
Abstract
Purpose
The oocyte-borne genetic causes leading to fertilization failure are largely unknown. We aimed to identify novel human pathogenic variants (PV) and genes causing fertilization failure.
Methods
We performed exome sequencing for a consanguineous family with a recessive inheritance pattern of female infertility characterized by oocytes with a thin zona pellucida (ZP) and fertilization failure in routine in vitro fertilization. Subsequent PV screening of ZP2 was performed in additional eight unrelated infertile women whose oocytes exhibited abnormal ZP and similar fertilization failure. Expression of ZP proteins was assessed in mutant oocytes by immunostaining, and functional studies of the wild-type and mutant proteins were carried out in CHO-K1 cells.
Results
Two homozygous s PV (c.1695-2A>G, and c.1691_1694dup (p.C566Wfs*5), respectively) of ZP2 were identified in the affected women from two unrelated consanguineous families. All oocytes carrying PV were surrounded by a thin ZP that was defective for sperm-binding. Immunostaining indicated a lack of ZP2 protein in the thin ZP. Studies in CHO cells showed that both PV resulted in a truncated ZP2 protein, which might be intracellularly sequestered and prematurely interacted with other ZP proteins.
Conclusion
We identified loss-of-function PV of ZP2 causing a structurally abnormal and dysfunctional ZP, resulting in fertilization failure and female infertility.
Similar content being viewed by others

Introduction
Female infertility is a worldwide health problem that affects an estimated 48 million women.1 A healthy oocyte is a major determinant of female fertility, defects in which usually lead to fertilization failure, as well as zygotic or embryonic arrest. However, it is very difficult to discover the oocyte anomalies via routine examinations until they are retrieved for in vitro fertilization (IVF).
Despite the improvement of IVF techniques in the past years, total fertilization failure (TFF) is still a recurrent problem for 5–10% of routine IVF cycles2 and produces no zygote. Although fertilization failure occurs at different IVF stages, defective sperm–zona binding and penetration is a chief cause.3,4 However, in cases for which fertilization fails at pre–sperm penetration stages, no genetic cause tied to oocytes has been identified.
In mammals, normal fertilization depends on structural and functional integrity of the zona pellucida (ZP), which is an extracellular matrix surrounding oocytes. In mice, the ZP consists of three glycoproteins: mZP1–mZP3. mZP2 and mZP3 are building blocks of ZP filaments and are sperm receptors during fertilization.5,6 Knockout of either Zp27 or Zp38 in female mice produced eggs without a ZP, as well as infertility. Whereas, knockout of Zp19, which crosslinks individual filaments, resulted in the formation of a loosely organized ZP and fertile, but less fecund, female mice.
The human ZP is composed of four glycoproteins: ZP1–ZP4.10,11 Until now, variants in ZP1 and ZP3 genes have been reported to be the causes for oocyte maturation defect (MIM: 615774 and 617712) and female infertility.12,13 In a consanguineous Chinese family, an autosomal recessive homozygous truncating mutation in ZP1 was identified in infertile females with abnormal oocytes lacking a ZP, and this was suggested to explain infertility.12 This suggests that ZP proteins play different roles across species but limited human studies suggest that this hypothesis warrants study. In addition, no definitely deleterious variant in either ZP2 or ZP4 gene had been found to be responsible for female infertility.
Here, we identified two homozygous truncating pathogenic variants (PV) in ZP2 (GenBank NM_003460.2) in two unrelated consanguineous families as likely causes of fertilization failure and infertility of the female members. We also assessed each for loss-of-function effects by in vitro studies.
Materials and methods
Study subjects
All infertile individuals and the control group included in this study were from the Reproductive and Genetic Hospital of CITIC-Xiangya. The control group consisted of 100 women with proven fertility and at least one child. All blood samples, immature or mature oocytes unfertilized after insemination were used with consent from subjects and spouses. Study ethical approval and oversight was obtained from our hospital (reference LL-SC-2016-003).
Transmission electron microscopy (TEM)
Normal control and patient oocytes were treated as described previously.14 Briefly, oocytes were fixed with glutaraldehyde (Sigma-Aldrich, St. Louis, MO, USA) and osmium tetroxide, postfixed with OsO4 and sucrose, dehydrated with graded concentrations of ethanol, and then embedded in Epon812, dodecenylsuccinic anhydride, methylnadic anhydride, and dimethylaminomethyl phenol. Ultrathin 70–90-nm-thick sections were contrasted with uranyl acetate and lead citrate and examined using a HT7700 Hitachi transmission electron microscope (Hitachi, Tokyo, Japan). MegaView III digital camera (Munster, Germany) was used to capture digital images.
Polarized light microscopy (polscope)
To image zona pellucida, each oocyte was placed in a 5-µl drop of G-MOPS (Vitrolife, Goteborg, Sweden) handling medium covered with paraffin oil in a glass-bottomed (0.17 mm thick) Petri dish (20 mm diameter; Delta TPG dish, Bioptechs, Butler, PA, USA). Dishes were maintained at 37 ℃ during examination with a thermal plate (IVF-1200, Pacific Contrast Co., San Diego, CA, USA) and objective heater (HT300, Minitube, Tiefenbach, Germany). Oocytes were examined with an inverted microscope (Nikon TE-2000U, Tokyo, Japan) equipped with Hoffman interference optics 20× objective lenses equipped with LC-Polscope filters, analog video camera (COHU, Cambridge Research and Instrumentation, Woburn, MA, USA), and a personal computer (Dell, Round Rock, TX, USA) running image analysis software (LC-Polscope pro 4.4; Cambridge Research and Instrumentation, Woburn, MA, USA).
Exome sequencing (ES) and variant analysis
Genomic DNA was purified from peripheral blood samples by means of DNA extraction kit (Qiagen, Hilden, Germany). ES and data analysis were performed by Beijing Genome Institute at Shenzhen as described previously.14 Briefly, exome capture and sequencing were carried out by the SureSelect Human All Exon V5 (Agilent, Santa Clara, CA, USA) and the HiSeq2000 sequencing platform (Illumina, San Diego, CA, USA). Sequenced reads were collected, filtered for quality, and aligned to the human reference sequence (University of California–Santa Cruz Genome Browser hg19) with the Burrows–Wheeler Aligner.15 Genotypes were called by Genome Analysis Toolkit (GATK).16 Sequence variants including single-nucleotide variants (SNVs) and small insertions or deletions (INDELs) were annotated by ANNOVAR software. Candidate variants were identified according to the following filtering criteria: (1) rare variants with a minor allele frequency below 1% in the three public databases: the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project Exome Variant Serve (EVS), 1000 Genomes, and the Exome Aggregation Consortium (ExAC) Browser; (2) exonic nonsynonymous or splice site variants, or coding INDELs; (3) due to the first cousin consanguinity of family 1, homozygous variants were prioritized; and (4) homozygous variants shared by both infertile women II-3 and II-4, but absent in two fertile women I-2 and II-2 in family 1. Meanwhile, homozygosity mapping was performed using HomozygosityMapper,17 and homozygous variants located in homozygous regions greater than 5.0 Mb were considered with priority.
Sanger sequencing
Each exon and its flanking intron sequence of the ZP2 gene were amplified by primers listed in Supplementary Table S1. polymerase chain reaction (PCR) products were analyzed by agarose gel electrophoresis, purified, and analyzed by bidirectional sequencing.
Single-cell reverse transcription and 3’ rapid amplification of cDNA end (RACE)
A single immature oocyte from patients or normal controls was dispensed in lysis buffer and complementary DNA (cDNA) libraries were generated using Smart-seq218. Briefly, following cell lysis, Poly-A(+) RNA was reverse transcribed by Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) and an oligo-dT adapter primer, utilizing a strand-switch reaction to add a reverse primer for the second-strand synthesis. The cDNA was amplified by PCR (18 cycles) using KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA, USA) and purified with Ampure XP beads (Beckman Coulter, Kraemer Boulevard Brea, CA, USA). The quantity and quality of the cDNA libraries were assessed using an Agilent 2100 BioAnalyzer. 3’ end of ZP2 cDNA, containing the PV sites, was then amplified by PCR using a sense primer specific to exon 14 (1935F) and a reverse primer (Adapter-R) that targets the adapter region of the oligo-dT adapter primer (Supplementary Table S1). Using the first-round PCR products, a nested second PCR was performed by using a sense primer specific to exon 15 (2087 F) and primer Adapter-R. The final PCR products were analyzed by agarose gel electrophoresis and Sanger sequencing.
Construction of expression plasmids
The expression plasmids encoding wild-type ZP1/2/3/4 (pENTER-ZP1/2/3/4) and mutant ZP2 proteins (pENTER-mut1ZP2: skipping exons 16 and 17, or pENTER-mut2ZP2: c.1691_1694dup) were constructed by Vigene Biosciences (Jinan, China). To construct plasmids encoding fluorescence fusion ZP proteins, cDNA encoding full-length ZsGreen or mcherry fluorescent proteins were inserted at the N-terminus of ZP2 or ZP3/4 in pENTER vectors, downstream the signal sequence (Supplementary Fig. S1).
Expression in heterologous cells
CHO-K1 cells (China Center for Type Culture Collection, Wuhan, China) were grown (37 °C, 5% CO2) to 70–80% confluence in F-12 medium supplemented with 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Transient transfections were performed with Lipofectamine 3000 (Thermo Fisher Scientific) in accordance with the manufacturer’s protocol. For each transfection, 6 µl Lipofectamine 3000 transfection reagent was added to 100 µl Opti-MEM (Gibco) predissolved with 2 µg template plasmid and incubated for 5 minutes at room temperature. The complex was added into the well with growing cells. Transiently transfected cells were harvested at 48 h for analysis.
Immunoblots
Cells were recovered 48 h after transfection, after which cells were homogenized with M-PER Mammalian Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL, USA) supplemented with HaltTM Protease Inhibitor Cocktail (Thermo Fisher Scientific) according to the manufacturer’s instructions. Following centrifugation (16,000g, 4 °C), cell lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membranes, which were probed with antibodies to ZP1 (Santa Cruz, Dallas, TX, USA), ZP2 (against amino acids 86–228 mapping near the N-terminus of human ZP2) (Sigma-Aldrich, St Louis, MO, USA), or β-actin (Santa Cruz) and detected by secondary antibodies (Santa Cruz) and ECL western blotting kit (Pierce Biotechnology).
Immunofluorescent staining
Oocytes from patients and controls that were immature or unfertilized after intracytoplasmic sperm injection (ICSI) were fixed in 2% paraformaldehyde (room temperature, 1 h), washed in phosphate buffer saline (PBS) (3×), permeabilized (PBS and Triton X-100, 5 minutes), blocked (PBS with bovine serum albumin [BSA], 1 h) and double-stained with rabbit polyclonal antibody to ZP2 (Sigma-Aldrich) or ZP3 (Santa Cruz), and goat polyclonal antibody to ZP1 (Santa Cruz) (1:100, overnight, 4 ℃). Antibodies binding were detected with Alexa-Fluor-488-conjugated donkey anti-goat and Alexa-Fluor-647-conjugated donkey antirabbit secondary antibodies (Life Technologies, Europe, Ghent, Belgium) (1:1000, room temperature, 1 h).
Transfected cells expressing fluorescent-tagged proteins were grown on coverslips for 48 h, fixed with 2% paraformaldehyde (room temperature, 15 minutes), washed in PBS (3×), and counterstained with DAPI (room temperature, 10 minutes). For ZP1 imaging, cells were fixed with 2% paraformaldehyde (room temperature, 15 minutes), washed in PBS (3×), permeabilized (PBS and Triton X-100, 5 minutes), blocked (PBS with BSA, 1 h), and incubated with mouse monoclonal antibody to ZP1 (Santa Cruz) (1:100, overnight, 4 ℃). Antibody binding was detected with Alexa-Fluor-594-conjugated donkey antimouse antibody secondary antibody (Life Technologies) (1:1000, room temperature, 1 h). Confocal images were obtained with a microscope (Olympus IX81 and (CLSM) Olympus FV1000, Tokyo, Japan).
Immunoprecipitation
For immunoprecipitation of ZP1, ZP2ZsGreen, and mutZP2ZsGreen proteins, a mouse monoclonal antibody specific to ZP1 (Santa Cruz) was incubated with protein A/G magnetic beads (Pierce Biotechnology) according to the manufacturer’s instructions. The complex was incubated with lysate from transfected cells and analyzed by immunoblot.
Results
Clinical characterization of patients with primary female infertility and fertilization failure in routine IVF
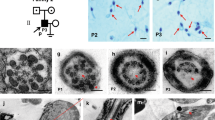
Two Chinese consanguineous families were included in this study (Fig. 1a). In family 1, the proband (II-4) was a 28-year-old woman with a 3-year history of unexplained primary infertility. She has two older sisters; one (II-3, 30 years old) also had a 10-year history of primary infertility, and the other (II-2, 31 years old) with normal fertility has her own children. Thus, two of the three sisters from a marriage of the first cousins in this family suffer from primary infertility. In family 2, the patient (II-1) was a 32-year-old woman with a 7-year history of unexplained primary infertility and her parents were also first cousins. For all infertile women, infertility-related assessments did not reveal any abnormalities, and their spouses had normal sperm counts, with normal motility and morphology (Supplementary Tables S2–S4).

a Pedigrees of two consanguineous families with inherited ZP2 pathogenic variants. ZP2 genotype for each individual is indicated, with N indicating a normal allele and a dash (-) indicating the mutant allele. b Homozygosity mapping of individuals II-3 and II-4 in family 1. The most promising homozygous regions are colored in red. The asterisk indicates the area where ZP2 is located. c Splice mechanisms of ZP2 exons associated with the variant c.1695-2A>G, which leads to a loss of the acceptor splice site of intron 15 and skipping of exons 16 and 17 in the messenger RNA (mRNA), and causes a frameshift and the formation of a premature termination codon (PTC) (p.C566Hfs*5). The frameshift variant c.1691_1694dup does not affect splicing, but introduces a PTC at the same site in mRNA (p.C566Wfs*5). d Domain organization of ZP2 (top) and mutant ZP2 (bottom). SS signal sequence, ZPD zona pellucida domain, CFCS consensus furin cleavage site, EHP external hydrophobic patch, TMD transmembrane domain. e Immunoblot analysis of wild-type (WT) and mutant ZP2 proteins (antibody: against amino acids 86–228 mapping near the N-terminus of human ZP2) expressed in CHO cells
Patient II-4 in family 1 (F1_II-4) underwent one IVF treatment cycle, while patient II-1 in family 2 (F2_II-1) received two IVF cycles in our center (Table 1). In the first cycle for F2_II-1, all of 17 oocytes failed in fertilization in routine IVF. In her second cycle and the first cycle for F1_II-4, all mature oocytes from the two patients were divided into two groups to prepare for IVF and ICSI. None of the oocytes in the IVF groups were fertilized 20 h after insemination. In the ICSI groups, all 8 (100%) oocytes from F1_II-4 and 12 of the 15 (80%) oocytes from F2_II-1 were fertilized. In F1_II-4, two fresh 8-cell embryos were transferred, but failed in implantation. Two blastocysts formed from the remaining embryos and were cryopreserved. In the following frozen embryo transfer cycle, the two vitrified blastocysts were thawed and transferred (Supplementary Fig. S2a), resulting in a singleton pregnancy delivered at an estimated the gestational age of 39 weeks. In F2_II-1, five zygotes developed to blastocysts on day 5 (Supplementary Fig. S2b), one of which was transferred in the first attempt, but failed in implantation.
Oocyte characteristics
At egg retrieval, 16 oocytes from F1_II-4 and 42 oocytes from F2_II-1 were obtained with cumulus complexes. After granulosa cell removal, oocytes had an abnormal ZP with a thinner matrix (F1_II-4: 7.2 ± 0.7 µm, n = 5; F2_II-1: 7.4 ± 1.2 µm, n = 5) and an enlarged perivitelline space (PVS) compared with normal oocytes (16.1 ± 1.7 µm, n = 34) (Fig. 2a). These abnormalities were defined ultrastructurally using TEM and a polscope. In normal oocytes, a fine network of filaments with different arrangements from the inner to the outer zona was observed (Fig. 2b, left panel), corresponding to the multilayered model of the structure of human ZP19 observed under the polscope (Fig. 2c, upper panel). For patient oocytes, the thin zona matrix had a comparatively irregular arrangement of filaments with large holes under TEM (Fig. 2b, right panel, arrows), and had only one layer similar to, but thinner than, the inner layer of normal zonae when imaged with the polscope (Fig. 2c, middle and lower panels).

a Representative images of oocytes with abnormal zona pellucida (ZP) appearance. Oocytes from controls and patients were separated from granulosa cells and imaged under visible inverted microscopy. ZP thickness of each oocyte was measured and computed as ZP = (Left + Right + Up + Down)/4. The lower image in each pair corresponds to a magnified view of the boxed region in the upper image. b Transmission electron micrographs of the patient’s ZP. Arrowheads indicate the ZP, and arrows indicate the large holes. c Polscope images of the patients’ ZP. OL ZP outer layer, ML ZP middle layer, IL ZP inner layer. d Defective sperm binding to patients’ oocytes observed 20 h after in vitro fertilization (IVF). The scale bars represent 50 μm in (a) and (d), and 10 μm in (b)
Oocytes from both patients were not fertilized with IVF, but ICSI solved this problem and resulted in a live birth (Table 1), indicating a major defect in gamete recognition. At 20 h after IVF, sperm–ZP binding was studied and in contrast to sperm tightly bound to or penetrating the ZP of normal oocytes (Fig. 2d, left panel), sperm bound loosely to the outer ZP surface (Fig. 2d, middle and right panels) and were more easily removed (Supplementary Video). These observations indicate defective sperm-binding to thin ZP of both patient’s oocytes, causing IVF failure.
Identification of ZP2 pathogenic variants
To determine the causative PV, we initially carried out ES in the two infertile (II-3 and II-4) and two healthy control (I-2 and II-2) individuals in family 1 (Supplementary Table S5). Because of the parental consanguinity, a recessive inheritance model was applied for the genetic analysis and homozygous variants were preferentially considered. By using the filtering criteria described in Materials and methods, we identified two homozygous variants (Supplementary Tables S6–7). The first is a homozygous missense PV in exon 4 in PDE6B (GenBank NM_001145292: c.G130A (p.G44S)). The second is a homozygous splicing PV in ZP2 (GenBank NM_003460.2: c.1695-2A>G) (Supplementary Table S7). Human PDE6B is mainly expressed in retina and brain20 but at a very low level in oocytes (according to previously reported data21 and our in-house database). Homozygous or compound heterozygous PV in this gene resulted in retinitis pigmentosa (OMIM 613801) in several families,22 notably, in one of which an affected woman carrying biallelic nonsense mutations was fertile and had her own children. Thus, PDE6B was ruled out as a candidate gene, and then the c.1695-2A>G in ZP2, a gene expressed specifically in oocytes, is the only survived variant most likely responsible for the phenotype in this pedigree. This ZP2 homozygous PV was confirmed by Sanger sequencing and found to cosegregate with female infertility in this family (Fig. 1a). Both infertile individuals carry the homozygous PV, whereas the unaffected parents and fertile sister have a heterozygous PV. Moreover, homozygosity mapping revealed that this variant in ZP2 is located within a large homozygous region with size of 6.6 Mb shared in both infertile individuals (Fig. 1b).
ZP2 encodes the ZP2 glycoprotein, which is highly expressed in human oocytes and acts as a sperm receptor during gamete recognition, as well as building blocks of human ZP filaments.10,23 Thus, we subsequently performed PV screening in each exon and its flanking sequence in ZP2 by using Sanger sequencing (primers for ZP2 were listed in Supplementary Table S1) in additional 8 independent infertile women whose oocytes showed an abnormal ZP and TFF in routine IVF despite using sperm with optimal concentration, normal motility and morphology, but were normally fertilized by ICSI. Another novel homozygous frameshift PV in ZP2 (c.1691_1694dup [p.Cys566Trpfs*5]) was identified in the affected woman in family 2 (Fig. 1a), which is predicted to result in a premature termination codon (PTC) (Fig. 1c). Family 2 is also consanguineous, and the unaffected parents were heterozygous carriers (Fig. 1a). Both of the ZP2 PV were absent in 100 population-matched control women with normal fertility and were not reported in the three public databases: the EVS, 1000 Genomes, or the ExAC Browser.
Characterization of mutant ZP2 mRNA and proteins
To investigate the effects of PV on messenger RNA (mRNA) integrity, we applied 3’ RACE methods for 3’ end amplification of the ZP2 transcript from an immature oocyte from patients and normal controls. After sequencing, a splicing alternation due to skipping exons 16 and 17 was found in ZP2 cDNA from the oocyte carrying variant c.1695-2A>G, and a 4-bp duplication was found at exon 15 end in ZP2 cDNA from the oocyte carrying variant c.1691_1694dup (Fig. 1c).
Human ZP2 protein consists of 745 amino acids and both PV disrupt the coding frame (Supplementary Fig. S3) and introduce downstream PTCs. The PTCs predict a truncated protein of 569 amino acids (Fig. 1c), which lacks C-terminal domains, including a ZP-C subdomain, a consensus furin cleavage site (CFCS), an external hydrophobic patch (EHP), and a transmembrane domain (TMD) (Fig. 1d). To test this prediction, expression plasmids carrying wild-type or mutant ZP2 cDNA (skipping exons 16 and 17 (p.C566Hfs*5) or c.1691_1694dup [p.C566Wfs*5]) were transfected into CHO cells and immunoblot data show a decrease of ~20 kDa in molecular mass in both mutant ZP2. Moreover, mutant proteins in CHO cells were greater than that of wild-type protein (Fig. 1e).
Absence of ZP2 in the ZP of mutant oocytes
ZP proteins in the zona matrix were assayed with immunofluorescent staining of oocytes from patients and normal controls. Clear and strong staining of ZP1 and ZP3 proteins occurred in the ZP of all subjects (Supplementary Fig. S4). However, ZP2 staining was completely absent in the ZP of patients with homozygous PV (Fig. 3), indicating that the thin ZP lacks ZP2 protein. In addition, cytoplasmic staining of ZP proteins in patient oocytes was stronger than in normal oocytes, suggesting that the truncated ZP2 was intracellularly sequestrated and might cause partially sequestration of the other ZP proteins.

Oocytes from control and patients were fixed and immunolabeled with the use of antibodies against ZP1 and ZP2. Fluorescent signals of ZP1 (green) and ZP2 (red) were imaged individually and merged under confocal laser scanning microscopy (600×). The scale bar represents 50 μm
Effects of ZP2 PV on intracellular trafficking of ZP proteins
To investigate the effects of ZP2 PV on intracellular trafficking of ZP proteins, expression plasmids encoding ZP2ZsGreen or mutZP2ZsGreen fusion protein (c.1691_1694dup [p.C566Wfs*5]) (Supplementary Fig. S1) were transfected into CHO cells and imaged under confocal fluorescent microscopy. Intact ZP2 protein was expressed and diffusely present in the cytoplasm, while truncated ZP2 protein clumped (Fig. 4a). In agreement with immunoblots (Fig. 1e), within cells, truncated ZP2 protein was greater than intact ZP2 protein (Fig. 4a), and flow cytometry confirmed this (Fig. 4b). These results might be a reflection of intracellular sequestration and aggregation of ZP2 protein caused by truncating PV.

a CHO cells transfected with ZP2ZsGreen or mutZP2ZsGreen (c.1691_1694dup [p.C566Wfs*5]) expression vectors encoding full-length or truncated proteins. Cells were fixed and imaged under confocal laser scanning microscopy (1000×). Higher-magnification inserts are images of aggregates with protein lacking C-terminal domains. b Flow cytometry analysis of ZP2ZsGreen or mutZP2ZsGreen fusion proteins in CHO cells. All values are means ± SD from six independent experiments. P = 0.00006 versus ZP2 wild type (WT). c ZP1, ZP3mcherry, or ZP4mcherry expression vectors were cotransfected with ZP2ZsGreen or mutZP2ZsGreen vectors into CHO cells. Cells were fixed and imaged under confocal laser scanning microscopy (1000×). ZP1 protein was imaged by immunofluorescent staining using anti-ZP1 antibody. d Cell lysates were immunoprecipitated with anti-ZP1 antibody. Resultant protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and analyzed by immunoblot using antibodies against ZP1 and ZP2. The scale bars in (a) and (c) represent 10 μm
Reductions in zona thickness (Fig. 2) and increased cytoplasmic staining of ZP1 and ZP3 of patient oocytes (Supplementary Fig. S4) suggests that extracellular secretion of ZP1 and ZP3 proteins was influenced by the truncated ZP2 protein. Thus, we studied a ZP2 PV and interactions between ZP2 and other ZP proteins using cotransfection of expression vectors (Supplementary Fig. S1) in CHO cells. Within cells, intact ZP2 appeared to traffic independently of ZP1, ZP3, and ZP4. However, most truncated ZP2 lacking the C-terminal domains colocalized with ZP3, especially ZP1 and ZP4 (Fig. 4c). Subsequently, a coimmunoprecipitation (co-IP) assay confirmed premature interactions of truncated ZP2 with ZP1 protein within cells prior to secretion (Fig. 4d).
Discussion
The mouse ZP consists of three glycoproteins—mZP1–mZP3—and first appears around growing oocytes in primary follicles, increasing to 7 µm surrounding fully grown, ovulated oocytes. Zp2-null mice can form a thin zona matrix in early follicles composed of mZP1 and mZP3. However, this matrix is not sustained in preovulatory follicles and ovulated oocytes are zona-free and fewer.7 Here, we identified two ZP2 PV in two unrelated women with a similar abnormal ZP phenotype and TFF in routine IVF. Both PV truncated ZP2 protein at the same site and caused a resultant thin ZP lacking ZP2. In contrast to Zp2-null mice, a thin ZP lacking ZP2 was observed in all mature oocytes from both patients and sustained until the blastocyst stage (Supplementary Fig. S2a). The ZP domain (ZPD) of human ZP2 is 40% identical with ZPD of human ZP4 protein, which is absent in mouse ZP, suggesting that ZP4 may substitute for ZP2 in forming a zona matrix as reported in mice.23 Moreover, retrieved oocyte numbers after stimulation were not affected by ZP2 PV, and more than 15 oocytes were obtained at each retrieval (Table 1).
Human and mouse ZP proteins share motifs (Supplementary Fig. S1), including a signal peptide (SS), a “ZP domain” (ZPD), a CFCS, and an EHP, which is followed by a TMD.6 Within the mouse oocyte, mZP proteins traffic separately.24,25 The cleavage of the C-terminal tail at the CFCS is required for secretion of the mZP proteins.26 Once secreted and released into the extracellular milieu, mZP2 and mZP3 polymerize and incorporate into fibrils and the zona matrix. With removal of cytoplasmic tails or additional removal of the TMD, secretion is not affected but failed incorporation of mZP2 and mZP3 into the ZP occurs.25,27,28 With further truncation of the C-terminus up to the CFCS, the proteins fail to be secreted and are retained within the cell.26 It was later found that the EHP sequence between CFCS and TMD is required to prevent premature polymerization of mZP proteins within cells.28 These findings reveal the essential roles of these C-terminus motifs in guaranteeing independent trafficking, secretion, and incorporation of ZP proteins into the ZP. Indeed, our in vitro studies suggested that separate trafficking of ZP proteins within cells was probably disturbed by a mutant ZP2 lacking these crucial C-terminus motifs. Within mutant patient oocytes, ZP2, ZP1, and ZP3 proteins were greater than in normal controls. Next, within transfected CHO cells, truncated ZP2 colocalized with other ZP proteins, but intact ZP2 did not. Moreover, truncated ZP2 coprecipitated with ZP1 using anti-ZP1 antibody, but intact ZP2 did not. Due to the thin ZP that positively stained for ZP1 and ZP3 surrounding mutant oocytes, we speculated that truncated ZP2 caused intracellular sequestration of ZP2, partial retention of the three ZP proteins, and a thin ZP. Previous mouse studies show that a 50% reduction of mZP3 protein assembled into ZP reduced by 50% oocyte ZP thickness.29
Although a thin ZP appeared around mature oocytes, it lacked ZP2 and was defective for sperm binding and penetration and IVF failed. These observations correlate with studies of egg coat–sperm recognition in which an intact ZP2 protein,30 especially its ZP-N1 domain that interacts with sperm, is necessary for human and mouse gamete recognition and penetration via the ZP.23,31
Using family pedigrees and fertile females (I-2 and II-2 in family 1 and I-2 in family 2) who are carriers of ZP2 PV, we proposed an autosomal recessive genetic mode of infertility and excluded the possibility that heterozygous PV in ZP2 cause disease. These data agree with earlier mouse studies that show the presence of a single Zp3 allele reduced by 50% the respective gene product in vivo, but had no effect on reproduction.29 A fertile male (II-2 in family 2) homozygous for ZP2 PV p.C566Wfs*5 confirmed that ZP2 gene biallelic loss-of-function PV did not affect male fertility.
In conclusion, a human ZP phenotype lacking ZP2 protein exists and causes female infertility characterized by morphologically abnormal ZP defective in gamete recognition due to recessive ZP2 loss-of-functionPV. This should facilitate diagnosis of oocyte-borne gamete interaction defects and promote therapeutic developments.
References
Mascarenhas MN, Flaxman SR, Boerma T, et al. National, regional, and global trends in infertility prevalence since 1990: a systematic analysis of 277 health surveys. PLoS Med. 2012;9:e1001356.
Mahutte NG, Arici A. Failed fertilization: is it predictable? Curr Opin Obstet Gynecol. 2003;15:211–8.
Liu DY, Baker HW. Defective sperm-zona pellucida interaction: a major cause of failure of fertilization in clinical in-vitro fertilization. Hum Reprod. 2000;15:702–8.
Rawe VY, Olmedo SB, Nodar FN, et al. Cytoskeletal organization defects and abortive activation in human oocytes after IVF and ICSI failure. Mol Hum Reprod. 2000;6:510–6.
Wassarman PM, Mortillo S. Structure of the mouse egg extracellular coat, the zona pellucida. Int Rev Cytol. 1991;130:85–110.
Gupta SK, Bhandari B, Shrestha A, et al. Mammalian zona pellucida glycoproteins: structure and function during fertilization. Cell Tissue Res. 2012;349:665–78.
Rankin TL, O’Brien M, Lee E, et al. Defective zonae pellucidae in Zp2-null mice disrupt folliculogenesis, fertility and development. Development. 2001;128:1119–26.
Liu C, Litscher ES, Mortillo S, et al. Targeted disruption of the mZP3 gene results in production of eggs lacking a zona pellucida and infertility in female mice. Proc Natl Acad Sci U S A. 1996;93:5431–6.
Rankin T, Talbot P, Lee E, et al. Abnormal zonae pellucidae in mice lacking ZP1 result in early embryonic loss. Development. 1999;126:3847–55.
Lefievre L, Conner SJ, Salpekar A, et al. Four zona pellucida glycoproteins are expressed in the human. Hum Reprod. 2004;19:1580–6.
Conner SJ, Lefievre L, Hughes DC, et al. Cracking the egg: increased complexity in the zona pellucida. Hum Reprod. 2005;20:1148–52.
Huang HL, Lv C, Zhao YC, et al. Mutant ZP1 in familial infertility. N Engl J Med. 2014;370:1220–6.
Chen T, Bian Y, Liu X, et al. A recurrent missense mutation in ZP3 causes empty follicle syndrome and female infertility. Am J Hum Genet. 2017;101:459–65.
Tan YQ, Tu C, Meng L, et al. Loss-of-function mutations in TDRD7 lead to a rare novel syndrome combining congenital cataract and nonobstructive azoospermia in humans. Genet Med. 2017. https://doi.org/10.1038/gim.2017.130
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Seelow D, Schuelke M. HomozygosityMapper2012--bridging the gap between homozygosity mapping and deep sequencing. Nucleic Acids Res. 2012;40:W516–520.
Picelli S, Faridani OR, Bjorklund AK, et al. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc . 2014;9:171–81.
Pelletier C,Keefe DL,Trimarchi JR. Noninvasive polarized light microscopy quantitatively distinguishes the multilaminar structure of the zona pellucida of living human eggs and embryos. Fertil Steril. 2004;81(suppl 1):850–6.
Collins C, Hutchinson G, Kowbel D, et al. The human beta-subunit of rod photoreceptor cGMP phosphodiesterase: complete retinal cDNA sequence and evidence for expression in brain. Genomics. 1992;13:698–704.
Yan L, Yang M, Guo H, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–9.
McLaughlin ME, Sandberg MA, Berson EL, et al. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4:130–4.
Avella MA, Baibakov B, Dean J. A single domain of the ZP2 zona pellucida protein mediates gamete recognition in mice and humans. J Cell Biol. 2014;205:801–9.
Hoodbhoy T, Aviles M, Baibakov B, et al. ZP2 and ZP3 traffic independently within oocytes prior to assembly into the extracellular zona pellucida. Mol Cell Biol. 2006;26:7991–8.
Jimenez-Movilla M,Dean J, ZP2 and ZP3 cytoplasmic tails prevent premature interactions and ensure incorporation into the zona pellucida. J Cell Sci. 2011;124:940–50.(Pt 6).
Williams Z, Wassarman PM. Secretion of mouse ZP3, the sperm receptor, requires cleavage of its polypeptide at a consensus furin cleavage-site. Biochemistry. 2001;40:929–37.
Jovine L, Qi H, Williams Z, et al. The ZP domain is a conserved module for polymerization of extracellular proteins. Nat Cell Biol. 2002;4:457–61.
Jovine L, Qi H, Williams Z, et al. A duplicated motif controls assembly of zona pellucida domain proteins. Proc Natl Acad Sci U S A. 2004;101:5922–7.
Wassarman PM, Qi H, Litscher ES. Mutant female mice carrying a single mZP3 allele produce eggs with a thin zona pellucida, but reproduce normally. Proc Biol Sci. 1997;264:323–8.
Gahlay G, Gauthier L, Baibakov B, et al. Gamete recognition in mice depends on the cleavage status of an egg’s zona pellucida protein. Science. 2010;329:216–9.
Raj I, Sadat Al Hosseini H, Dioguardi E, et al. Structural basis of egg coat-sperm recognition at fertilization. Cell. 2017;169:1315–26 e1317.
Acknowledgments
This work was financially supported by grants from the National Key Research and Development Program of China (2016YFC1000200 to G.L.), the National Natural Science Foundation of China (81222007 and 81471510 to G.L., and 81372627 to L.H.), and the Program for New Century Excellent Talents in University (907010003 to G.L.). We thank the patients and their families and the staff at the Reproductive and Genetic Hospital of CITIC–Xiangya for their assistance. We are grateful to Wen-Bin He at the Institute of Reproductive and Stem Cell Engineering, Central South University, for discussion of molecular genetics experiments. We thank Ju Peng at the Reproductive and Genetic Hospital of CITIC–Xiangya for technical assistance with confocal microscopy.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
DISCLOSURE
The authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Dai, C., Hu, L., Gong, F. et al. ZP2 pathogenic variants cause in vitro fertilization failure and female infertility. Genet Med 21, 431–440 (2019). https://doi.org/10.1038/s41436-018-0064-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0064-y
Keywords
This article is cited by
-
Advances in the study of genetic factors and clinical interventions for fertilization failure
Journal of Assisted Reproduction and Genetics (2023)
-
Effect of In Vitro Maturation of Human Oocytes Obtained After Controlled Ovarian Hormonal Stimulation on the Expression of Development- and Zona Pellucida-Related Genes and Their Interactions
Reproductive Sciences (2023)
-
A novel homozygous C-terminal deletion in BTG4 causes zygotic cleavage failure and female infertility
Journal of Assisted Reproduction and Genetics (2023)
-
Dynein axonemal heavy chain 10 deficiency causes primary ciliary dyskinesia in humans and mice
Frontiers of Medicine (2023)
-
Development and validation of a conventional in vitro total fertilization failure prediction model
Journal of Assisted Reproduction and Genetics (2023)
