
Abstract
Purpose
To perform comprehensive genotyping of TSC1 and TSC2 in a cohort of 94 infants with tuberous sclerosis complex (TSC) and correlate with clinical manifestations.
Methods
Infants were enrolled at age <4 months, and subject to intensive clinical monitoring including electroencephalography (EEG), brain magnetic resonance imaging (MRI), and neuropsychological assessment. Targeted massively parallel sequencing (MPS), genome sequencing, and multiplex ligation-dependent probe amplification (MLPA) were used for variant detection in TSC1/TSC2.
Results
Pathogenic variants in TSC1 or TSC2 were identified in 93 of 94 (99%) subjects, with 23 in TSC1 and 70 in TSC2. Nine (10%) subjects had mosaicism. Eight of 24 clinical features assessed at age 2 years were significantly less frequent in those with TSC1 versus TSC2 variants including cortical tubers, hypomelanotic macules, facial angiofibroma, renal cysts, drug-resistant epilepsy, developmental delay, subependymal giant cell astrocytoma, and median seizure-free survival. Additionally, quantitative brain MRI analysis showed a marked difference in tuber and subependymal nodule/giant cell astrocytoma volume for TSC1 versus TSC2.
Conclusion
TSC2 pathogenic variants are associated with a more severe clinical phenotype than mosaic TSC2 or TSC1 variants in TSC infants. Early assessment of gene variant status and mosaicism might have benefit for clinical management in infants and young children with TSC.
Similar content being viewed by others

INTRODUCTION
Tuberous sclerosis complex (TSC) is an autosomal dominant disease affecting multiple organs that is due to pathogenic variants in either of the tumor suppressor genes TSC1 and TSC2.1,2 These genes encode hamartin and tuberin, respectively, proteins that form the TSC protein complex along with TBC1D7. The complex functions as a GTPase activating protein for the ras-family member RHEB, which regulates mammalian target of rapamycin complex 1 (mTORC1).3,4,5 Second hit loss of either TSC1 or TSC2 has been shown in multiple kinds of TSC lesions, in the same gene as the germline pathogenic variant, consistent with function of these genes as tumor suppressors, following the classic Knudson model.1,2
Clinically, the natural history of TSC is characterized by predominant neurological and cardiac manifestations (cardiac rhabdomyoma) in infancy and early childhood, followed by development of tumors in multiple organ systems including renal angiomyolipoma (AML), facial angiofibroma, ungual fibroma, subependymal giant cell astrocytoma (SEGA), and pulmonary lymphangioleiomyomatosis (LAM).1,2 Brain pathology commonly seen in TSC includes the hallmark cortical tubers, subependymal nodules (SEN), radial migration lines, and less commonly white matter cysts, calcification, and other white matter abnormalities. These structural anomalies, as well as the effects of haploinsufficiency due to reduced expression of either TSC1 or TSC2 in brain cells, are likely responsible for the significant and variable neurological manifestations, including epilepsy, infantile spasms, autistic spectrum disorder, neurodevelopmental delay, and intellectual disability.
It is well known that TSC displays a variable natural history, with some patients being mildly affected, and others showing a severe phenotype with early onset, refractory seizures, and developmental disorders.1,2 Past and recent studies have shown that mosaicism is relatively common in TSC, seen in 10–15% of patients, and is associated with a milder phenotype in multiple respects.6,7,8,9,10,11 TSC1 pathogenic variants have also been associated with a milder phenotype than TSC2 pathogenic variants in multiple past reports.12,13,14,15,16,17
EPISTOP was a long-term, prospective study assessing clinical and molecular biomarkers of epileptogenesis in TSC, which included a randomized trial of early intervention with vigabatrin in those infants in whom a high risk of seizure development was expected (NCT02098759). Children diagnosed with TSC were enrolled prior to the age of 4 months and were followed until the age of 2 years. Here we report on the assessment of pathogenic variants in TSC1 and TSC2 in the EPISTOP cohort, and correlation of variant findings with multiple clinical manifestations through age 2 years.
MATERIALS AND METHODS
Patient recruitment and sample collection
The study was approved by all local Ethics Committees at the clinical sites for EPISTOP: Komisja Bioetyczna przy Instytucie “Pomnik-Centrum Zdrowia Dziecka” at Children’s Memorial Health Institute in Warsaw, Comitato Etico Independente Università degli Studi di Roma “Tor Vergata” at Tor Vergata University of Rome, Etické komise pro multicentrické klinické hodnocení Fakultní nemocnice v Motole at University Hospital Motol in Praque, Children’s Health Queensland Hospital and Health Service Human Research Ethics Committee at the Queensland Children’s Hospital in South Brisbane, “Ethikkommission der Charité” Charite–Universitaetsmedizin Berlin at Charité University Medicine Berlin, Medisch Ethisze Toetsingscommissie (METC) UMC Utrecht at University Medical Center Utrecht, Commissie Medische Ethiek UZ Brussel at Vrije Universiteit Brussel, Ethikkommission der Medizinischen Universität Wien at the Medical University of Vienna, and Comité de Protection des Personnes Ile de France VIII; CPP IDF 8 at Paris Descartes University. Caregivers of all participants signed informed consent before enrollment. EPISTOP subjects were enrolled if they met the following criteria: age ≤4 months, definite TSC diagnosis according to consensus criteria,18 no prior seizures, and no clinical seizures on baseline video electroencephalogram (EEG). Exclusion criteria included any condition considered by the investigator to hinder participation in the study or affect primary outcome. At the ages of 6, 12, 18, and 24 months subjects had neurodevelopmental testing with the Bayley Scales of Infant Development (BSID-III) and the Autistic Diagnostic Observation Schedule 2nd edition (ADOS-2). Subjects in EPISTOP were part of either a randomized controlled trial of early intervention for epilepsy with vigabatrin, or a similar nonrandomized open label trial of vigabatrin, at the time of detection of epileptiform activity on serial EEG versus conventional treatment with vigabatrin started after seizures. The sample size was designed to have 80% power to detect a 50% improvement in the primary study endpoint of time from birth to the first clinical seizure. Intensive clinical follow-up was used for EPISTOP subjects including visits every 4–8 weeks for EEG, seizure, and developmental assessment, and comprehensive scans at age 2 years. Drug-resistant epilepsy was defined as failure of two trials of antiepileptic drugs, either as monotherapies or in combination, to achieve sustained seizure freedom. Autistic features were assessed according to ADOS-2 at age 2 years, and neurodevelopmental delay was assessed as a BSID cognitive score <70 at age 2 years.
Blood samples from EPISTOP infants were subjected to DNA extraction using Qiagen Clotspin Baskets and the Gentra Puregene Blood Kit. One subject also provided a buccal swab for DNA extraction using Qiagen QIAamp DNA Mini Kit.
Targeted massive parallel sequencing and analysis
All genetic analyses were performed blinded to all clinical features of the subjects.
Targeted massive parallel sequencing (MPS) was performed using a custom bait set (Agilent platform) that covers the entire TSC1 and TSC2 genes including 10 kb upstream and downstream and all coding exons and introns, as described previously.7,8 The bait set also covered all coding exons and adjacent introns of 48 other genes involved in mTOR pathway signaling and/or TSC. DNA was subjected to fragmentation using Covaris sonication to an average size of 250 nucleotides. The fragmented DNA was purified using Agencourt AMPure XP beads and ligated to dual indexed adapters for Illumina sequencing. A MiSeq run was performed to quantify each library. Libraries were then pooled in equal mass and captured using the custom bait set using Agilent SureSelect hybrid capture kit. The captured libraries were then sequenced on either the HiSeq 2500 or the HiSeq 3000 instrument.
The sequencing output was deconvoluted into individual sample reads and sorted using Picard tools. Reads were aligned to the hg19 reference sequence using bwa, and duplicate reads were identified and marked using Picard tools. Alignments were further refined using the GATK tool for localized realignment around indel sites and recalibration of the quality scores was also performed using GATK tools. Pathogenic variant analysis for single-nucleotide variants (SNVs) was performed using MuTect v1.1.4 and annotated by Variant Effect Predictor (VEP). Insertions and deletions were called using IndelLocator and SomaticIndelDetector. MuTect was run in paired mode using a CEPH cell line sample as a control, and a germline variant filter was then applied. Both SNVs and small indels were compared with the ExAC and gnomAD databases.
The mean target coverage for the TSC1/TSC2 regions ranged from 327× to 1614×, with a median of 716×.
A second custom approach was used in parallel to analyze the sequence data in greater detail for TSC1 (NM_000368.4) and TSC2 (NM_000548.4).7,8 Capture of read calls at all positions was done using SAMtools Pileup, followed by custom processing in Python and Matlab to determine base call frequency at each position in each read orientation. All variant read calls seen in both read orientations were retained for further review, and an output file of potential significant variants was generated with the following information: chromosome, nucleotide (nt) position, reference nt, variant nt, maximum allele frequency of the variant among all samples analyzed, number of samples with variants at this nt position, population allele frequency of the variant from ExAC and gnomAD, exon, position, complementary DNA (cDNA) number, amino acid number, reference amino acid, variant amino acid, a flag for occurrence near a polyA run, and the variant allele frequency for each sample under analysis. A similar file was generated for indel variants.
This variant read call list was then manually reviewed. Single-nucleotide polymorphisms (SNPs) seen at any allele frequency in ExAC or gnomAD were segregated and examined for distortion in allele frequency from the expected 50% as a potential marker of mosaic large deletion. Variants seen in many samples or occurring near polyA sequences were eliminated as artifacts. All SNVs or indels remaining and observed at an allele frequency >0.5% were directly reviewed using Integrative Genomics Viewer (IGV), to help identify bona fide variants and exclude sequencing or alignment artifacts. Potential pathogenic variants were also examined in the TSC1 and TSC2 LOVD database (https://databases.lovd.nl/shared/genes/). Probable pathogenic SNVs and indels were confirmed by Sanger sequencing if present at allele frequency >10%; and by amplicon MPS if present at allele frequency <10%.
Detection of large genomic deletion
Examination for large genomic deletions in TSC1 and TSC2 was performed by four methods. (1) Multiplex ligation-dependent probe amplification (MLPA) was performed using SALSA MLPA probe mixes TSC1 P124-C1 and TSC2 P337-B1 with reagents from MRC-Holland (Amsterdam, The Netherlands), according to the manufacturer's recommendations. The products of the MLPA reactions were separated by size with capillary electrophoresis (ABI3730xl Genetic Analyzer apparatus; Applied Biosystems, Carlsbad, CA, USA), and electropherograms analyzed using GeneMarker software (version 2.2.0; SoftGenetics, State College, PA, USA). The normalized signal of each probe (peak height divided by the average peak height of the control probes) was divided by the corresponding signal in a reference sample and multiplied by 2. The obtained values that correspond to the copy number of particular exons/regions were visualized in bar graphs. (2) Read depth was determined at every nucleotide position in the genomic regions of TSC1/TSC2 and compared computationally among different samples in 100 nucleotide windows to identify potential genomic deletion regions. (3) Using paired-end read information from the bam files, reads with unexpectedly large inserts (> 1000 nt) were identified and tabulated as markers of potential genomic deletion events. Output files were manually examined to identify consistent patterns suggestive of large genomic deletions. (4) Genome sequencing was performed using the NEBNext® Ultra DNA Library Prep kit for Illumina (catalog number NEB #E7370S/L, New England Biolabs, Ipswich, MA) using 100 ng input DNA. The samples were paired-end sequenced on an Illumina HiSeq 4000, with 150 cycles per read, according to manufacturer protocol. After sequencing, the resulting reads were trimmed for adapter sequences using Trimmomatic v0.30.19 To remove noise introduced by sequencing errors, Sickle v1.3.3 was utilized with default settings.20 The remaining reads were mapped to the human reference hg19. The mapping was performed with the BWA-MEM v0.7.4b aligner based on Burrows–Wheeler Transform.21 Default settings and mismatch rates were used. Duplicate reads were marked and removed with MarkDuplicates of the Picard v2.13.2 package (Picard Toolkit, 2019). After duplicate removal, base recalibration and variant calling was performed based on the best practices of GATK using GATK HaplotypeCaller v3.7.22 Single-nucleotide variants and small indels in the sample data were detected when compared with the reference and reported in (g)VCF format.
Findings among these four methods of analysis were typically concordant and provided additional detail and confirmation of findings. However, for cases of low allele fraction mosaicism, the third method was most sensitive. All findings indicative of probable large genomic deletions were confirmed by polymerase chain reaction (PCR) amplification across putative breakpoints, followed by gel electrophoresis and Sanger sequencing.
Pathogenic variant data was deposited in the TSC1 and TSC2 LOVD variant database (https://databases.lovd.nl/shared/genes/).
Quantitative analysis of brain magnetic resonance imaging
Magnetic resonance images (MRIs) were performed at inclusion and at the age of 24 months, on 1.5 or 3 T MRI scanners (Philips, Siemens, and GE), with minimum of 8-channel head coil. MRI acquisition was harmonized across centers, with a predefined MRI protocol. MRI sequences included T1 SE 3 mm, T2 TSE_3D Vista, axial T2 fluid-attenuated inversion recovery (FLAIR) 4 mm, T1W_3D_ISO. For lesion detection and segmentation all MRIs were visually evaluated for presence of tubers, radial migration lines (RML), white matter abnormalities (WMA) other than RML, cysts, calcifications, SEN, and SEGA (defined as subependymal lesions exceeding 1 cm or showing growth on serial MRI). Subsequently, lesions were manually segmented on the best possible T2W sequence in infants aged 24 months, using in-house developed software based on MeVisLab (MeVis Medical Solutions AG, Bremen, Germany).23 All manual segmentations were performed by the same researcher (H.M.H.), trained and supervised by a pediatric neuroradiologist and a pediatric neurologist (F.E.J.). Total brain volumes were calculated using FMRIB Software Library (FSL). Lesion volumes were expressed as the proportion of the brain occupied by the respective lesion (tuber, RML, WMA, etc.).
Statistical analysis
Statistical analysis was performed using GraphPad PRISM 8 (GraphPad, San Diego, CA, USA) software to assess the Mann–Whitney and log-rank (Mantel–Cox) tests. Significance was assessed for contingency tables (Tables 1 and S2) using Fisher’s exact test (http://vassarstats.net/tab2x2.html). Differences were considered statistically significant when p < 0.05, and all tests were two-sided.
RESULTS
Clinical cohort
One hundred and one (101) subjects were enrolled into EPISTOP. Three were excluded soon after enrollment due to TSC1 or TSC2 variant reclassification and clinical reassessment. Four were excluded when clinical evaluation at age 2 years indicated they did not meet TSC diagnostic criteria. The remaining 94 EPISTOP subjects, with a definite TSC diagnosis following consensus criteria, are reported here (Table S1).
Detection of TSC1 and TSC2 pathogenic variants
Probable pathogenic variants in TSC1 or TSC2 were identified in 93 of 94 (99%) subjects (Table S1) thanks to the high sensitivity of the sequencing methods. All variant findings were confirmed by secondary analyses as described (“Materials and Methods”). The pathogenic variant spectrum in TSC1/TSC2 was 8 (9%) insertions, 17 (18%) deletions, 18 (19%) splice, 20 (22%) missense, 21 (23%) nonsense, 7 (8%) large deletions, and 2 (2%) in-frame deletions (Fig. S1).
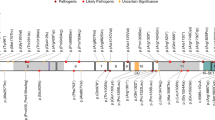
Twenty-three of 93 (25%) variants were identified in TSC1, of which 22 were small truncating (nonsense, splice, or out of frame indels) variants and one was a large deletion of the entire TSC1 gene comprising 284,752 nt (Figs. 1a, S1B, S2A). Of 93 pathogenic variants, 70 (75%) were identified in TSC2, which consisted of 22 (31%) missense, 40 (57%) truncating (nonsense, splice, or out of frame indels), 2 (3%) in-frame deletions, and 6 (9%) large deletions, ranging in size from 1003 to 175,789 nt (Figs. 1b, S1C, S2B–G). Three of the six large deletions in TSC2 extended into PKD1, but only one of these affected the coding region of PKD1 (Figs. 1b, S2B–G).

(a, b) Position of variants of various kinds are indicated for each gene with frequency indicated by height of the lollipop. Multiple variants in the same position are shown in one color arbitrarily selected by Mutational Mapper in cBioPortal. Large genomic deletion variants are shown at the bottom, with red indicating the region of the gene that is deleted, and gray meaning that the deletion extends beyond the gene in that direction. (c) Pie chart showing relative numbers of TSC1, TSC2, and no variant identified (NMI) subjects.
Of 94 subjects, 9 (10%) had mosaic variants, at allele frequencies ranging from 0.7% to 32% (0.7%, 2.0%, 3.0%, 6.0%, 7.0%, 11%, 17%, 18%, 32%, Table S1). All of these mosaic variants were seen in TSC2. Three of the six large genomic deletion variants in TSC2 were mosaic. Notably, one of these mosaic variants (TSC2 c.3284+1G>A) was identified in a buccal swab sample from an EPISTOP subject whose blood DNA sample had shown no evidence of this variant. This is similar to previous observations we have made that there can be variable allele frequency of mosaic variants in different tissue samples (skin, saliva, blood) from TSC subjects.6,7,8 No pathogenic variant was identified in one EPISTOP subject.
Genotype–phenotype correlation
We examined the correlation between clinical manifestations in this cohort, and occurrence of pathogenic variants in TSC1 versusTSC2, including all 20 clinical features that were scored in this population both at study enrollment and at the age of 2 years, the end of the study. Because mosaic pathogenic variants are associated with milder phenotype severity,6,7,8 we excluded those samples with allele frequency below 30% from the comparison between TSC1 and TSC2 variants. There was a significantly larger number of clinical features present in subjects with TSC2 variants versus those with TSC1 variants, both at study entry and at the age of 2 years (Fig. 2a), although this difference was modest. We also compared the number of features present in subjects with mosaic variants (excluding the subject with mosaic variant frequency of 32%) and those with nonmosaic variants. There was no significant difference seen at either study entry or at the age of 2 years, which may partially reflect the limited number of mosaic subjects (n = 8) for this comparison (Fig. 2b).

(a, b) The total number of clinical features present per subject is shown both at study entry, and at the age of 2 years according to TSC1 vs. TSC2 variant status (a), and according to mosaic vs. nonmosaic (Het) pathogenic variant status (b). Significant differences in number of pathogenic variants are seen for the TSC1 vs. TSC2 comparison (a), **p < 0.01, ***p < 0.001, Wilcoxon rank sum test. Bar is drawn at the median value.
Only 2 of 20 clinical features assessed at study entry were seen significantly less frequently in subjects with pathogenic variants in TSC1 versus TSC2. Cortical tubers were present on initial brain MRI in 12 of 23 (53%) in TSC1 versus 52 of 62 (84%) in TSC2 (p = 0.0045); multiple renal cysts (but still much milder than classic polycystic kidney disease) were seen in 0 of 19 (0%) in TSC1 versus 20 of 59 (34%) in TSC2 (p = 0.0049). None of the 20 clinical features were significantly different in frequency in those with mosaic (n = 8) versus heterozygous (n = 85) variants at study entry (Table S2).
In the comparison made at the age of 2 years, four additional clinical manifestations were included, which had been assessed following enrollment. These were seizure occurrence at any time, development of drug-resistant epilepsy, developmental delay, and risk of autism spectrum disorder; all of which were assessed at the age of 2 years.
In contrast to the findings at study enrollment, 8 of 24 clinical features assessed at the age of 2 years were seen more frequently in TSC2 versus TSC1 subjects. These eight features were cortical tubers, facial angiofibroma (>3) or fibrous cephalic plaque, hypomelanotic macules (>3, 5 mm diameter), two or more renal cysts, SEGA, seizure occurrence, development of drug-resistant epilepsy, and developmental delay (Table 1). All but one of these showed a >20% difference between the two groups, including facial angiofibroma and renal cysts, which were not seen at all in the TSC1 variant subjects, compared with 21% and 29% in the TSC2 variant subjects, respectively. Seven clinical features were not recorded in any EPISTOP subject at either enrollment or 24 months, including bone cysts, gingival fibroma (>1), hamartomatous rectal polyps, lymphangioleiomyomatosis, dental enamel pits (>3), nonrenal hamartomas, and ungual fibromas (>2).
Quantitative brain MRI assessment
Quantitative assessment of brain MRI scans at the age of 2 years was performed for 68 EPISTOP subjects enabling more detailed comparison of differences according to genotype (Table 1). Both tubers and WMA, other than radial RML, were seen at a significantly lower frequency in TSC1 variant subjects compared with those with TSC2 variants (14/18 [78%] vs. 44/45 [98%], p = 0.0208; 1/18 [6%] vs. 23/45 [51%], p = 0.0011; respectively, Table 1). Analysis of tuber volume, normalized for brain volume, showed a dramatic difference with a median value of 0.00050 for TSC1 variant subjects compared with 0.01725 in those with TSC2 variants (Fig. 3c). Similar dramatic differences were seen in the relative volume of subependymal nodules + subependymal giant cell astrocytoma, and in the total brain lesion fractional volume (Fig. 3d, e). There was a range of ages for the timing of the 2-year-old brain MRI, but this did not differ significantly according to gene variant (Fig. 3a). Total brain volume was smaller in the TSC2 variant subjects compared with those with TSC1 variants (Fig. 3b), which we suspect may have been due to differences in seizure rates and occurrence of drug-resistant epilepsy. However, we cannot exclude an important influence from polygenic factors since head circumference of parents was not available. Quantitative brain MRI data were available for only five subjects mosaic for TSC2 variants, preventing formal statistical analysis, and these subjects had variable levels for all of these measures (Fig. 3).

All data shown is from an MRI brain scan done at age 2 years. (a) Ages in days of subjects at the time of the scan. (b) Total brain volume in cm3. (c) Tuber volume divided by brain volume. (d) SEN + SEGA volume divided by brain volume. (e) Total lesion volume divided by brain volume. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by Wilcoxon rank sum test. Bar is drawn at the median value. SEGA subependymal giant cell astrocytoma, SEN subependymal nodule.
Seizure onset and seizure-free survival
Seizure-free survival at 2 years of age was significantly better in the TSC1 variant population (12/23, 52%) than in those with TSC2 variants (8/60, 13%) (p = 0.0011, log-rank test), with median seizure-free survival greater than 2 years and 162 days, for those with TSC1 and TSC2 variants, respectively (Fig. 4a). In addition, improved seizure-free survival was observed in subjects with mosaic TSC2 variants compared with all subjects with heterozygous variants (p = 0.0422, Fig. 4b), with median seizure-free survival greater than 2 years versus 227 days, respectively.

(a) Seizure-free survival by pathogenic variant in TSC1 vs. TSC2. (b) Seizure-free survival by mosaic vs. heterozygous pathogenic variant in either gene. Significance was assessed by log-rank (Mantel–Cox) test.
In addition, 9 of 20 (45%) of subjects with TSC1 variants and full developmental evaluation had no seizures and normal development; this occurred for only 5 of 51 (10%) subjects with a TSC2 variant (p = 0.0018) (Table 1).
DISCUSSION
Many past studies have noted a milder phenotype in TSC individuals with TSC1 versus TSC2 pathogenic variants, based predominantly on analyses of clinic-based or genetic testing based mixed-age populations.12,13,14,15,16,17
This is one of the first age-based cohort studies in which comprehensive clinical characterization and quantitative brain MRI analysis has been performed, along with sensitive variant detection enabling a detailed analysis of correlations between genotype and clinical manifestations in TSC. We are aware of only one other recent report of genetic analysis in a cohort of TSC infants.24 There were several important observations in the present genetic analysis of the EPISTOP cohort.
First, comprehensive pathogenic variant analysis by MPS enables pathogenic variant detection in nearly all newborns with a clinically definite diagnosis of TSC, 93 of 94 (99%) in this cohort. This was achieved through use of MPS focused on the genomic extent of TSC1 and TSC2, providing high read depth, as the primary approach to variant detection, and our custom analytic pipeline for low allele frequency variant detection. The high read depth achieved with MPS is critical for this sensitivity.
Second, mosaicism is surprisingly common in this population of TSC infants, seen in 9 of 94 (10%) subjects. Although TSC1 variants and mosaicism are both enriched in TSC populations who are diagnosed at older ages,25 these observations indicate that mosaicism for a TSC2 pathogenic variant commonly occurs in TSC children, at a rate of 9 of 70 (13%) of all pathogenic TSC2 variants seen. Interestingly, the mosaic allele frequency seen here, 0.7–32%, with median 7.0%, was significantly higher than what we have seen in a population of mildly affected TSC adults seeking genetic guidance for family planning, i.e., range 0–8.4%, median 2.0%.7 This may reflect the tendency for higher levels of mosaicism to be associated with more prominent clinical features, enabling diagnosis in infancy, in contrast to lower levels of mosaicism. In adults, there is more opportunity for additional clinical manifestations to be found, including multiple skin features, renal angiomyolipoma, and pulmonary lymphangioleiomyomatosis that show age related expression and are rare in young TSC children.2
Third, there was a very strong correlation between phenotype and gene with pathogenic variant in this series. TSC1 variant bearing subjects had a milder phenotype for all clinical manifestations. Arguably most striking among these differences between subjects with TSC1 versus TSC2 variants at the age of 2 years were a much lower frequency of developmental delay, 3/20 (15%) vs. 22/51 (43%); absence of facial angiofibroma, 0/23 (0%) vs. 12/58 (21%); absence of renal cysts, 0/21 (0%) vs. 16/56 (29%); a lower median value for fraction of tuber to brain volume of 0.00050 vs. 0.01680; a lower median value for fraction of SEN + SEGA to brain volume of 0 vs. 0.00020; and a longer median seizure-free survival greater than 2 years vs. 162 days, p = 0.0054.
Mosaicism was also associated with a milder phenotype, including seizure-free survival greater than 2 years in mosaic cases compared with 227 days for all heterozygous cases (Fig. 4b, p = 0.0421), and a trend toward a smaller number of clinical features compared with heterozygous cases (Fig. 2b). It is also notable that all mosaic cases had TSC2 variants. It is likely that the combination of a milder phenotype associated with TSC1, and further phenotype attenuation associated with mosaicism, make it unlikely that a TSC1 mosaic infant would meet TSC diagnostic criteria.
We note that the frequency of cardiac rhabdomyoma was very high in this cohort (91/94, 97%), reflective of its key contribution to diagnosis in TSC infants. This suggests that this population may have been enriched for more severe TSC overall and makes the finding of mosaicism in 10% even more remarkable. Subependymal nodules (86/94, 91%) and cortical tubers (70/94, 74%) were also common in these subjects. The most common combination of clinical features establishing the diagnosis was cardiac rhabdomyoma, subependymal nodules, and cortical tubers seen in 14 of 94 (15%) subjects at study entry.
In summary, these observations provide further quantitative support for the important difference in phenotype associated with TSC1 versus TSC2 pathogenic variants and indicate that this difference is seen in TSC infants. They suggest that infants with TSC with a mild phenotype should be expected to be enriched for both TSC1 and mosaic TSC2 variants. More importantly, they suggest that early assessment of gene variant and presence or absence of mosaicism may have benefit for prognostication and multiple aspects of management and guidance of TSC infants and young children, including risk of seizure development and need for intensive electroencephalogram (EEG) monitoring. In particular, they suggest that infants diagnosed with TSC and found to have a TSC2 pathogenic variant should be considered for early intervention with disease modifiers/therapies in future clinical trials.
Code availability
Code for performing this custom analysis for variants is available on request.
References
Salussolia CL, Klonowska K, Kwiatkowski DJ, Sahin M. Genetic etiologies, diagnosis, and treatment of tuberous sclerosis complex. Annu Rev Genomics Hum Genet. 2019;20:217–240.
Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers. 2016;2:16035.
Kwiatkowski DJ, Manning BD. Molecular basis of giant cells in tuberous sclerosis complex. N Engl J Med. 2014;371:778–780.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat Rev Cancer. 2020;20:74–88.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976.
Treichel AM, Hamieh L, Nathan NR, et al. Phenotypic distinctions between mosaic forms of tuberous sclerosis complex. Genet Med. 2019;21:2594–2604.
Giannikou K, Lasseter KD, Grevelink JM, et al. Low-level mosaicism in tuberous sclerosis complex: prevalence, clinical features, and risk of disease transmission. Genet Med. 2019;21:2639–2643.
Tyburczy ME, Dies KA, Glass J, et al. Mosaic and intronic mutations in TSC1/TSC2 explain the majority of TSC patients with no mutation identified by conventional testing. PLoS Genet. 2015;11:e1005637.
Verhoef S, Bakker L, Tempelaars AM, et al. High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet. 1999;64:1632–1637.
Rose VM, Au KS, Pollom G, Roach ES, Prashner HR, Northrup H. Germ-line mosaicism in tuberous sclerosis: how common? Am J Hum Genet. 1999;64:986–992.
Kwiatkowska J, Wigowska-Sowinska J, Napierala D, Slomski R, Kwiatkowski DJ. Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N Engl J Med. 1999;340:703–707.
van Eeghen AM, Black ME, Pulsifer MB, Kwiatkowski DJ, Thiele EA. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet. 2012;20:510–515.
Jansen FE, Braams O, Vincken KL, et al. Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology. 2008;70:908–915.
Au KS, Williams AT, Roach ES, et al. Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med. 2007;9:88–100.
Sancak O, Nellist M, Goedbloed M, et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype—phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet. 2005;13:731–741.
Dabora SL, Jozwiak S, Franz DN, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68:64–80.
Jones AC, Shyamsundar MM, Thomas MW, et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999;64:1305–1315.
Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol. 2013;49:243–254.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120.
Joshi N, Fass J. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. 2011.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303.
Kuijf HJ. Image processing techniques for quantification and assessment of brain MRI (Doctoral dissertation, 10 December 2013, Utrecht University, Utrecht, the Netherlands). https://dspace.library.uu.nl/handle/1874/287431.
Farach LS, Pearson DA, Woodhouse JP, et al. Tuberous sclerosis complex genotypes and developmental phenotype. Pediatr Neurol. 2019;96:58–63.
Treichel AM, Kwiatkowski DJ, Moss J, Darling TN. A diagnostic algorithm for enhanced detection of mosaic tuberous sclerosis complex in adults. Br J Dermatol. 2020;182:235–237.
Acknowledgements
We thank all the patients and their families who participated in this study, all medical staff members who contributed to collection of clinical information, Karthik V. Karnik for his help with the customized computational code for variant detection and reporting, and The Center for Cancer Genomics at Dana-Farber Cancer Institute for help with targeted capture sequencing. This work was supported by the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement number 602391, Polish Ministerial funds for science (years 2014–2019) for the implementation of international co-financed projects, and the Engles Family LAM/TS Research Fund.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ogórek, B., Hamieh, L., Hulshof, H.M. et al. TSC2 pathogenic variants are predictive of severe clinical manifestations in TSC infants: results of the EPISTOP study. Genet Med 22, 1489–1497 (2020). https://doi.org/10.1038/s41436-020-0823-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0823-4
Key words
This article is cited by
-
Is tuberous sclerosis complex-associated autism a preventable and treatable disorder?
World Journal of Pediatrics (2024)
-
Clinical practice recommendations for kidney involvement in tuberous sclerosis complex: a consensus statement by the ERKNet Working Group for Autosomal Dominant Structural Kidney Disorders and the ERA Genes & Kidney Working Group
Nature Reviews Nephrology (2024)
-
Molecular EPISTOP, a comprehensive multi-omic analysis of blood from Tuberous Sclerosis Complex infants age birth to two years
Nature Communications (2023)
-
Subependymal Giant Cell Astrocytomas (SEGAs): a Model of Targeting Tumor Growth and Epilepsy
Current Treatment Options in Neurology (2021)
-
Precision Therapy for Epilepsy Related to Brain Malformations
Neurotherapeutics (2021)

{kind=link}
{kind=link}
{kind=link}
{kind=link}