
Abstract
Purpose
Roughly 70% of suspected Mendelian disease patients remain undiagnosed after genome sequencing, partly because knowledge about pathogenic genes is incomplete and constantly growing. Generating a novel pathogenic gene hypothesis from patient data can be time-consuming especially where cohort-based analysis is not available.
Methods
Each patient genome contains dozens to hundreds of candidate variants. Many sources of indirect evidence about each candidate may be considered. We introduce InpherNet, a network-based machine learning approach leveraging Monarch Initiative data to accelerate this process.
Results
InpherNet ranks candidate genes based on orthologs, paralogs, functional pathway members, and colocalized interaction partner gene neighbors. It can propose novel pathogenic genes and reveal known pathogenic genes whose diagnosed patient-based annotation is missing or partial. InpherNet is applied to patient cases where the causative gene is incorrectly ranked low by clinical gene-ranking methods that use only patient-derived evidence. InpherNet correctly ranks the causative gene top 1 or top 1–5 in roughly twice as many cases as seven comparable tools, including in cases where no clinical evidence for the diagnostic gene is in our knowledgebase.
Conclusion
InpherNet improves the state of the art in considering candidate gene neighbors to accelerate monogenic diagnosis.
Similar content being viewed by others
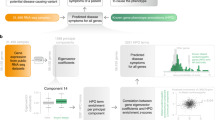
INTRODUCTION
Every year, approximately 7 million newborns worldwide are affected by severe, possibly Mendelian, diseases [1]. Mendelian diseases are most often monogenic, caused by 1–2 highly penetrant variants in a single gene. Using genome sequencing, diagnosing such monogenic conditions can be done by identification of the causative gene against the current body of biomedical knowledge. This is a time-consuming task for clinicians, since sequencing can result in dozens to hundreds of candidate causative genes that contain variants rarely found in the unaffected population [2,3,4,5]. As sequencing technology becomes more time- and cost-efficient, the number of patients being sequenced for genetic disease diagnosis is expected to grow to the millions by 2025 [6].
Numerous tools that automate and therefore speed up various aspects of the diagnosis pipeline for patients with suspected Mendelian disease have been developed. For example, ANNOVAR [7] annotates variants with various relevant attributes, and tools like M-CAP [8] and S-CAP [9] help assess variant pathogenicity. ClinPhen [10] helps extract patient phenotypes from their free text medical records, and candidate causative gene prioritization tools such as Phevor [11], PhenIX [12], Phrank [13], and AMELIE [14] improve diagnosis efficiency by prioritizing a patient’s candidate genes for their likelihood of causing the patient’s set of phenotypes.
Hundreds of novel Mendelian pathogenic genes are discovered each year [15,16,17,18]. After a novel pathogenic gene is proven, it is conceptually moved from the research realm into the clinic, where diagnoses are best done by alignment to clinical evidence from previously diagnosed patients (Fig. 1). However, while a gene’s pathogenicity is still being evaluated, researchers will consult literature in search of indirect evidence that makes the gene a plausible hypothesis for a particular patient’s case. For example, one of the patient candidate genes, yet undocumented as causing a monogenic disease in human, may have an ortholog known to cause similar phenotypes in a model organism. Similarly, a candidate gene may be in the same functional pathway as known relevant pathogenic genes or have an obligate interaction partner known to explain the patient’s set of phenotypes.

Patient sequencing data are first assessed against human clinical data, where one is able to match a candidate variant or gene in the current patient to previously diagnosed patients with very similar phenotypic abnormalities. When clinical evidence cannot be found, the case moves to the research realm where indirect evidence is sought to suggest a novel causative gene candidate. InpherNet aims to accelerate this discovery process by offering researchers its most appealing testable hypothesis through indirect evidence. Consequently, InpherNet can also aid in cases where a clinical diagnosis exists but is not well represented in one’s knowledgebase.
This open-ended search for the most plausible hypothesis is very time-consuming. Computational inference tools like Phevor [11], Phive [12], and hiPhive [12] have been developed to help accelerate the discovery of testable research hypotheses. Such tools perform cross-species and gene product interaction-based inference to prioritize candidate genes beyond patient-based phenotypic knowledge.
Here we propose InpherNet, a network-based machine learning gene prioritization method that leverages the Monarch Initiative knowledgebase [19] to improve the discovery of appealing novel pathogenic gene hypotheses. To predict causative genes using non-patient-derived information, InpherNet considers variant-based information and four sources of indirect evidence, or gene neighbors: phenotypes associated with orthologs (i.e., the same gene in a related organism), paralogs (i.e., another gene from the same gene family member), members of the same functional pathway, and expression colocalized interaction partners. Using real patient data, we show that InpherNet improves on previous similar tools in ranking causative genes based on indirect evidence, which is helpful both in cases where the disease is novel and cases where the phenotype annotations are currently lacking.
MATERIALS AND METHODS
InpherNet graph
Ensembl genes
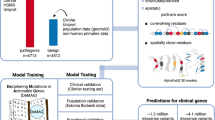
Ensembl [20] is a consortium that develops and curates many comparative genomics resources. We anchor the InpherNet graph (Fig. 2 and Table S1) on their human, mouse, and zebrafish gene sets.

We base our network on the Ensembl gene sets for human, mouse, and zebrafish. We augment it with human, mouse, and zebrafish (in-species) paralogs, and human–mouse and human–zebrafish orthologs, also from Ensembl. To these we add pathway, interaction, anatomical localization, phenotypes, and monogenic disease relationships from Monarch Initiative’s graph database.
Mendelian subgraph of Monarch Initiative’s multispecies biological network
The Monarch Initiative [19] is an effort to develop a comprehensive biological database incorporating numerous entities (e.g., genes and phenotypes) and relationships among these entities from multiple existing databases. Since InpherNet aims to prioritize candidate genes in patients affected with Mendelian diseases, we picked a subset of the Monarch database that is both relevant to Mendelian disease inference and annotated with sufficient data (Supplementary Methods). Organismal and cellular level gene phenotype and functions are obtained from Monarch’s unified phenotype ontology, Upheno [19]. We focus on the three species for which annotations are by far most abundant—human, mouse, and zebrafish—taking the cross-species Gene Ontology [21] (GO) along with three species specific phenotypic databases: the Human Phenotype Ontology [22] (HPO), Mouse Genome Informatics (MGI) phenotype [23], and the Zebrafish Information Network [24] (ZFIN). Cross-species pathway and gene expression anatomical localization data is obtained from Reactome [25] and Uberon [19], respectively. Potential human protein–protein interaction (PPI) data is obtained from BioGRID [26], and monogenic human disease information is obtained from OMIM [27] and Orphanet [28]. In total, we selected 9 ontology sources (Table S1) encompassing 1,231,846 attributes of human, mouse, and zebrafish genes (Fig. 2).
Gene orthology and paralogy mappings from Ensembl
We used Ensembl’s human, mouse, and zebrafish within species gene paralogs relationships, as well as mouse and zebrafish to human gene orthology relationships (Table S1) to complete the InpherNet knowledge graph (Fig. 2). Extending observations associated with human genes through paralogs and their mouse and zebrafish orthologs enables hypothesis generations on many more human genes [16]. For example, only 3,438, or 17.8%, of human protein-coding genes in our graph are annotated with direct human patient-derived phenotypes. However, after projecting mouse and zebrafish phenotypes to their orthologous human genes, over 56.3% (3.2× more) of human genes can be phenotypically annotated (Fig. 3). If we also consider information from human, mouse, and zebrafish paralogous genes, coverage rises to 71.8% (4.04× more). Overall, in our data set, 17,784 (91.9%) of 19,343 total human genes have orthologous genes in either mouse or zebrafish (not all of which are currently phenotypically annotated), 13,315 (68.8%) have human (in-)paralogous genes, and 13,189 (68.2%) have mouse or zebrafish (out-)paralogous genes.

HPO-A contains human gene–phenotype relationships for about 3,400 out of 20,000 human protein-coding genes. Thus, clinical evidence-based gene prioritization methods that only use known patient phenotype associations cannot prioritize 82.4% of human protein-coding genes. However, many unannotated human genes have functionally annotated orthologs in mouse and zebrafish that can be combined via Monarch Initiative’s Upheno cross-species phenotype ontology to triple the annotation coverage to 56% of human protein-coding genes compared to the original 17.5%.
Gene scoring by means of a supervised machine learning algorithm
The goal of candidate gene-ranking tools is to rank the true causative gene at the top to allow clinicians to find diagnoses, or enable researchers to propose a novel hypothesis, after reviewing as few candidate genes as possible. Our machine learning classifier takes a vector of scalar values (called “features”) as input, and outputs a score between 0 and 1, indicating the classifier’s assessment of whether the input should be classified as positive (here, indicating that the indirect evidence suggests a gene is causative for a patient) or negative (here, that the indirect evidence does not support a match). InpherNet uses a Gradient Boosting Tree classifier [29], a supervised machine learning classifier, to assign such a score to each candidate gene (Fig. S1 and Supplementary Methods).
InpherNet feature set
Candidate genes are genes that contain at least one candidate causative variant in the patient (Supplementary Methods). We associated each candidate gene with a vector of 15 features derived from information about the candidate gene’s neighbors (orthologs, paralogs, pathways, and interaction partners) and the candidate variants (Fig. S1).
Gene neighbors feature set
Mouse ortholog
Orthologs are similar genes in two different species related via a speciation event and often have similar functions [30]. For each patient candidate gene, we computed a phenotypic match score between the mouse ortholog–associated phenotypes from the MGI phenotype database [23] and the patient phenotypes in HPO terms using a Phrank [13]-based phenotype match score that we call “Phranken” (for Phrank-Normalized). The Phranken score takes two sets of phenotype terms and an underlying phenotype directed acyclic graph (DAG) as inputs and returns a match similarity score (Supplementary Methods). In InpherNet, we took the phenotype DAG and gene’s phenotype annotations from Upheno [19], which organizes hierarchical relationships among phenotype terms about multiple species. For candidates with more than one Ensembl mouse ortholog, the highest Phranken score among all orthologs is selected as the value for the “mouse ortholog” feature. If a candidate gene has no mouse ortholog or none of the mouse orthologs has any annotations, −1 is assigned. The same convention is used repeatedly to compute the other neighbor feature values described below.
Zebrafish ortholog
Defined as the highest Phranken match score between zebrafish ortholog–associated phenotypes from ZFIN and the patient’s phenotypes.
Human in-paralog
In-paralogs are genes found in the same species that are in the same gene family (related via duplication). For human patient-derived phenotypes in HPO-A, their phenotype abnormalities are linked through a disease term from OMIM [27] or Orphanet [28] (Fig. 2). Therefore, for human genes, instead of calculating the max Phranken score per gene, we computed the Phranken score for all diseases known to be caused by each candidate gene and select the max score (Supplementary Methods).
Mouse out-paralog
For each candidate gene, we collected all mouse in-paralogs of the candidate gene’s mouse ortholog, which are also known as mouse out-paralogs. The highest Phranken match score between mouse out-paralog-associated phenotypes and the patient’s phenotypes is picked.
Zebrafish out-paralog
We similarly used the candidate gene’s zebrafish out-paralogs.
Pathway
For each candidate gene, we collected all human, mouse, and zebrafish genes that are in the same Reactome [25] pathways as the patient candidate gene. For human genes, we also collected diseases they are known to cause and their related phenotypes. The highest Phranken match score between the patient’s phenotypes and any pathway gene’s phenotypes for mouse and zebrafish genes or pathway gene’s disease phenotypes for human genes is then used.
Interaction partner
For each candidate gene, we retrieved a set of interaction partners supported by both a human protein–protein interaction (PPI) BioGRID [26] network and human gene expression anatomical localization Uberon [19] data (Supplementary Methods). Intuitively, we limited ourselves to genes whose protein products may interact with the candidate genes in human cells. We picked the highest Phranken score between the patient’s phenotypes and the phenotypes related to the diseases the interaction partners are known to cause.
Candidates in 1-hop neighborhood
For each candidate gene, we defined a 1-hop neighborhood as a set of genes that can be reached through 1-hop interaction links from the BioGRID by Uberon human PPI subnetwork defined above (i.e., their gene products can interact directly in human cells). We counted how many other candidate genes are in this neighborhood, and this count is reported as the “candidates in 1-hop neighborhood” feature.
Candidates in 2-hop neighborhood
For the “candidates in 2-hop neighborhood” feature, we repeated the step above but looked for 2-hop neighborhood instead. The 2-hop neighborhood excludes all genes in the 1-hop neighborhood.
Variant-based feature set
Candidate variants count
This is the number of variants in the candidate gene.
M-CAPgene
M-CAP [8] is a pathogenicity score that assigns a number between 0 (likely benign) and 1 (possibly pathogenic) to rare human missense variants. We calculated an M-CAP-based feature for each gene as the maximum M-CAP score of all candidate variants in the candidate gene. A candidate variant that did not have an M-CAP score was assigned the maximum M-CAP score in a window of −50, +50 basepairs adjacent to that variant.
M-CAP100
We calculated the highest M-CAP score in a window of −50, +50 basepairs adjacent to all candidate variants in the candidate gene, then select the maximum value.
RVIS score
This is the RVIS [31] gene mutability score of the candidate gene.
pLI score
This is the pLI [32] haploinsufficiency score of the candidate gene.
Average ExAC allele count
The average Exome Aggregation Consortium [32] (ExAC) allele count of all candidate variants in the candidate gene is used.
For M-CAP, RVIS, pLI, and ExAC, if the original resource did not offer relevant values, we assigned a default “null” value (Supplementary Methods).
Other gene prioritization tools
We compared InpherNet’s performance to seven other gene prioritization tool configurations, in two conceptual sets. PhenIX [12], Phrank_HPOA [13], and Phranken_HPOA target genes that have patient-based Mendelian disease associations, while Phevor [11], Phive [12] and hiPhive [12], similar to InpherNet, use additional non-patient-based information and also infer novel pathogenic candidates (Fig. 1). Phrank_HPOA ranks candidate genes by their Phrank match score using patient-derived gene annotations from the HPO-A database. Similarly, Phranken_HPOA normalizes the Phrank score using the maximal possible Phrank score between the patient and gene phenotype sets (Supplementary Methods). PhenIX ranks candidate genes by their phenotype annotations similarities with the patient’s phenotypes both in HPO and the candidate variants’ pathogenicity. Phevor [11] combines multiple ontologies to rank patient candidate genes. We used Phevor in two ways: “Phevor_HPOA” uses only HPO-A similar to other clinical tools, and “Phevor_all” uses additional non-patient-based ontologies similar to the other inference tools. Phive ranks candidate genes using mouse phenotypic data, and most comparable to InpherNet, hiPhive combines functional data derived from human, mouse, and zebrafish genes and the candidate genes’ relatedness in a PPI network to the suspected causative gene to rank candidate genes. We took great care to compare the causative gene-ranking performance of all methods on equal footing (Supplementary Methods).
InpherNet training set
We constructed a set of synthetic patients to train InpherNet’s Gradient Boosting Tree classifier to conserve all real patient data for testing. For this process, we used 2,504 sequenced individuals from the 1,000 Genomes Project (KGP) [33], Mendelian pathogenic variants with an OMIM disease identifier from ClinVar [34], and HPO-A phenotypes associated with each OMIM disease. To construct each synthetic patient, we took a KGP genome, added a randomly selected pathogenic variant from ClinVar for a known OMIM disease, and associated the patient with a subset of noisily sampled and augmented HPO-A disease-associated phenotypes, mimicking imperfect prediagnosis clinical annotations (Supplementary Methods). We also ensured that no causative gene in the training set is the causative gene of any real patient used for testing or validation. Using this method, we generated 2,504 different synthetic patients with an average of 9.1 phenotypes and 300.4 candidate genes per patient.
InpherNet test set
We tested InpherNet’s performance on real singleton patients with their prediagnosis phenotypes and clinician-verified Mendelian diagnoses (Supplementary Methods). Since InpherNet is meant to complement clinical evidence-based tools, we created test sets containing diagnosed patient cases whose causative genes were incorrectly given low priority by clinical evidence gene-ranking tools. This helps to examine InpherNet’s ability to augment clinical evidence, where such augmentation is most needed. We first ran Phrank_HPOA and PhenIX on all available 255 real patients to find a cohort of patients where Phrank_HPOA failed to rank the causative gene in top 10 (Phrank > 10), and a cohort of patients where PhenIX failed to rank the causative gene in top 10 (PhenIX > 10; Table S2). Phrank > 10 contains 70 patients (with an average of 8.6 phenotypes and 321.0 candidate genes per patient), and PhenIX > 10 contains 115 patients (with an average of 7.8 phenotypes and 309.0 candidate genes per patient). Assuming the top 10 genes from these clinical tools would already have been scrutinized by a clinician and discarded as noncausative, we removed the top 10 Phrank_HPOA- or PhenIX-ranked genes from the candidate gene list for each patient in each test cohort, respectively.
InpherNet’s feature ablation analysis
We analyzed which above listed features contribute most to the accuracy of the model by performing a feature ablation test. We iteratively removed each feature, retrained the model, then recorded how the causative genes’ rankings changed. We used the same 2,504 synthetic patients for training and the same two test sets, Phrank > 10 and PhenIX > 10, for testing. The importance of each feature was measured using the average causative gene rankings.
Interpreting InpherNet gene rankings
To provide human-interpretable explanations for InpherNet’s gene rankings, we included a ranked list of each candidate gene’s neighbors ordered by the Phranken phenotype match score between the patient’s phenotypes and neighbor-associated phenotypes (Fig. S1). This list helps researchers see which neighbor is phenotypically most similar to the patient’s phenotypes, and via the InpherNet graph (Fig. 2) and Monarch, link back to the original databases and publications supporting these claims.
RESULTS
InpherNet outperforms existing phenotype ranking-based methods
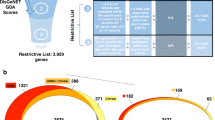
First, of the two clinical data based tools we use to examine their top 10 genes, Phrank and PhenIX, we see that Phrank leaves over 40% fewer cases unresolved (70 vs. 115) and performs much better on the PhenIX > 10 set than PhenIX does on the Phrank > 10 set (Fig. 4), in concordance with earlier tool evaluations [13]. After removing the clinical evidence-based top 10 genes from consideration (guaranteed to not include the correct causative gene), InpherNet offers a very significant improvement on all tools for the Phrank > 10 set, with over three times as many cases where the causative gene ranks 1, and almost twice as many cases where it ranks 1–5 as all other tools (Fig. 4b). InpherNet also offers a marked improvement over the weaker (larger) PhenIX > 10 set, with at least 10 more cases ranked 1–5 compared to all other tools (Fig. 4c).

(a) We took 255 real patients with diverse prediagnosed conditions and first used two patient evidence-based methods to highlight cases where either patient evidence is missing or differs significantly from the current patient. In 115 cases the causative genes were not among the top 10 PhenIX-ranked genes, and in ~40% fewer cases (70), the causative gene was not in the Phrank_HPOA top-ranked 10 genes. Each case is then reviewed by 8 inference tools after removing the highest ranked 10 genes that were already determined as not causative. (b) InpherNet offers a large improvement on all tools for the tougher (preferred, smaller) Phrank > 10 set, with over three times as many cases where the causative gene ranks 1, and almost twice as many cases where it ranks 1–5 as all other tools. (c) InpherNet still outperforms all other tools over the larger (weaker) PhenIX > 10 set, ranking 10 or more causative genes among top 1–2, 1–3, and 1–5 as all other tools. (d,e) InpherNet also outputs a list of gene neighbors ranked by their relevance to the patient phenotype. In both test sets we see that all 4 types of indirect evidence (i.e., orthology, paralogy, pathways, and interactions) contribute the most to the correct gene being ranked on top for different candidates.
InpherNet ranks candidate genes that lack patient phenotype annotations
Ranking candidate genes that lack any patient phenotype annotations is critical for the discovery of novel pathogenic genes and helpful where one’s knowledgebase may be missing clinical annotations for a known causative gene. In our real patient test cohort Phrank > 10, an average of 79% of patient candidate genes (7,749 unique genes across all 70 patients) do not have any HPO annotations and are therefore automatically ranked at the bottom by methods that rely exclusively on HPO-A clinical evidence (Supplementary Methods). But InpherNet can rank a causative gene high even in the absence of any direct clinical evidence. For example, in our Phrank > 10 test set, patient 122 causal gene BPTF is one of 246 patient candidate gene for which our knowledgebase contains no human patient phenotypic evidence, but InpherNet ranks BPTF top 10. For patient 104, the causative gene PTCHD1 is one of 303 genes with no patient phenotypes, yet InpherNet ranks it 10.
Feature ablation test
We performed feature ablation as described in “Materials and Methods.” All ablated models performed worse compared to the full model (i.e., InpherNet). InpherNet’s average causative gene ranking was 8.67 and 6.98 for Phrank > 10 and PhenIX > 10, respectively while the ablated models’ averages increased up to 13.78 and 13.63, respectively. The most informative features were average ExAC allele count, interaction partner, and mouse ortholog (Fig. S2).
Interpretability of InpherNet’s prediction process
All four neighbor types (orthology, paralogy, pathways, and interactions) contribute to InpherNet’s performance. We observe that all categories of neighbors are represented as the most important contributor in our top rankings (Fig. 4d, e). Table 1 provides an example each where the correct causal gene ranking is best supported by each evidence type.
For example, in patient 096, the causative gene CTNNB1 is ranked 1 by InpherNet, most strongly supported through its mouse ortholog Ctnnb1. The patient presented six phenotypes including Achilles tendon contracture (HP:0001771) and childhood-onset truncal obesity (HP:0008915). In our HPO-A, the causative gene CTNNB1 is annotated by some relevant phenotypes including abnormality of the hair (HP:0001595), abnormality of skin pigmentation (HP:0001000), and obesity (HP:0001513). However, its annotation does not explain the patient’s phenotypes fully and lacks a clear match, resulting in a lower Phrank_HPOA rank of 22 among 364 candidate causative genes. However, InpherNet ranks CTNNB1 at the very top because its mouse ortholog, Ctnnb1, is annotated with more relevant phenotypes such as abnormal forelimb morphology (MP:0000550) and abnormal melanogenesis (MP:0005077) [35].
Similarly, InpherNet ranks the causative gene KCNA2 for patient 128 at the top of 389 candidate genes, best supported by information about its in-paralog, KCNA1. Human patients with rare variants in KCNA1 have shown phenotypes similar to this patient’s phenotypes including abnormality of movement (HP:0100022), global developmental delay (HP:0001263), hearing impairment (HP:0000365), hypotonia (HP:0001252), intellectual disability (HP:0001249), and seizures (HP:0001250). The patient’s actual causative gene, KCNA2, lacks clinical motor- or hearing-related patient-based phenotype annotations in our database resulting in low rank (53 for Phrank_HPOA and 121 for PhenIX) for the clinical ranking tools, while inference tool hiPhive ranks this gene 31.
In the case of patient 123, the causative gene identified is GNB1. Phrank_HPOA ranks this gene at 26, and hiPhive 25. InpherNet ranks this gene at the top among 312 candidate causative genes, supported predominantly through ITPR, which is in the same Ca2+ pathway [25] as GNB1.
Finally, the connection between interaction partners PHF8 and TAF1 bring the correct causative gene PHF8 for patient 073 to the top, compared to Phrank_HPOA at 29 and hiPhive at 11 among 321 candidate genes. While rare variants in both PHF8 and TAF1 are known to cause X-linked mental retardation, the phenotypic abnormalities associated with each of these genes in our knowledgebase differ. PHF8 is associated with phenotypic abnormalities unobserved in this patient such as cleft upper lip (HP:0000204). However, its well-known interaction partner TAF1 is associated with much more relevant phenotypes including delayed gross motor development (HP:0002194), delayed speech and language development (HP:0000750), microcephaly (HP:0000252), and neurodevelopmental delay (HP:0012758).
DISCUSSION
To make a conclusive diagnosis, the desired outcome is finding one or more rare functional variants in a patient’s genome that match the disease state of previously diagnosed patients. This however only currently happens in 30% of analyzed cases. [4, 36] Furthermore, to decrease the cost of genomic diagnosis, one would like to use computational tools to effectively prioritize candidate variants so that clinicians use their time efficiently. However, as our body of knowledge is constantly growing [14], it is impossible to guarantee that any knowledgebase we provide our tools will be comprehensive. Here we show that InpherNet, by using indirect evidence—namely, evidence other than previous human patients-derived phenotypes known to be tied to the candidate gene—is able to help in both scenarios. By leveraging the phenotypes associated with orthologs, paralogs, pathway members, and potential interaction partners, InpherNet is able to rank the causative gene high both when few or no human phenotypes are known that match the patient phenotypes or in cases the knowledgebase is lacking a full annotation. InpherNet has shown to improve performance on previous tools that have been devised for the same purpose such as Phevor [11], Phive [12], and hiPhive [12]. We carefully ensure a fair comparison including testing InpherNet, Phrank_HPOA, Phranken_HPOA, and all Exomiser tools on the same clinical annotation set (HPO-A), and only querying Phevor (web API access only) several months later. InpherNet leverages a potent combination of resources available from Ensembl [20] and the Monarch Initiative [19]. InpherNet uses a Phrank [13] based metric to measure set similarity, filters protein interactions by anatomical colocalization, adds variant-related features, and applies a gradient boosting tree classifier. Of particular value are Monarch’s cross-species anatomy and phenotype ontologies, Uberon and Upheno, respectively, which allow one to compare model organism annotations, in a structured way, with those of human patients.
The boosting tree model, which is at the heart of InpherNet, allowing it to assign different weights to different evidence combinations, makes it robust. For example, we see no correlation across our real patients test sets between the InpherNet rank of the causative gene and the relative number of disease-associated paralogs each of the patient candidate gene has (Fig. S3). InpherNet currently uses only mouse and zebrafish annotations because of the richness of annotations these two species offer. For example, our graph includes 184,313 mouse-based gene–phenotype relationships and 42,367 zebrafish-based gene–phenotype relationships. The next best vertebrate species, rat, has only 1,231 relationships. However, we show that using these two species the number of phenotypically annotated human genes increases fourfold. As additional species’ functional data grows, they can be easily integrated into InpherNet’s flexible gradient boosting tree model. And while InpherNet’s overall performance is high, we see that it performs better over dominant cases than recessive cases (Fig. S4) suggesting that featurizing and learning candidate genes’ zygosity may further improve its performance.
Our testing methodology currently advocates a hybrid approach. Scan the first few computationally ranked candidates based on clinical patient data. Should no quick diagnosis be found that way, we show that it is beneficial to switch to prioritizing indirect gene evidence from a variety of sources. It is interesting to note that of the hundreds of real patient cases we start with, a large 34–45% of cases are better served by this strategy. It would be beneficial in future work to build a method unifying both strategies that may boost performance even further. Meanwhile, we have improved the way to leverage indirect evidence, from both human and model organisms, and accelerate the detection of a causative gene where direct information about it is lacking or missing from our knowledgebase or indeed from the body of scientific knowledge.
Data availability
Relevant data used in this project are available at https://doi.org/10.6084/m9.figshare.14058113.v1.
Code availability
The snapshot accompanying the paper is in https://doi.org/10.6084/m9.figshare.14058113.v1, and live code base is available at https://bitbucket.org/bejerano/inphernet/.
References
Church G. Compelling reasons for repairing human germlines. N Engl J Med. 2017;377:1909–11.
Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–11.
Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7.
Dragojlovic N, Elliott AM, Adam S, van Karnebeek C, Lehman A, Mwenifumbo JC, et al. The cost and diagnostic yield of exome sequencing for children with suspected genetic disorders: a benchmarking study. Genet Med. 2018;20:1013–21.
Kingsmore SF, Cakici JA, Clark MM, Gaughran M, Feddock M, Batalov S, et al. A randomized, controlled trial of the analytic and diagnostic performance of singleton and trio, rapid genome and exome sequencing in ill infants. Am J Hum Genet. 2019;105:719–33.
Stark Z, Dolman L, Manolio TA, Ozenberger B, Hill SL, Caulfied MJ, et al. Integrating genomics into healthcare: a global responsibility. Am J Hum Genet. 2019;104:13–20.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Jagadeesh KA, Wenger AM, Berger MJ, Guturu H, Stenson PD, Cooper DN, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet. 2016;48:1581–6.
Jagadeesh KA, Paggi JM, Ye JS, Stenson PD, Cooper DN, Bernstein JA, et al. S-CAP extends pathogenicity prediction to genetic variants that affect RNA splicing. Nat Genet. 2019;51:755–63.
Deisseroth CA, Birgmeier J, Bodle EE, Kohler JN, Matalon DR, Nazarenko Y, et al. ClinPhen extracts and prioritizes patient phenotypes directly from medical records to expedite genetic disease diagnosis. Genet Med Off J Am Coll Med Genet. 2019;21:1585–93.
Singleton MV, Guthery SL, Voelkerding KV, Chen K, Kennedy B, Margraf RL, et al. Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet. 2014;94:599–610.
Smedley D, Jacobsen JOB, Jäger M, Köhler S, Holtgrewe M, Schubach M, et al. Next-generation diagnostics and disease-gene discovery with the Exomiser. Nat Protoc. 2015;10:2004–15.
Jagadeesh KA, Birgmeier J, Guturu H, Deisseroth CA, Wenger AM, Bernstein JA, et al. Phrank measures phenotype sets similarity to greatly improve Mendelian diagnostic disease prioritization. Genet Med. 2019;21:464–70.
Birgmeier J, Haeussler M, Deisseroth CA, Steinberg EH, Jagadeesh KA, Ratner AJ, et al. AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Sci Transl Med. 2020;12. https://doi.org/10.1126/scitranslmed.aau9113
Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209–14.
Bamshad MJ, Nickerson DA, Chong JX. Mendelian gene discovery: fast and furious with no end in sight. Am J Hum Genet. 2019;105:448–55.
Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020. https://doi.org/10.1007/s00439-020-02199-3. Published online June 28.
Landrum MJ, Chitipiralla S, Brown GR, Chen C, Gu B, Hart J, et al. ClinVar: improvements to accessing data. Nucleic Acids Res. 2020;48:D835–44.
Mungall CJ, McMurry JA, Köhler S, Balhoff JP, Borromeo C, Brush M, et al. The Monarch Initiative: an integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Res. 2017;45:D712–22.
Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–61.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29.
Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, et al. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res. 2014;42:D966–74.
Eppig JT, Smith CL, Blake JA, Ringwald M, Kadin JA, Richardson JE, et al. Mouse genome informatics (MGI): resources for mining mouse genetic, genomic, and biological data in support of primary and translational research. Methods Mol Biol Clifton NJ. 2017;1488:47–73.
Howe DG, Bradford YM, Conlin T, Eagle AE, Fashena D, Frazer K, et al. ZFIN, the zebrafish model organism database: increased support for mutants and transgenics. Nucleic Acids Res. 2013;41:D854–60.
Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018;46:D649–55.
Chatr-Aryamontri A, Oughtred R, Boucher L, Rust J, Chang C, Kolas NK, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–79.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43(D1):D789–98.
Rath A, Olry A, Dhombres F, Brandt MM, Urbero B, Ayme S. Representation of rare diseases in health information systems: the Orphanet approach to serve a wide range of end users. Hum Mutat. 2012;33:803–8.
Hastie T, Tibshirani R, Friedman J (eds). The Elements of Statistical Learning - Data Mining, Inference, and Prediction, 2nd Edn. Springer. Accessed June 12, 2019. https://www.springer.com/us/book/9780387848570.
Koonin EV. Orthologs, paralogs, and evolutionary genomics. Annu Rev Genet. 2005;39:309–38.
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic Intolerance to Functional Variation and the Interpretation of Personal Genomes. PLOS Genet. 2013;9:e1003709.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, et al. Interactions between Sox9 and beta-catenin control chondrocyte differentiation. Genes Dev. 2004;18:1072–87.
Tan TY, Dillon OJ, Stark Z, Schofield D, Alam K, Shrestha R, et al. Diagnostic impact and cost-effectiveness of whole-exome sequencing for ambulant children with suspected monogenic conditions. JAMA Pediatr. 2017;171:855–62.
Acknowledgements
We thank members of the Bejerano laboratory, particularly Yosuke Tanigawa, Heidi Chen, Karthik Jagadeesh, Cole Deisseroth, Ethan Dyer, Ethan Steinberg, and Surag Nair for technical advice and valuable feedback; Monarch Initiative members including Christopher Mungall and Kent Shefchek for providing access to and guidance in using Monarch Initiative data; Maria Haanpaa, as well as Jennefer Kohler, Devon Bonner, and their colleagues for advice. Funding: Bio-X SIGF fellowship to J.B., DARPA (G.B.), the Stanford Pediatrics Department (J.A.B., G.B.), a Packard Foundation Fellowship (G.B.), a Microsoft Faculty Fellowship (G.B.), and the Stanford Data Science Initiative (G.B).
Author information
Authors and Affiliations
Contributions
Conceptualization: B.Y., J.B., J.A.B., G.B. Data curation: B.Y. Formal analysis: B.Y., J.A.B., G.B. Funding acquisition: G.B. Investigation: B.Y. Methodology: B.Y., G.B. Software: B.Y., J.B. Supervision: G.B. Visualization: B.Y. Writing – original draft: B.Y. Writing – review & editing: B.Y., J.B., J.A.B., G.B.
Corresponding author
Ethics declarations
Ethics Declaration
This study was approved by Stanford Institutional Review Board (IRB) protocol #28362. Informed consent was obtained by us for all Stanford participants per our Stanford IRB protocol. The DDD data we use has U.K. Research Ethics Committee (REC) approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12, granted by the Republic of Ireland REC).
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yoo, B., Birgmeier, J., Bernstein, J.A. et al. InpherNet accelerates monogenic disease diagnosis using patients’ candidate genes’ neighbors. Genet Med 23, 1984–1992 (2021). https://doi.org/10.1038/s41436-021-01238-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01238-2
