with an osteogenesis imperfecta/Ehlers–Danlos phenotype. Clin Genet. 2008;73:97–101.
Colombi M, Dordoni C, Venturini M, Zanca A, Calzavara-Pinton P, Ritelli M. Delineation of Ehlers–Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: Report on a three-generation family without cardiovascular events, and literature review. Am J Med Genet. 2017;173:524–530.
Duong J, et al. A family with Classical Ehlers–Danlos syndrome (cEDS), mild bone fragility and without vascular complications, caused by the p.Arg312Cys mutation in COL1A1. Eur J Med Genet. 2020;63:103730.
Nistala H, Makitie O, Juppner H. Caffey disease: new perspectives on old questions. Bone. 2014;60:246–251.
Marini JC, et al. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–221.
Canty EG, Kadler KE. Procollagen trafficking, processing and fibrillogenesis. J Cell Sci. 2005;118:1341–1353.
Toroian D, Price PA. The essential role of fetuin in the serum-induced calcification of collagen. Calcif Tissue Int. 2008;82:116–126.
Hoornaert KP, et al. The phenotypic spectrum in patients with arginine to cysteine mutations in the COL2A1 gene. J Med Genet. 2006;43:406–413.
Ueda K, et al. Cortical hyperostosis following long-term administration of prostaglandin E1 in infants with cyanotic congenital heart disease. J Pediatr. 1980;97:834–836.
Kinoshita A, et al. Domain-specific mutations in TGFB1 result in Camurati–Engelmann disease. Nat Genet. 2000;26:19–20.
Geneviève D, et al. Thromboxane synthase mutations in an increased bone density disorder (Ghosal syndrome). Nat Genet. 2008;40:284–286.
Heyman E, Laver J, Beer S. Prostaglandin synthetase inhibitor in Caffey disease. J Pediatr. 1982;101:314.
Jahnen-Dechent W, Heiss A, Schafer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res. 2011;108:1494–1509.
Demetriou M, Binkert C, Sukhu B, Tenenbaum HC, Dennis JW. Fetuin/alpha2-HS glycoprotein is a transforming growth factor-beta type II receptor mimic and cytokine antagonist. J Biol Chem. 1996;271:12755–12761.
Szweras M, et al. alpha 2-HS glycoprotein/fetuin, a transforming growth factor-beta/bone morphogenetic protein antagonist, regulates postnatal bone growth and remodeling. J Biol Chem. 2002;277:19991–19997.
Takeuchi Y, Nakayama K, Matsumoto T. Differentiation and cell surface expression of transforming growth factor-beta receptors are regulated by interaction with matrix collagen in murine osteoblastic cells. J Biol Chem. 1996;271:3938–3944.
Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E. Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect Tissue Res. 2003;44:109–116.
Kawelke N, Vasel M, Sens C, Au A, Dooley S, Nakchbandi IA. Fibronectin protects from excessive liver fibrosis by modulating the availability of and responsiveness of stellate cells to active TGF-beta. PLoS One. 2011;6:e28181.
Basha S, Surendran N, Pichichero M. Immune responses in neonates. Expert Rev Clin Immunol. 2014;10:1171–1184.
Acknowledgements
We thank all the patients and the referring physicians. D.S. and F.M. are fellows of the Research Foundation, Flanders (FWO). T.D. has a BOF doctoral fellowship from Ghent University. This study was financially supported by Ghent University (Methusalem grant BOFMET2015000401) and with funds from the Collagen Diagnostic Laboratory, Department of Pathology, University of Washington.
Author information
Authors and Affiliations
Contributions
CRediT category: Conceptualization: T.D., D.S., P.H.B., F.M. Data curation: D.S., S.S. Formal analysis: T.D., S.S. Funding Acquisition: P.H.B., F.M. Investigation: T.D., D.S., T.H., I.H., S.S. Methodology: T.D., D.S., S.S., F.M. Project administration: D.S., S.S., F.M. Resources: G.M., J.Z., P.H.B., F.M. Supervision: S.S., P.H.B., F.M. Validation: T.D., D.S., S.S.,; Visualization: T.D. Writing—original draft: T.D., F.M. Writing—review & editing: T.D., D.S., T.H., I.H., G.M., J.Z., S.S., P.H.B., F.M.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This study was approved by the Ethics Committee of the Ghent University Hospital and conducted according to the Declaration of Helsinki. For the study done at the University of Washington the samples were part of an Institutional Review Board reviewed Research Repository for Heritable Disorders of Bone, Blood Vessels, and Skin at the University of Washington. The parents of the probands of families 19 and 28 consented to the publication of the radiographic images via an opt-in for the submission of the samples in the Repository. For the other families, only de-identified summarized clinical data are presented. The (parents of the) probands of families 1 and 26 signed an informed consent form for taking a skin biopsy.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dhooge, T., Syx, D., Hermanns-Lê, T. et al. Caffey disease is associated with distinct arginine to cysteine substitutions in the proα1(I) chain of type I procollagen. Genet Med 23, 2378–2385 (2021). https://doi.org/10.1038/s41436-021-01274-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01274-y
