
Abstract
Purpose
The high percentage of patients carrying germline mutations makes pheochromocytomas/paragangliomas the most heritable of all tumors. However, there are still cases unexplained by mutations in the known genes. We aimed to identify the genetic cause of disease in patients strongly suspected of having hereditary tumors.
Methods
Whole-exome sequencing was applied to the germlines of a parent–proband trio. Genome-wide methylome analysis, RNA-seq, CRISPR/Cas9 gene editing, and targeted sequencing were also performed.
Results
We identified a novel de novo germline mutation in DNMT3A, affecting a highly conserved residue located close to the aromatic cage that binds to trimethylated histone H3. DNMT3A-mutated tumors exhibited significant hypermethylation of homeobox-containing genes, suggesting an activating role of the mutation. CRISPR/Cas9-mediated knock-in in HeLa cells led to global changes in methylation, providing evidence of the DNMT3A-altered function. Targeted sequencing revealed subclonal somatic mutations in six additional paragangliomas. Finally, a second germline DNMT3A mutation, also causing global tumor DNA hypermethylation, was found in a patient with a family history of pheochromocytoma.
Conclusion
Our findings suggest that DNMT3A may be a susceptibility gene for paragangliomas and, if confirmed in future studies, would represent the first example of gain-of-function mutations affecting a DNA methyltransferase gene involved in cancer predisposition.
Similar content being viewed by others


Introduction
Approximately 75% of neural crest-derived pheochromocytomas (PCCs) and paragangliomas (PGLs) (OMIM 171300) carry mutually exclusive germline (~40%) or somatic (~35%) mutations affecting 1 of at least 20 genes.1 The high percentage of patients for which germline mutations have been found in recent years has turned PCCs and PGLs into the most heritable of all tumors. In addition to the known hereditary cases, there is still a subset of PCC/PGL patients showing clinical features of heritability without germline mutations in the susceptibility genes identified so far. The presence of multiple tumors in some of these patients, in the absence of a family history of the disease, strongly suggests the presence of de novo alterations, recessive inheritance, imprinting, or somatic mosaicism involving precursor cells.
More than one-third of the PCC/PGL disease-causing genes (SDHA, SDHB, SDHC, SDHD, SDHAF2, FH, MDH2 and IDH1) encode Krebs cycle enzymes. Recently, gene expression and methylation profiling have shown that tumors carrying mutations in Krebs cycle-related genes exhibit an identifiable CpG island methylator phenotype (CIMP), caused by the accumulation of specific metabolites.2 These oncometabolites lead to inactivation of DNA demethylases such as TET2 and lysine demethylases, giving rise to epigenetic alterations in the genome that cause global gene expression changes similar to those found in glioblastomas carrying mutations in IDH1 and IDH2.3 In addition, mutations affecting chromatin remodeling genes have recently been found either in the germline or in tumor cells of patients with PCC/PGL.4 However, though global methylation changes are closely related to the development of a substantial subset of PCCs and PGLs, to date no alterations in DNA methyltransferase genes, which are frequently disrupted in cancer, have been reported in PCC/PGL.
In the present study, we identified a de novo germline mutation in the DNA (cytosine-5-)-methyltransferase 3 alpha (DNMT3A) gene in a patient with multiple (n = 7) PGLs. The same mutation was also found subclonally in six additional PGLs. A second germline mutation was identified in a patient with a family history of the disease. The altered methylation profile exhibited by DNMT3A-mutated tissues (both PGLs and lymphocytes) and HeLa cells carrying one of the mutations, and the location of the altered amino acids close to the aromatic cage responsible for binding trimethylated histone H3, together suggest a gain of alternative activities (referred to as gain-of-function) caused by these alterations.
Materials and Methods
Index patient
Whole-exome sequencing was applied to DNA samples from a parent–proband trio. The index case, a 22-year-old woman, was referred to hospital because of persistent dysphonia for 3 years. Multiple laryngeal PGLs (T1) and a right carotid PGL (T2) were detected by single photon emission computed tomography (CT), magnetic resonance imaging (MRI), and octreoscan, and the patient underwent surgery at the age of 23 years. Four additional PGLs were diagnosed subsequently, two jugulotympanic, one in the left carotid, and one normetanephrine-producing paracardiac tumor presenting with impaired fasting glucose and high blood pressure. One jugulotympanic PGL was treated with radiosurgery at age 28 years, and the other was embolized, surgically removed (T3), and treated with radiosurgery at ages 29 and 30 years. The paracardiac and left carotid PGLs were treated with external radiation therapy at ages 30 and 32 years, respectively. The patient reported no family history of PCC or PGL.
Whole-exome sequencing analysis
Whole-exome sequencing was carried out at the National Centre for Genomic Analysis (CNAG) using germline DNA samples obtained from the patient and her parents using FlexiGene DNA kit (Qiagen). Briefly, the Covaris S2 System (Covaris) was used for DNA fragmentation. Exome capture was performed using the SureSelect XT HumanAllExon 50-Mb kit (Agilent Technologies). Exome sequencing at a mean coverage >50× was performed by 75-bp paired-end technology using a HiSeq2000 sequencer (Illumina). The GEM5 and BFAST programs were used to align the reads against the whole human genome (hg19 assembly). To identify single-nucleotide substitutions and small insertions and deletions (indels) the SAMtools program was used (http://samtools.sourceforge.net). Filtering was applied to exclude noncoding substitutions and variants in genomic regions with low mappability, with low depth readings, with the alternative allele present in <20% of reads, or with the alternative allele present only in forward or reverse reads. Variants with genotype quality (GQ) lower than 90% in any of the three exomes were also excluded. The GQ for a variant is a measure, derived from the Phred quality scores, of the probability that the genotype is correct. The higher the GQ value (highest possible value = 99%), the more likely it is that the genotype is correct. Three inheritance models were assessed: de novo, recessive, and dominant with incomplete penetrance. Manual curation using Integrative Genomics Viewer (IGV) software was also applied in the de novo analysis to exclude variants present in at least one of the progenitors or caused by a sequencing artifact. The PredictSNP consensus classifier6 was used to predict the effect of the only nucleotide substitution that passed all filtering steps.
Extended study of PCC/PGL patients
Sanger sequencing of the two exons (8 and 9) encompassing the complete proline–tryptophan–tryptophan–proline (PWWP) domain of DNMT3A was applied to screen for additional germline mutations in a selected series of patients (n=35) without germline alterations in known susceptibility genes and who were strongly suspected of having hereditary PCC/PGL due to the presence of multiple tumors and/or familial antecedents of the disease. The Instituto de Salud Carlos III (ISCIII) ethics committee approved the study, and all the patients provided written informed consent. Also studied for mutations within the PWWP domain of DNMT3A were 95 tumors (52 head and neck PGLs, 28 PCCs, and 15 thoracic–abdominal PGLs) that tested negative for mutations in the known PCC/PGL susceptibility genes and, when it could be assessed, positive for SDHB immunostaining (to rule out the presence of hidden mutations affecting the SDH genes). To test for subclonal DNMT3A mutations, targeted deep sequencing (Illumina) of an amplicon containing the c.896A>T mutation was applied to the 95 tumor DNA samples. Once the library was prepared following the manufacturer’s instructions, next-generation sequencing was performed using the MiSeq desktop sequencer (Illumina). Sequence alignment was carried out using MiSeq Reporter and Illumina VariantStudio software (Illumina). Variant calling was performed using GATK7 and Somatic Variant Caller (Illumina) and identified variants were filtered considering mapping quality, variant score, depth, strand bias, and annotation quality.
DNA methylation assay
DNA from peripheral blood leukocytes and formalin-fixed paraffin-embedded (FFPE) PCCs/PGLs was obtained using the DNeasy kit (Qiagen), following the manufacturer’s instructions. Bisulfite conversion of DNA was performed using the EZ DNA Methylation Kit (Zymo Research) and genome-wide DNA methylation was assayed using the Infinium MethylationEPIC BeadChip (Illumina) at the Centro Nacional de Genotipado (CEGEN-ISCIII) (www.cegen.org), as previously described.8 This BeadChip interrogates over 850,000 methylation sites per sample. Beta values for interrogated CpGs were assigned using the Genome Studio Methylation module and transformed into M values by applying the formula: log2[Beta value/(1-Beta value)]. Negative M values indicate less than 50% methylation and positive M values indicate more than 50% methylation. M values were used for all statistical analyses. The Infinium MethylationEPIC BeadChip (Illumina) was also used to identify specific methylated probes related to the DNMT3A genetic alteration. Three c.896A>T DNMT3A-mutated PGLs (T1, T2, and T3) from the index patient were used for supervised analysis, along with 17 controls (5 SDHB-, 2 HRAS-, 1 NF1-, 1 RET-, 1 VHL-, and 1 IDH3B-mutated PCCs/PGLs, and 6 tumors without a known mutation). Differentially methylated probes were identified using a t test (limma) carried out as implemented in POMELO II.9 To account for multiple hypothesis testing, the significance level (P) was adjusted using Benjamini’s FDR correction.10 Probes with an FDR < 0.05 were considered differentially methylated between classes. Gene functional annotation analysis was performed by applying the web-based applications DAVID (the Database for Annotation, Visualization, and Integrated Discovery).11 and Enrichr.12 to the genes annotated to significantly methylated probes obtained in the supervised analysis. After accounting for duplicate genes (i.e., various probes targeting the same gene), 1,285 individual human genes were recognized by DAVID and included in further analysis. Hierarchical clustering, performed using GeneCluster 2.013 on the 3,092 probes found to be significantly differentially methylated in the supervised analysis, was used to interrogate first the EPIC methylation data from the three DNMT3A-mutated tumors (T1–T3) from the index patient, the c.952C>T DNMT3A-mutated PGL plus the 17 control tumors previously described; and second, EPIC methylation data from lymphocyte DNAs from the c.896A>T DNMT3A-mutated carrier (average of two samples) and her parents (I1 and I2), the c.952C>T DNMT3A-mutated patient, and eight controls (C1–C8). Additionally, we used the same list of probes to interrogate Illumina 450k methylation data from 26 acute myeloid leukemia (AML) samples carrying DNMT3A mutations compared with 30 DNMT3A wild-type (WT) cases.14 Finally, supervised analysis was performed to identify differentially methylated probes between HeLa knock-in (c.896A>T) and HeLa WT cells. The reliability of the clusters obtained with DNMT3A-mutated and no-mutated PCCs/PGLs was verified by consensus clustering, a method that obtains the consensus across multiple runs of a clustering algorithm and assess the stability of the discovered clusters by using resampling techniques.
Reverse-transcriptase PCR and qPCR
Total RNA was isolated from peripheral blood lymphocytes from both DNMT3A-mutation carriers and the parents of the index case using the TriReagent kit (MRC), following the manufacturer’s instructions. First strand complementary DNA (cDNA) was synthesized from 1 μg of total RNA by oligo (dT) 14 primer reverse transcription using Superscript II Reverse Transcriptase (Invitrogen) following the manufacturer’s instructions. Three genes, HOXD13, IRX5, and NKX2-4, were selected for messenger RNA (mRNA) quantification because several CpGs annotated to them were found amongst the differentially methylated in DNMT3A-mutated tumors. HOXD13, IRX5, and NKX2-4 mRNA levels were determined by quantitative polymerase chain reaction (PCR) on a 7500 fast real-time PCR system (Applied Biosystems) using the Universal ProbeLibrary set (https://www.roche-applied-science.com), as described by the manufacturer. Relative mRNA levels were estimated using the 2-CT method15 and normalized using β-glucuronidase (GUS) and β-actin (ACTB) as housekeeping genes. The results are shown as mean ± s.d. (n ≥ 2).
CRISPR/Cas9
A HeLa cell line (provided by Flow Cytometry Core Unit, CNIO, Madrid) was authenticated by short tandem repeat profiling (GenePrint 10 System, Promega) and periodically confirmed to be mycoplasma-free by qPCR. HeLa cells were cultured in Dulbecco’s modified Eagle medium Gluta MAX (DMEM; Invitrogen); supplemented with 10% (v/v) fetal bovine serum (FBS, PAA Laboratories), 1% (v/v) penicillin/streptomycin, and 0.6% (v/v) Fungizone (Gibco); and maintained at 37 °C in a humidified incubator with 5% CO2. Recombinant Cas9 protein and two single-guide RNAs (sgRNAs) (sgRNA-1: AACTGCGGGGCTTCTCCTGGTGG; sgRNA-2: CTGGTGTGGGGGAAACTGCGGGG) were used as previously described.16 Briefly, sgRNAs were designed using the CRISPR MIT software tool (http://crispr.mit.edu) and transcribed in vitro through runoff reactions by T7 RNA polymerase using the HiScribe T7 high-yield RNA synthesis kit (NEB). Transcribed sgRNAs were purified with a column purification kit (RNeasy mini kit, Qiagen, Hilden, Germany), quantified, and quality-checked by denaturing RNA gel electrophoresis. RNP complexes were generate by premixing Cas9 protein (1–8 μg, Integrated DNA Technologies, CA) with in vitro transcribed sgRNA (0.5–8 μg) for 10 min at room temperature. A single-stranded oligodeoxynucleotide with the nucleotide change c.896A>T was purchased from IDT as an ultramer oligo (available upon request). For electroporation, we employed the Neon Transfection System (ThermoFisher Scientific, Waltham, MA). Confluent HeLa cells were trypsinized and washed twice with DPBS, counted, and resuspended in 10 µl Buffer T (ThermoFisher Scientific) per well. In each reaction approximately 100,000 cells were electroporated under the following conditions: 2 pulses of 35-ms width and 1,005 V. Cells were then seeded in a 96-well plate with prewarmed medium. The following day the media was replaced with fresh DMEM. After culture expansion, cells were sorted to a 96-well plate to establish single-cell clones. Detection of knock-in cell clones was performed using Sanger sequencing. Genomic DNA extraction, PCR amplification, and purification of PCR product were performed following standard procedures.
RNA sequencing (RNA-seq)
PolyA+ fraction from total RNA samples was purified and randomly fragmented, converted to double-stranded cDNA, and processed using the TruSeq Stranded mRNA Library Prep Kit (Illumina). Adapter-ligated library was completed by PCR and the resulting purified cDNA library was applied to a flow cell for cluster generation and sequenced on a HiSeq 2500 instrument, following manufacturer protocols. Image analysis, basecalling, and quality score assignment was performed with Real Time Analysis software (Illumina), and Illumina2bam tool, BamToFastq (Bedtools), and SAMtools (http://samtools.sourceforge.net/) were used to convert the files to BAM, FASTQ, and SAM format, respectively. FASTQ files containing the Illumina reads (length = 50) of the samples were submitted to the Nextpresso v.1.8 pipeline using the default parameters. Clustering average for linkage and Euclidean as the distance measure were applied in the hierarchical clustering using Genecluster 2.0, and a standard deviation of 0.5 was assumed.
Immunohistochemistry
An automated immunostaining platform was used (Ventana Discovery XT, Roche) for immunohistochemistry of H3(K9) in 3-μm FFPE sections from the DNMT3A-mutated tumors. Antigen retrieval was first performed with RiboCC (pH6) and endogenous peroxidase was blocked (peroxide hydrogen at 3%). Then, slides were incubated with rabbit polyclonal anti-H3(K9) (1/500; Abcam, ab8898) and following with the corresponding visualization system (OmniMap antirabbit, Ventana, Roche) conjugated with horseradish peroxidase. Immunohistochemical reaction was developed using 3,30-diaminobenzidine tetrahydrochloride (DAB) (ChromoMap DAB, Ventana, Roche). Nuclei were counterstained with Carazzi’s hematoxylin. Finally, the slides was dehydrated, cleared, and mounted with a permanent mounting medium for microscopic evaluation.
Results
Exome sequencing findings
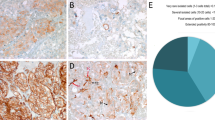
First, we focused on heterozygous gene-coding single-nucleotide substitutions and small insertions and deletions that were present in blood DNA from the patient but absent in germline DNA from both of her parents, following a de novo model. The filtering process applied uncovered a single de novo missense variant (c.896A>T; p.Lys299Ile) in DNMT3A, which was subsequently validated by Sanger sequencing (Fig. 1a). The substitution was not found in the >120,000 exomes in the gnomAD database (http://gnomad.broadinstitute.org/), 570 population-matched unrelated Spanish exomes from the CIBERER Spanish Variant Server (http://csvs.babelomics.org/), or the database of Epigenetic Modifiers (dbEM; http://crdd.osdd.net/raghava/dbem/). The lysine residue affected by the mutation (Lys299) is located within the highly conserved PWWP domain of the protein (Fig. 1b), and all in silico algorithms predict the change to be deleterious (Supplementary Figure S1). In addition, high levels of H3K9me3 were observed in the c.896A>T DNMT3A-mutated tumor compared with controls (Fig. 1c). Though recessive and autosomal dominant with incomplete penetrance models of inheritance were also considered, we gave priority to the de novo model because we found a single mutation in a gene previously associated with various cancers.

(a) Schematic representation of the coding variants passing all filtering steps (with a genotype quality ≥90) that were found in the father (orange square), the mother (green circle), and the patient (blue circle) exomes. Manual curating by the integrative genomic viewer (IGV) excluded 4 of the 5 variants because they were either found in at least one of the progenitors or because they were probably the result of a sequencing artifact. The single de novo variant found (c.896A>T in DNMT3A) was confirmed by Sanger sequencing. (b) PyMOL representation of the interactions between the PWWP domain of DNMT3B and trimethylated K36 in histone H3 (structure deposited in the Protein Data Base with accession code 5CIU). The histone peptide is shown in yellow and the aromatic cage residues in blue. The represented residues are fully conserved in DNMT3A. The lysine residue 232 is equivalent to lysine 299 in DNMT3A and is represented in magenta. The dashed line depicts a hydrogen bond interaction between Lys232 and the carbonyl oxygen of a phenylalanine within the same loop. (c) Highly positive immunohistochemical staining for H3K9me3 in one representative c.896A>T DNMT3A-mutated tumor from the de novo patient (upper panel) compared to a DNMT3A wild-type tumor staining negative (lower panel). The scale bars represent 50 µm
Extended study of PCC/PGL patients and tumors
Considering the recurrence of activating mutations affecting oncogenes, Sanger sequencing–based screening for germline mutations was performed on the sequence encompassing the PWWP domain of DNMT3A. A missense mutation (c.952C>T; p.Arg318Trp) (Supplementary Figure S2A) affecting a highly conserved residue was identified in germline DNA from a woman (54 yr) with two head and neck PGLs and a family history of the disease (her mother was diagnosed with bilateral head and neck PGL). The arginine 318 amino acid is highly conserved, and the mutation (predicted to be deleterious) (Supplementary Figure S2B) introduces a tryptophan that could alter the binding of DNMT3A to trimethylated histone H3 (Fig. 2a). Further study of the PWWP domain in a series of 94 tumors identified three cases with a small mutation peak affecting the same nucleotide as that mutated in the original patient (Table 1). Subsequent analysis by targeted deep sequencing showed a clonal effect, ranging between 14 and 36% of reads, in all three cases, and identified three additional carriers that hadn’t been detected by Sanger sequencing. All c.896A>T DNMT3A mutations were found in head and neck PGLs. The analysis of the corresponding germline DNA showed in all available cases that the mutation was somatic. All available DNMT3A-mutated PGLs showed high immunostaining of H3K9me3.

(a) PyMOL representation of the interactions between the PWWP domain of DNMT3B and trimethlyated K36 in histone H3 (similar to the one depicted in Fig. 2a). The histone peptide is shown in yellow and the aromatic cage residues in blue. The tryptophan that substitutes the arginine 318 amino acid (equivalent to lysine 251 in DNMT3B) in the mutated Arg318Trp-DNMT3A sample is represented in dark red. (b) Hierarchical clustering using methylation data from the 3,092 probes significantly differentially methylated (FDR <0.05) between c.896A>T DNMT3A-mutated and nonmutated tumors. T1–T3: c.896A>T DNMT3A-mutated PGLs. H&N head and neck, PGL paraganglioma, PCC pheochromocytoma, TAP thoracic, abdominal or pelvic
Methylation and expression profiles of DNMT3A-mutated tissues
Supervised DNA methylation analyses revealed 3,092 probes differentially methylated (FDR <0.05) in tumors carrying the c.896A>T DNMT3A mutation, compared with controls. Amongst the differentially methylated probes, an enrichment of homeobox-containing genes (n = 90), dopaminergic neurogenesis, neural crest differentiation, and pattern specification and embryonic morphogenesis processes (Supplementary Table S1), was observed. Probes from 26 Hox genes were amongst those methylated in c.896A>T DNMT3A-mutated tumors. Twenty-one probes targeting the HOXD9-13locus, nine of them targeting HOXD13, were significantly more methylated (Supplementary Figure S3). Downregulation of three of the differentially methylated homeobox-containing genes (HOXD13, NKX2-4, and IRX5) was found by qPCR in c.896A>T DNMT3A-mutated lymphocytes compared with controls (Supplementary Figure S4A). Hierarchical clustering, using data from the 3,092 probes significantly differentially methylated, grouped all the tumors carrying DNMT3A mutations together; the three c.896A>T-mutated tumors, previously used in the supervised comparison, and the tumor carrying the c.952C>T mutation (Fig. 2b). Additionally, following a similar profiling, lymphocyte DNAs from the two DNMT3A mutation carriers were also clustered together and separated from different controls (Supplementary Figure S4B). Moreover, a different global expression profile was observed in c.896A>T DNMT3A-mutated lymphocytes compared with controls by RNA-seq (Supplementary Figure S4C). On the whole, these results demonstrate that both DNMT3A mutations led to a similar and characteristic methylation profile, being independent of the tissue of origin. Finally, based on the same list of differentially methylated probes, all but two AML samples carrying the recurrent DNMT3A mutation affecting p.Arg882 (Supplementary Figure S5A) clustered together. This result suggests that either c.896A>T or c.952C>T and p.Arg882 DNMT3A mutations cause opposite effects on DNA methylation in PCC/PGL and AML, respectively.
The p.Lys299Ile mutation increases DNMT3A DNA methylation capabilities in HeLa cells
CRISPR/Cas9-mediated knock-in introduction of the DNMT3A p.Lys299Ile mutation in HeLa cells caused genome-wide alterations in DNA methylation. Thus, more than 16,000 probes, targeting more than 6,000 different genes, were found differentially methylated (FDR <0.05; fold change >|3.5|) in HeLa cells carrying the mutation compared with WT controls (represented in Supplementary Figure S5B). Amongst probes that were differentially hypermethylated upon the introduction of the mutation, an enrichment of those targeting homeobox-containing genes (n = 52), nervous system development, neuronal system, and signaling genes was observed compared to WT HeLa cells (adjusted p < 0.05) (Supplementary Table S2).
Discussion
DNMT3A encodes one of the two de novo DNA methyltransferases, DNMT3A and DNMT3B, that establish DNA methylation patterns during embryonic development and gametogenesis in mammals.17 Somatic inactivating mutations in DNMT3A were first described causing global hypomethylation in acute myeloid leukemia (AML),18 and later most types of hematological malignancies.19 Other recurrent somatic alterations affecting genes involved in epigenetic regulation are common in myeloid malignancies.20 In fact, the crosstalk between genomic alterations and deregulation of epigenetic pathways is a well-known cause of cancer that is not limited to hematologic malignancies. Thus, alterations in genome-wide gene expression caused by somatic disruption of epigenetic modifying genes lead to the development of several solid tumors, including PCC/PGL.4 However, to date no constitutional alteration of an epigenetic regulator has been reported as a cause of hereditary cancer. As far as we know, the case described herein might be the first example of a Mendelian disorder caused by an alteration of the epigenetic machinery involved exclusively in cancer predisposition. In addition, our finding of subclonal somatic mutations in a substantial number of cases points to the importance of DNMT3A alterations in PGL, especially for tumors located in the head and neck region. However, due to the low allele frequencies of the DNMT3A mutation, it is unlikely that this mutation is the cancer initiating event in these sporadic cases.
Variant p.Lys299Ile affects a highly conserved residue within the PWWP motif of DNMT3A that is involved in both DNA binding and targeting the protein to trimethyl lysine marks (on lysine 36 and 9) in histone H3 (H3K36me3 and H3K9me3, respectively).21,22 The lysine 299 residue is one of the most conserved amino acids within the PWWP domain.23 It is part of the positively charged surface of the protein that interacts with DNA22 It also interacts with the aromatic cage of DNMT3A involved in binding methyllysine histones,22,23 presumably through a hydrogen bond with one of the amino acids. NMR perturbation experiments performed on histone-binding PWWP motifs of two proteins, Pdp1 and BRPF1, showed a high chemical shift for the equivalent lysine residue in both.24,25 The specific methylation of homeobox-containing genes observed in c.896A>T DNMT3A–mutated tissues (PGLs and blood from the patient), further suggests that, instead of a loss of function, the p.Lys299Ile variation is associated with different DNMT3A-related DNA methylation capabilities. The high level of H3K9me3 observed in DNMT3A-mutated tumors, something also found in SDH- and FH-mutated PCC/PGL,26 provides further evidence for the transcriptional repression role of the mutation. The effect on DNA methylation caused by the introduction of the p.Lys299Ile mutation in HeLa cells supports the role of this mutation in changing DNMT3A function, which is consistent with the hypermethylated profile observed in PCC/PGL.
Germline de novo loss-of-function mutations in DNMT3A cause overgrowth syndrome with intellectual disability,27 while inherited compound heterozygous or homozygous mutations in DNMT3B are the most common cause of the ICF (immunodeficiency, centrosome instability, and facial anomalies) syndrome in humans.28 The patient described herein did not have any clinical feature related to the overgrowth or ICF syndromes. Overgrowth syndromes are also caused by constitutional mutations in two well-known AML-related genes that encode important histone methyltransferases: EZH2 (Weaver syndrome)29 and NSD1 (Sotos syndrome and Beckwith–Wiedemann syndrome).30 Moreover, germline de novo inactivating mutations in ASXL1, which are frequently found in myelodysplastic syndromes and in AML, also cause a growth disorder called Bohring–Opitz syndrome.31 In all cases of these syndromes, genome-wide hypomethylation was observed and no cancer development was reported. Taken together, our data suggest that, unlike constitutional inactivation of DNMT3A, DNMT3A gain-of-function might be associated with PCC/PGL development. Interestingly, both gain-of-function and loss-of-function EZH2 mutations occur in cancers with distinct subtypes,32 including PCC.4
Several Hox family genes, which encode crucial players during embryogenesis, neural crest patterning, and hematopoietic development, are hypomethylated and upregulated in hematological malignancies carrying somatic DNMT3A mutations,33,34 and have been proposed as targets for DNMT3A-dependent methylation. However, Hox genes could be important for both oncogenesis and tumor suppression, depending on the context. Thus, hypermethylation of several Hox genes has been widely found in breast, lung, prostate, urothelial, and ovarian tumors,35 and methylation of HOXA9 has been found in both neuroblastoma and PCC.36 The presence of methylation of 26 Hox genes in DNMT3A-mutated tumors suggests that these transcription factors may have a tumor suppressor role in PCC/PGL. Our results are also suggestive of a critical role of methylation of homeobox-containing genes in PCC/PGL development. So far, ~300 homeobox genes have been identified in the human genome and the aberrant expression of several members of this superfamily of transcription factors has been implicated in tumor initiation and metastasis in many human tumors.37 Both losses and gains of homeobox gene expression are related to tumorigenesis, and DNMT3A mutations are known to be associated with altered expression of homeobox-containing genes.38 Of note, this latter is also observed in our model of HeLa cells carrying the DNMT3A p.Lys299Ile mutation. Interestingly, 65 differentially methylated homeobox-containing genes found in c.896A>T DNMT3A-mutated tumors were found differentially methylated in FLT3-ITD/DNMT3A-mutated compared with FLT3-ITD/DNMT3A-WT AMLs.38 The methylation described herein in DNMT3A-mutated PGLs and blood samples of mammalian key neural crest determination genes, such as PAX3, FOXD3, WT1, and the ZIC genes, stresses the relevance of repression of autonomic lineage cell differentiation in PCC/PGL, which is also observed in HIF2A-mutated PCCs.39
In summary, we describe herein the first example of gain-of-function mutations affecting a DNA methyl transferase gene potentially involved in cancer predisposition. More cases and further studies, such as mutation segregation analysis and additional functional assays, are necessary to more precisely determine the role of DNMT3A alterations in PCC/PGL susceptibility.
References
Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14:108–19.
Letouze E, Martinelli C, Loriot C, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–52.
Noushmehr H, Weisenberger DJ, Diefes K, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–22.
Toledo RA, Qin Y, Cheng ZM, et al. Recurrent mutations of chromatin-remodeling genes and kinase receptors in pheochromocytomas and paragangliomas. Clin Cancer Res. 2016;22:2301–10.
Marco-Sola S, Sammeth M, Guigo R, Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat Methods. 2012;9:1185–8.
Bendl J, Stourac J, Salanda O, et al. PredictSNP: robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput Biol. 2014;10:e1003440.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Bibikova M, Le J, Barnes B, et al. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1:177–200.
Morrissey ER, Diaz-Uriarte R. Pomelo II: finding differentially expressed genes. Nucleic Acids Res. 2009;37:W581–586.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–84.
Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128.
Reich M, Ohm K, Angelo M, Tamayo P, Mesirov JP. GeneCluster 2.0: an advanced toolset for bioarray analysis. Bioinformatics. 2004;20:1797–8.
Ley TJ, Miller C, Ding L, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8.
Torres-Ruiz R, Martinez-Lage M, Martin MC, et al. Efficient recreation of t(11;22) EWSR1-FLI1+in human stem cells using CRISPR/Cas9. Stem Cell Rep. 2017;8:1408–20.
Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395–402.
Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33.
Yang L, Rau R, Goodell MA. DNMT3A in haematological malignancies. Nat Rev Cancer. 2015;15:152–65.
Shih AH, Abdel-Wahab O, Patel JP, Levine RL. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612.
Dhayalan A, Rajavelu A, Rathert P, et al. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem. 2010;285:26114–20.
Rondelet G, Dal Maso T, Willems L, Wouters J. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J Struct Biol. 2016;194:357–67.
Wu H, Zeng H, Lam R, et al. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE. 2011;6:e18919.
Vezzoli A, Bonadies N, Allen MD, et al. Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol. 2010;17:617–9.
Qiu Y, Zhang W, Zhao C, et al. Solution structure of the Pdp1 PWWP domain reveals its unique binding sites for methylated H4K20 and DNA. Biochem J. 2012;442:527–38.
Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget. 2015;6:38777–88.
Tatton-Brown K, Seal S, Ruark E, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014;46:385–8.
Hansen RS, Wijmenga C, Luo P, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 1999;96:14412–7.
Tatton-Brown K, Hanks S, Ruark E, et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget. 2011;2:1127–33.
Baujat G, Rio M, Rossignol S, et al. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am J Hum Genet. 2004;74:715–20.
Hoischen A, van Bon BW, Rodriguez-Santiago B, et al. De novo nonsense mutations in ASXL1 cause Bohring-Opitz syndrome. Nat Genet. 2011;43:729–31.
Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34.
Yan XJ, Xu J, Gu ZH, et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309–15.
Qu Y, Lennartsson A, Gaidzik VI, et al. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics. 2014;9:1108–19.
Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nat Rev Cancer. 2010;10:361–71.
Margetts CD, Morris M, Astuti D, et al. Evaluation of a functional epigenetic approach to identify promoter region methylation in phaeochromocytoma and neuroblastoma. Endocr Relat Cancer. 2008;15:777–86.
Abate-Shen C. Homeobox genes and cancer: new OCTaves for an old tune. Cancer Cell. 2003;4:329–30.
Meyer SE, Qin T, Muench DE, et al. Dnmt3a haploinsufficiency transforms Flt3-ITD myeloproliferative disease into a rapid, spontaneous, and fully-penetrant acute myeloid leukemia. Cancer Discov. 2016;6:501–15.
Toledo RA, Qin Y, Srikantan S, et al. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2013;20:349–59.
Acknowledgements
This work was supported by the Instituto de Salud Carlos III (ISCIII), through the “Acción Estratégica en Salud” (AES) (projects PI15/00783 to A.C., PI14/00240 to M.R., and PI14/01884 to S.R.-P., cofounded by the European Regional Development Fund (ERDF)). M.C.-F. is a predoctoral fellow of the Severo Ochoa Program. We thank Antonio Galarreta for his help with the validation of the exome sequencing findings. We thank Maria Jesús Artiga and Manuel Morente for their help in obtaining tumor samples, collected from Spanish hospitals through the Spanish National Tumor Bank Network (CNIO).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Remacha, L., Currás-Freixes, M., Torres-Ruiz, R. et al. Gain-of-function mutations in DNMT3A in patients with paraganglioma. Genet Med 20, 1644–1651 (2018). https://doi.org/10.1038/s41436-018-0003-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0003-y
Keywords
This article is cited by
-
Research in the genetics of pheochromocytoma and paraganglioma: a bibliometric analysis from 2002 to 2022
Clinical and Experimental Medicine (2023)
-
Methylation of recombinant mononucleosomes by DNMT3A demonstrates efficient linker DNA methylation and a role of H3K36me3
Communications Biology (2022)
-
Mechanisms of chromatin-based epigenetic inheritance
Science China Life Sciences (2022)
-
Identification of three novel DNMT3A mutations with compromising methylation capacity in human acute myeloid leukaemia
Molecular Biology Reports (2022)
-
Germline Abnormalities in DNA Methylation and Histone Modification and Associated Cancer Risk
Current Hematologic Malignancy Reports (2022)
