
Abstract
Purpose
With the description of circulating fetal DNA in maternal blood, noninvasive prenatal diagnostics became theoretically possible. As the presence of background maternal DNA interferes with the detection of fetal DNA, analytical methods require genetic markers capable of distinguishing by quantitative or targeted approaches the minor population of DNA molecules of the fetus. Here we evaluate the feasibility of analyzing fetal DNA with novel DIP-STR genetic markers, designed for the investigation of forensic mixed biological evidence.
Methods
The DIP-STR molecular approach is based on sequence-specific analysis of paternally inherited fetal alleles. These sequences are biallelic deletion/insertion polymorphisms (DIPs) located very close to short tandem repeat (STR) markers, for combined analysis. In this study, 48 women were tested with 28 DIP-STRs during the first, second, and third trimester of pregnancy.
Results
Positive results were obtained across markers, including longer ones (386 base-pairs) and with blood samples collected during early pregnancy, such as 10 weeks of gestational age.
Conclusion
These data show that DIP-STR markers can be used to amplify specific genomic regions of circulating fetal DNA to obtain targeted genetic information. This method may contribute to developments in noninvasive prenatal paternity testing and diagnosis of certain genetic diseases.
Similar content being viewed by others


Introduction
In obstetric care, prenatal screening and diagnosis allows women to make informed choices about the continuation of a pregnancy affected by genetic conditions such as sex-linked disorders,1 fetal RhD status,2,3 β-thalassemia,4,5,6 congenital adrenal hyperplasia,7 chromosomal aneuploidy,8,9 preeclampsia,10,11 and others.12 In forensic science, prenatal paternity testing aims at helping those pregnant women who have been victims of sexual assault and are unclear about the paternity of their unborn child.
Unfortunately, many definitive prenatal diagnoses of diseases including reliable prenatal paternity tests require the analysis of fetal genetic material, obtained through procedures performed at advanced gestational age (about 15 weeks) and that are associated with a certain degree of harm to the mother and the fetus. For instance, amniocentesis carries a miscarriage risk of 1 in 300 to 1 in 500 (ref.13) and a small risk of other complications,14,15 and direct chorionic villus sampling carries a similar miscarriage risk in addition to a 1 in 3000 risk of fetal limb reduction defects, especially when performed before the 10th week of gestation.16,17
New possibilities of developing prenatal diagnostic methods that are noninvasive emerged with the discovery of the presence of fetal DNA in maternal blood by Lo and collaborators.18 The continuous remodeling of the placenta, with trophoblast cells undergoing apoptotic events, is responsible for the releasing of cell-free fetal DNA (cffDNA) in the bloodstream of the mother. Because of its apoptotic origin, cffDNA is mostly short (about 200 base-pairs), it appears at around 6 weeks of pregnancy, and it constitutes approximately 10% of the total cell-free plasma DNA,19,20,21 thus generating an in vivo DNA mixture characterized by a large excess of maternal DNA (DNA microchimerism).22,23 Conventional DNA sequencing, single-nucleotide polymorphisms (SNPs), and short tandem repeat (STR) markers based on polymerase chain reaction (PCR) and capillary electrophoresis are not sensitive enough for characterizing a DNA contributing less than 10% to a mixed sample, and sex-specific Y-STR markers cannot be used in case of baby girls and to distinguish paternally related alleged fathers. Unless progress is made in methods for the enrichment of the fetal DNA fraction,24 one possibility for the analysis of the DNA microchimerism in pregnancy is the use of high-precision approaches. Digital PCR,25 large SNP array,26 and massive parallel sequencing (MPS),27 for example, enable to detect the small amount of sequences from a particular locus, even when applied to a sample in which fetal DNA only represents a small fraction.
Few studies evaluated the feasibility of noninvasive prenatal paternity testing by high-throughput SNP genotyping.28,29 Ryan et al.28 used an array of about 300,000 SNPs, optimized to detect cytogenetic abnormalities most relevant to human disease, and used this to establish the genotype of the plasma DNA. The genetic information of the mother (reference sample) is used to identify a series of SNP alleles transmitted by the father and with a “paternal support algorithm’” it is possible to estimate the probability of paternity of the alleged father. In addition to genotyping, this technique can be used to provide the relative quantity of alleles at each SNP to detect the presence or absence of fetal aneuploidies. Limitations of this approach sometimes include a low signal-to-noise ratio because the fetal DNA is neither enriched nor specifically targeted. Large-scale clinical trials are also needed to more accurately establish the specificity and sensitivity of the test.
Recently, MPS has also been used to analyze cffDNA in maternal plasma.30 As above, the use of the maternal DNA as reference sample enables the identification of the fetal DNA and the maternal versus paternal DNA inheritance. Applications focused so far on the detection of aneuploidies, because the quantitative detection of the chromosomal dosage over an extended region is more feasible than single pathogenic variant analysis. For paternity testing,31,32,33,34 the method currently lacks informatics-based analysis necessary to evaluate the accuracy with which paternity is confirmed. Signal-to-noise ratio is also an issue with many informative paternally transmitted alleles filtered out after quality controls.
Both of these procedures, SNP array and MPS, are labor intensive and require dedicated equipment. Another limitation associated with the genome-wide screening of fetal DNA is that the data gathered provide information beyond the specific genetic question investigated and may generate unclear and/or undesired diagnostics.
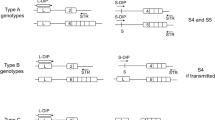
To circumvent these problems, we developed a PCR-based method that uses allele-specific primers to target DNA sequences that are unique to the fetal DNA (transmitted by the father).35,36 These sequences are biallelic deletion/insertion polymorphisms (DIPs) of several nucleotides, mostly between 3 and 15. The two possible alleles are referred to as the long allele (L) and short allele (S). Because biallelic markers have reduced information content, we proposed the selection of DIP markers that are physically very close (linked) to another STR marker, for combined analysis. The newly generated compound markers are termed DIP-STRs (Fig. 1). The multiallelic haplotype composed of both DIP and STR alleles is analyzed by using PCR primers overlapping the deleted/inserted sequence (S-DIP, L-DIP primers) on one side and downstream the STR region on the other side (STR primer). The sequence difference between S- and L-specific primers of several nucleotides allows the selective (allele-specific) amplification of the fetal DNA contributor in the presence of large quantities (more than 1000-fold excess) of maternal DNA background. In this way, it is possible to target those paternally transmitted alleles that are not shared with the maternal DNA.

Markers of type A, the mother and the father are homozygous for different DIP alleles. With this marker, the paternally transmitted DIP-STR haplotype can be targeted in maternal plasma using allele-specific primers. Markers of type B, the mother is DIP homozygous and the father is DIP heterozygous. With this marker, the paternally transmitted DIP-STR haplotype can be targeted in maternal plasma only if the transmitted haplotype carries a DIP allele not shared with the mother. Markers of type C, the mother and the father are homozygous for the same DIP allele. With this marker, no paternally transmitted DIP-STR haplotype should be detected in maternal plasma
In this study, we determined whether the DIP-STR markers can be used to analyze cffDNA in maternal plasma for noninvasive prenatal diagnosis. The results from testing this new method on 48 pregnant women with 95 samples collected at different gestational ages show that DIP-STRs can contribute to prenatal care, especially considering the potential for improvement in marker selection, multiplexing, and quantity of maternal plasma to be used for testing.
Materials and methods
Sample collection
Inclusion criteria for enrolled couples were singleton pregnancies with known paternity. Maternal blood samples (10 ml) were drawn longitudinally from 48 women at 8–13 weeks (first trimester), 14–26 weeks (second trimester), and 27–40 weeks (third trimester) of amenorrhea. Samples collected during the first, second, and third trimester are from 27, 28, and 40 pregnant women, respectively. See Table 1 for details on longitudinal sample collection. Venous blood samples were drawn into ethylenediaminetetraacetic acid (EDTA) blood collection tubes. Plasma was separated from the blood cells via double centrifugation (1600g for 10 min, tube transfer, and centrifugation at 28,000g for 10 min) within 2 h from blood drawn. Four to five aliquots of 1 ml were stored at –20 °C until further processing.37
DNA samples from both parents of the developing baby were collected by buccal swab. Such reference samples are necessary for searching DIP-STR markers showing informative genotypes. Written informed consent was obtained from all participants, and genetic samples were collected under an institutional review board–approved research protocol.
DNA extraction
Cell-free circulating DNA was extracted in duplicate from 2 ml of plasma by using the QIAamp Circulating Nucleic Acid Kit (Qiagen AG, Basel, Switzerland) according to the manufacturer’s protocol. Absorbed DNA was eluted with 60 µl of provided elution buffer. The synthetic DNA RT-SPCY-T02 (Eurogentec, Angers, France) was added to the plasma to function as positive control for circulating DNA extraction. According to the manufacturer’s protocol, 2 µl of a 10-fold diluted RT-SPCY-T02 was added to 2 ml of plasma. Reference buccal samples were extracted using the QIAamp DNA Mini Kit (Qiagen) according to the manufacturer’s protocol and eluted in 100 µl final volume. Both genomic and circulating DNA samples were stored at –20 °C.
Fetal DNA quantity and percent was measured in the subset of plasma samples from pregnancies of male fetuses. The forensic kit Investigator Quantiplex HYresTM assay (Qiagen) and an AB 7500 Real-Time PCR system (Life Technologies Europe, Zug, Switzerland) was used according to the manufacturer’s protocol; data were analyzed using the HID Real-Time PCR Analysis Software v1.2 (Life Technologies Europe).
DIP-STR genetic markers
The DIP-STR markers genotyped for this study include 18 markers previously published35,36 and 10 newly developed (Supplementary Table S1).
PCR reactions for the 10 new markers were performed as previously published DIP-STR genotyping protocols35,36 using 10 µl of plasma DNA. Primer sequences are reported in Supplementary Table S2. S- and L-DIP-STR specific amplifications were done in singleplex according to published protocols.35,36 For the 10 newly developed markers primers were used at 100 nM and 34 cycles of PCR. Annealing temperatures varied: rs71725104-STR, rs72534187-STR, rs139592446-STR, rs36194161-STR, rs138331044-STR were amplified at 58 °C, MID473-STR, MID2538-STR, MID1739-STR, MID2824-STR at 55 °C, and MID73-STR at 59 °C. To identify informative markers for plasma DNA analysis, reference DNA samples from the mothers and the fathers were first genotyped for 28 DIP markers using four multiplex reactions as described in Supplementary Table S3. For all markers PCR thermal cycling conditions were as follows: 5 min at 95 °C, 1 min at 94 °C, 1 min at annealing temperature specific to the markers set to be genotyped, 1 min at 72 °C for a number of PCR cycles that also varied across multiplex, and a final extension of 30 min at 72 °C.
PCR fragments were separated by capillary electrophoresis after adding 1 μl PCR amplicon to 8.5 μl deionized formamide HI-DI (Life Technologies Europe) and to 0.5 μl 600 LIZ size standard (Life Technologies Europe). Capillary electrophoresis was performed using an ABI PRISM 3130xl Genetic Analyzer (Life Technologies Europe) according to the manufacturer’s instruction and analyzed using the GeneMapper® ID v3.2.1 software (Life Technologies Europe), with a minimum peak height threshold of 50 relative fluorescence unit (RFU). The commercial DNA CEPH 1347-02 (Life Technologies Europe) was used as positive control of amplification and internal standard for allele designations. For standard PCR reactions (28–30 cycles) 0.5 ng of CEPH 1347-02 DNA was used, for all PCR reactions with increased number of cycles (34–36) 0.0125 ng of CEPH 1347-02 DNA was used.
Results
Type of informative markers
As described above, the target analysis of circulating fetal DNA is based on allele-specific amplifications of those haplotypes showing DIP differences between the mother and the paternally transmitted DNA. To do so, primers are selected based on the DIP genotype of the mother. Markers informative for the genotypes of the fetus are DIP homozygous in the mother (SS or LL) and should be analyzed using primers specific to the opposite DIP allele (L- and S-primers, respectively). The fetal allele is then targeted if a DIP allele, different from the mother, is transmitted by the father. Considering all possible genotype assortments, three types of informative markers exist (Fig. 1). With markers of type A, the father is homozygous for the DIP allele that is not shared with the mother; in this case the paternal haplotype can be targeted in maternal plasma regardless of which one is transmitted to the fetus. With markers of type B, the father is heterozygous at the DIP locus; in this case the paternal DNA can be targeted in maternal plasma if the transmitted haplotype carries the DIP allele not shared with the mother. Finally, with markers of type C, the father is homozygous at the DIP locus for the same allele of the mother; in this case no paternal haplotype can be targeted in maternal plasma.
Supplementary Table S4 shows the number of each type of informative marker per family. Most of the parental genotypes we studied showed two and three markers of type A, about eight of type B, and six of type C. Interestingly, the few parents (ID 6,15, and 18) who showed no markers of type A had between 5 and 10 choices of type B markers that allowed in practice the detection of the paternally transmitted DNA.
Detection of cffDNA by selected DIP-STRs
To test if DIP-STR markers could be used to detect fetal DNA across families and time points, each plasma sample was genotyped for at least one marker of type A. When many informative markers were available, the shortest was selected for this pilot study. As described above, families 6, 15, and 18 showed no marker of type A, therefore markers informative of the paternally transmitted alleles were identified by testing several markers of type B.
To further check for PCR specificity, we first genotyped plasma samples where the fathers were heterozygous for the STR composing the DIP-STR haplotype. The results of a few representative samples are illustrated in Fig. 2. Samples relative to family 2 never worked because of insufficient blood collection (2.5 ml instead of 10 ml) (Table 2). Seven plasma samples gave positive results after modification of the PCR protocol to include 36 cycles and eight plasma samples never worked with the selected DIP-STRs. This represents 6.9% of false negative results considering 8 fetal DNA amplifications of 116 PCR tests performed. The markers associated with these weak and negative results are all characterized by low PCR efficiency as previously reported;35,36 these are L-rs35032587-STR, S- and L-rs71070706-STR, and L-rs111478323-STR and L-rs112604544-STR. To report on the signal-to-noise ratio of the assay, Supplementary Fig. S1 shows the electropherogram results of cffDNA detection in maternal blood for the samples collected during the first and second trimester that showed low RFU values (<100 RFU). Overall, some markers appeared more sensitive than others, but the quantity of the PCR product obtained did not correlate with the time in pregnancy.

Electropherogram results of the positive detection of the paternally transmitted alleles in maternal blood collected during the first and second or third trimester. The detected allele can be compared with the paternal genotype indicated below. The STR heterozygosity of the father allows to confirm the target amplification of the paternal haplotype transmitted to the fetus. Primers are selected to be specific to the paternally transmitted alleles of the cell-free fetal DNA (cffDNA), therefore as expected no polymerase chain reaction (PCR) product is detected when using the reference DNA of the mother. a Family 51, marker rs36194161-STR; b family 31, marker MID473-STR; c family 27, marker MID473-STR
Detection of cffDNA using long DIP-STRs
The use of long DIP-STR markers on a reduced number of samples also showed positive results. Alleles of 300, 305, 335, 337, 348, 360, and 386 base-pairs were successfully amplified in nine different families (ID 30, 32, 42, 47, 48, 25, 8, 13, 31) using different markers and samples collected at both early and late pregnancy. The results of a few representative samples are illustrated in Fig. 3a. These families were selected solely based on the occurrence of long DIP-STR informative markers of type A, without other specific selecting criteria.

Electropherogram results of the positive detection of the paternally transmitted alleles in maternal blood using long DIP-STR markers. a Above, family 13, marker L-rs11277790-D10S530. Below, family 31, marker L-rs67842608-D5S468. b Electropherogram results showing one case of positive detection of the paternally transmitted allele in maternal plasma collected at 10 weeks of pregnancy. Family 49, marker S-MID1739-STR
Detection of cffDNA during early pregnancy
The blood samples collected during the first trimester of pregnancy were mostly drawn between 12 and 13 weeks of gestational age. One sample (ID 49) was collected at 10 weeks and 2 days of amenorrhea and could be used to test the DIP-STR based fetal DNA detection in early pregnancy. The only marker tested with this sample, S-MID1739-STR, gave a positive result (Fig. 3b).
Correlation between fetal DNA quantity and DIP-STR results
The subset of plasma samples collected from women bearing male fetuses during the first and second trimester were selected to quantify the fetal DNA fraction and to investigate a possible correlation to DIP-STR results. The data reported in Supplementary Table S5 indicate that positive results are obtained using as low as 0.029 ng of fetal DNA representing 3.5% of plasma DNA. The strength of the signal (RFUs) varies depending on the sensitivity of the markers used as previously reported. Note that samples showing false negative results (NR) do not show the lowest fetal DNA quantity or fraction.
DIP-STR markers not used for cffDNA detection
Because of the specific occurrence of informative markers in these families, not all available DIP-STRs have been used for cffDNA detection. Markers MID1013-D5S490, MID1950-D20S473, rs60194384-D15S1514, rs72534187-STR, rs138331044-STR, and MID73-STR were not tested in this study. Markers MID1107-D5S1980, rs35708668-D5S2045, and rs10564579-D3S1282 were sometimes among the informative markers but they were not selected because of their long size (600–700 bp). Finally, some markers were tested for only one DIP-STR allele such as L-rs67842608-D5S468, S-rs66679498-D2S342, L-rs10564579-D3S1282, L-rs72406828-STR, L-rs145423446-STR, L-rs139592446-STR, S-rs36194161-STR, and S-MID1739-STR.
Discussion
Our results show that cell-free fetal DNA in maternal plasma can be detected by applying novel DIP-STR genetic markers that we originally developed for the analysis of DNA mixtures encountered in forensic trace analysis. The results showed positive detection of fetal DNA, regardless of sex type, in the plasma of 48 women whose blood was collected during the first, second, and third trimester of pregnancy. With few exceptions, all tested markers could detect the fetal DNA, including longer ones targeting DNA fragments up to 386 base-pairs. Positive results were also obtained with one sample collected during early pregnancy at 10 weeks of gestational age. Yet, more samples collected between 6 and 10 weeks are necessary to validate the method for early noninvasive diagnostic purposes.
Overall, we observed 6.9% (8/116 PCR assays) of false negative results. The plasma samples (N = 31) in which the fetal DNA fraction could be quantified by Y-chromosome markers showed values from 3.5 to 19.2% and no correlation between low fetal DNA quantity or fraction and DIP-STR results. No issue of fetal DNA extraction efficiency was reported by the internal control of extraction. Moreover, families with false negative results showed low marker performance longitudinally during the first, second, and third trimester (see Table 2 for ID 17, 19, 21, 35, 37, and 42) and often with the same few markers, such as rs717070706-STR. Based on the observations, we interpreted the negative PCR results as mainly due to few low sensitive markers. On the other hand, no false positive results were generated by the large excess of maternal DNA, in agreement with our marker validation results that assessed allele specificity up to a mixture ratio of 1:1000. These results confirm the sensitivity and specificity of the DIP-STRs for targeting the minor DNA component of plasma DNA, even when fetal DNA molecules are of reduced numbers, such as in early pregnancy or because of the use of long markers.
These data provide the basis for the development of a noninvasive prenatal paternity test that is rapid, cost-effective, and easy to implement (one or two PCR multiplex) in any forensic or medical genetics laboratory. Such molecular approach would offer several advantages with respect to currently developing methods: (1) a targeted approach that guarantees a careful separation between paternity tests and disease diagnoses (aneuploidies and pathogenic variant discoveries), (2) a molecular system that is shared with other forensic and medical applications where the risk for false positive and negative result is carefully measured and the appropriate statistical framework is available (Bayesian approach), and (3) the choice of different type of informative markers (type A, B, and C) provides a useful flexibility in testing different scenarios including one, multiple, or unknown alleged fathers that can be associated to various degree of statistical power.
As explained in the introduction and previous publications, the number of markers capable of targeting the fetal DNA based on allele difference between maternal DNA and paternally transmitted alleles, is expected to vary across cases. As observed here with 49 families, 28 DIP-STRs generate on average 3 markers of type A, where the parents are opposite homozygous for the DIP allele and at least one paternal allele is detected; 7 markers of type B, which means one paternal allele could be detected if transmitted; and 7 markers of type C, which are mainly used for exclusion because negative results are expected. To bring our method closer to medical practice, we plan to first develop the statistical framework necessary for the analysis of DIP-STR genotypes in the context of paternity testing. Bayesian models will be used as they are generally accepted in forensic science to evaluate the strength of DNA evidence. Second, based on the model above, we will study how likelihood ratios values vary depending on the number and types of informative markers considering a set of simulated families.
Our results also showed that the sensitivity of the technique may vary across DIP-STRs and we recorded 4 DIP-STRs (of 19 markers tested on clinical samples) not sensitive enough for a positive/robust amplification of circulating fetal DNA in the conditions tested. Therefore, a suitably designed multiplex for amplification is needed including careful measurements of marker sensitivity and specificity, especially to assist the interpretation of results from markers of type B and C. Ideally, in case of negative results one should have a universal fetal DNA marker to demonstrate the presence of amplifiable fetal DNA. Moreover, we plan to explore the use of a larger (>2 ml) volume of plasma for DNA extractions. Note that the drawing of more blood (>10 ml) is possible without adducing important discomfort to the patient. Moreover, Streck BCT blood collection tubes could be used to facilitate the sample collection within a clinical environment. These tubes reduce the need for immediate plasma preparation by stabilizing nucleated blood cells at temperatures between 6 and 37 °C for several days. Although we achieve high levels of allele specificity with the DIP-STR genotyping approach, (markers are specific up to 1:1000 DNA ratio) and we didn’t observe issue of maternal DNA amplification, it is still possible that reducing the quantity of background maternal DNA the performance of less sensitive markers improves. This strategy could be the solution for early pregnancy tests, if studies on a larger cohort indicates that marker sensitivity is limiting. However, it should be noted that several reports indicated that the quantity of cffDNA varies little during pregnancy.
Finally, of interest is an initial comparison of this method to published noninvasive prenatal paternity tests even at this early stage of development (no calculation of paternity index for the described cases) where “inconclusive results” are due to false positive and false negative markers. Ou’s group published several articles32,33,34 where they use SNP panels for the analysis of paternally inherited alleles in maternal plasma using MPS techniques. These studies show that, although results change with the specific technology used (sequencing technology, library preparation, coverage, number of SNPs), a panel of 720 SNPs has 11% of poorly performing markers that need to be eliminated and a low signal-to-noise ratio that requires considering samples with a minimum fetal DNA fraction of 2%. When this threshold is used and suboptimal SNPs are eliminated, 10 to 50% of paternally transmitted alleles from the subset of informative SNPs (about 40% on average) are filtered out, and 0 to 11% when samples are collected during the second trimester. Events of allele drop-in are also observed by using MPS with three extra alleles in 20 families reported. With respect to studies using SNP arrays,28 the authors declare that each individual SNP measurement is noisy but technical false positive and false negative rates are not reported. Conversely, the 384-SNP method by allele-specific PCRs of Guo et al.29 indicated 13.3% false positive and 2.7% false negative rates. In our study the false negative rate is 6.9% including poorly performing DIP-STRs (4 markers) that were not eliminated and false positive results were not observed. Indeed, interrogating a large number of markers, as for MPS or SNP arrays, offers the advantage of reducing the issue of false positive and false negative results; however an appropriate signal quality control and statistical framework for results interpretation needs to be developed.
Therefore, of interest is the possibility of integrating novel large-scale technologies, although keeping a targeted approach. The use of high-throughput sequencing, instead of capillary electrophoresis, for fragment analysis will offer an improved capacity in the number of markers analyzed and more flexibility in primer design. The requirement of this sophisticated technology will be elucidated after measuring the statistical power of this initial set of DIP-STR markers on data from a large number of families simulated for paternity tests.
In summary, we show that the use of DIP-STR markers for targeting the analysis of fetal DNA circulating in the blood of a pregnant woman is possible and can be used for noninvasive prenatal paternity testing. Further study is warranted to determine the number of informative markers necessary for obtaining conclusive results on paternity across parental genotypes. Conversely, for diagnostic purposes entirely new DIP-STRs should probably be developed depending on the genomic region of interest. In this regard, we previously estimated that several thousands of DIP-STRs exist35 in the genome and that their availability should not limit the adaptation of this technique to specific needs.
Change history
11 March 2019
In the original version of this Article, the data for Mother ID 8; 13; 25; 31; and 47 of Table 2 was merged. This has now been corrected in both the PDF and HTML versions of the Article.
References
Costa JM, Benachi A, Gautier E. New strategy for prenatal diagnosis of X-linked disorders. N Engl J Med. 2002;346:1502.
Finning KM, Martin PG, Soothill PW, Avent ND. Prediction of fetal D status from maternal plasma: introduction of a new noninvasive fetal RHD genotyping service. Transfusion. 2002;42:1079–1085.
Lo YM, Hjelm NM, Fidler C, et al. Prenatal diagnosis of fetal RhD status by molecular analysis of maternal plasma. N Engl J Med. 1998;339:1734–1738.
Chiu RW, Lau TK, Leung TN, et al. Prenatal exclusion of beta thalassaemia major by examination of maternal plasma. Lancet. 2002;360:998–1000.
Ding C, Chiu RW, Lau TK, et al. MS analysis of single-nucleotide differences in circulating nucleic acids: application to noninvasive prenatal diagnosis. Proc Natl Acad Sci USA. 2004;101:10762–10767.
Li Y, Di Naro E, Vitucci A, et al. Detection of paternally inherited fetal point mutations for beta-thalassemia using size-fractionated cell-free DNA in maternal plasma. JAMA. 2005;293:843–849.
Chiu RW, Lau TK, Cheung PT, et al. Noninvasive prenatal exclusion of congenital adrenal hyperplasia by maternal plasma analysis: a feasibility study. Clin Chem. 2002;48:778–780.
Lo YM, Lau TK, Zhang J, et al. Increased fetal DNA concentrations in the plasma of pregnant women carrying fetuses with trisomy 21. Clin Chem. 1999;45:1747–1751.
Zhong XY, Burk MR, Troeger C, et al. Fetal DNA in maternal plasma is elevated in pregnancies with aneuploid fetuses. Prenat Diagn. 2000;20:795–798.
Levine RJ, Qian C, Leshane ES, et al. Two-stage elevation of cell-free fetal DNA in maternal sera before onset of preeclampsia. Am J Obstet Gynecol. 2004;190:707–713.
Lo YM, Leung TN, Tein MS, et al. Quantitative abnormalities of fetal DNA in maternal serum in preeclampsia. Clin Chem. 1999;45:184–188.
Lo YM. Fetal DNA in maternal plasma: progress through epigenetics. Ann N Y Acad Sci. 2006;1075:74–80.
American College of Obstetricians and Gynecologists. ACOG practice bulletin no. 88. December 2007. Invasive prenatal testing for aneuploidy. Obstet Gynecol. 2007;110:1459–1467.
Seeds JW. Diagnostic mid trimester amniocentesis: how safe? Am J Obstet Gynecol. 2004;191:607–615.
Elchalal U, Shachar IB, Peleg D, Schenker JG. Maternal mortality following diagnostic 2nd-trimester amniocentesis. Fetal Diagn Ther. 2004;19:195–198.
Brambati B, Simoni G, Travi M, et al. Genetic diagnosis by chorionic villus sampling before 8 gestational weeks: efficiency, reliability, and risks on 317 completed pregnancies. Prenat Diagn. 1992;12:789–799.
Firth HV, Boyd PA, Chamberlain PF, et al. Analysis of limb reduction defects in babies exposed to chorionic villus sampling. Lancet. 1994;343:1069–1071.
Lo YM, Corbetta N, Chamberlain PF, et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997;350:485–487.
Barrett AN, McDonnell TC, Chan KC, Chitty LS. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin Chem. 2012;58:1026–1032.
Lun FM, Chiu RW, Chan KC, et al. Microfluidics digital PCR reveals a higher than expected fraction of fetal DNA in maternal plasma. Clin Chem. 2008;54:1664–1672.
Wang E, Batey A, Struble C, et al. Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat Diagn. 2013;33:662–666.
Lo YM, Chiu RW. Prenatal diagnosis: progress through plasma nucleic acids. Nat Rev Genet. 2007;8:71–77.
Chitty LS, Lo YM. Noninvasive prenatal screening for genetic diseases using massively parallel sequencing of maternal plasma DNA. Cold Spring Harb Perspect Med. 2015;5:a023085.
Lo YM. Fetal nucleic acids in maternal plasma. Ann N Y Acad Sci. 2008;1137:140–143.
Lo YM, Lun FM, Chan KC, et al. Digital PCR for the molecular detection of fetal chromosomal aneuploidy. Proc Natl Acad Sci USA. 2007;104:13116–13121.
Johnson DS, Gemelos G, Baner J, et al. Preclinical validation of a microarray method for full molecular karyotyping of blastomeres in a 24-h protocol.Hum Reprod. 2010;25:1066–1075.
Chiu RW, Lo YM. Noninvasive prenatal diagnosis empowered by high-throughput sequencing. Prenat Diagn. 2012;32:401–406.
Ryan A, Baner J, Demko Z, et al. Informatics-based, highly accurate, noninvasive prenatal paternity testing. Genet Med. 2013;15:473–477.
Guo X, Bayliss P, Damewood M, et al. A noninvasive test to determine paternity in pregnancy. N Engl J Med.2012;366:1743–1745.
Lo YM, Chan KC, Sun H, et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus.Sci Transl Med. 2010;2:61ra91.
Jiang H, Xie Y, Li X, et al. Noninvasive prenatal paternity testing (NIPAT) through maternal plasma DNA sequencing: a pilot study. PLoS ONE . 2016;11:e0159385.
Qu N, Xie Y, Li H, et al. Noninvasive prenatal paternity testing using targeted massively parallel sequencing. Transfusion. 2018. https://doi.org/10.1111/trf.14577.
Yang D, Liang H, Gao Y, et al. Noninvasive fetal genotyping of paternally inherited alleles using targeted massively parallel sequencing in parentage testing cases. Transfusion. 2017;57:1505–1514.
Yang D, Liang H, Lin S, et al. An SNP panel for the analysis of paternally inherited alleles in maternal plasma using ion Torrent PGM. Int J Legal Med. 2018;132:343–352.
Castella V, Gervaix J, Hall D. DIP-STR: highly sensitive markers for the analysis of unbalanced genomic mixtures. Hum Mutat . 2013;34:644–654.
Oldoni F, Castella V, Hall D. A novel set of DIP-STR markers for improved analysis of challenging DNA mixtures. Forensic Sci Int Genet. 2015;19:156–164.
Chan KC, Yeung SW, Lui WB, et al. Effects of preanalytical factors on the molecular size of cell-free DNA in blood. Clin Chem. 2005;51:781–784.
Acknowledgements
We thank the participants who volunteered for the study. Prof. Olivier Irion, Prof. Marie Cohen, and Mrs. Véronique Othenin-Girard of the Department of Obstetrics and Gynecology of the University Hospital of Geneva for helping us with volunteer recruitment and sample collection. This work was financially supported by funding from the University Center of Legal Medicine of the University Hospital of Lausanne and the Faculty of Biology and Medicine of the University of Lausanne (Pro-Femmes fellowship to Diana Hall).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Moriot, A., Hall, D. Analysis of fetal DNA in maternal plasma with markers designed for forensic DNA mixture resolution. Genet Med 21, 613–621 (2019). https://doi.org/10.1038/s41436-018-0102-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0102-9
Keywords
This article is cited by
-
Early noninvasive prenatal paternity testing by targeted fetal DNA analysis
Scientific Reports (2023)
