
Abstract
Background/objectives
To compare the clinical and optical coherence tomography (OCT) characteristics of autosomal dominant optic atrophy (ADOA) and normal tension glaucoma (NTG) in Chinese patients.
Subjects/methods
Twenty-four unrelated patients with ADOA and 21 unrelated patients with NTG, younger than 30 years, were enrolled in this study. Data regarding the demographic and clinical characteristics of the patients were collected, and their peripapillary retinal nerve fibre layer (RNFL) and macular ganglion cell complex (GCC) thicknesses were evaluated using OCT. Sequencing of genes associated with neuro-ophthalmic disorders was performed for all patients.
Results
The average age at onset of the ADOA group (13.92 ± 10.73 years) was significantly younger than that of the NTG group (23.67 ± 4.98 years, P = 0.002). Best-corrected visual acuity was significantly poorer in the ADOA group (0.75 ± 0.32) than in the NTG group (0.16 ± 0.19, P < 0.001). The average peripapillary RNFL thickness and the RNFL thicknesses in the temporal upper, temporal lower, and nasal lower sectors were significantly thinner in the ADOA group than in the NTG group (all P < 0.05). Moreover, the macular GCC thickness of the ADOA group was significantly thinner than that of the NTG group (P < 0.001). Twenty-three OPA1 variants (11 novel OPA1 variants) and one OPA3 variant were detected in 24 patients with ADOA.
Conclusions
Our study revealed a distinct difference between the patterns of RNFL and GCC loss in ADOA and NTG, which will help to differentiate ADOA from NTG in young patients. Additionally, this study expanded the genetic spectrum of ADOA.
Similar content being viewed by others
Introduction
Optic atrophy is a common finding in many optic neuropathies, including hereditary optic neuropathies and glaucoma. Autosomal dominant optic atrophy (ADOA, OMIM: 165500) is one of the most common types of hereditary optic neuropathies [1]. ADOA typically presents as insidious and progressive vision loss, with temporal optic disc pallor, colour vision deficits, and central or paracentral visual field defects [2, 3]. The prevalence of ADOA is estimated to be between 1:10,000 and 1:30,000 worldwide [4]. OPA1 (OMIM: 605290) is the main causative gene in ADOA and is responsible for ~60–80% of all cases of ADOA [4].
Normal tension glaucoma (NTG) is a type of primary open angle glaucoma characterised by glaucomatous disc changes, progressive peripapillary retinal nerve fibre layer (RNFL) thinning, typical visual field defects, open anterior chamber angles, and intraocular pressure (IOP) ≤ 21 mmHg [5]. The estimated prevalence of NTG ranges from 0.36% to 1.98% worldwide [6].
As both ADOA and NTG are characterised by the loss of retinal ganglion cells (RGCs) and their axons, they have many overlapping clinical manifestations and are often misdiagnosed in clinical practice [7]. Clinically, ADOA is differentiated from NTG based on its young age at onset, central vision loss sparing of the peripheral fields, and family history [7, 8]. However, some patients with ADOA may experience vision loss in adulthood, whereas some patients with NTG may also present with visual field problems at a young age [9]. Disc excavation of the optic nerve head (ONH) is a typical feature of glaucoma; however, it can be also observed in ADOA [8, 10]. Furthermore, both NTG and ADOA are characterised by diffuse visual field loss in the advanced stages. Therefore, ADOA is one of the most important differential diagnoses of NTG [7, 11].
Optical coherence tomography (OCT) is a common imaging method used to investigate the loss of RGCs and their axons. Due to the differences in mechanisms of damage to RGCs in ADOA and NTG, the patterns of RNFL and ganglion cell complex (GCC) thinning observed in ADOA and NTG are different. Thus, aside from evaluating visual field defects, ONH morphology, age at onset, and genetic testing, OCT examination may help distinguish ADOA from NTG.
In this study, we compared the clinical manifestations and OCT features of young patients (<30 years old) with NTG with those of patients with ADOA. In addition, we evaluated the genetic spectrum of Chinese ADOA patients.
Methods
Participants and examinations
We recruited patients diagnosed with ADOA or NTG at the Ophthalmology Department of Eye & ENT Hospital of Fudan University between 2013 and 2020. This study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Review Board of Eye & ENT Hospital of Fudan University. Written informed consents were obtained from all patients or their legal guardians.
All patients with ADOA included in this study met the following criteria [3, 11, 12]: (1) gradual, bilateral vision loss, dyschromatopsia, (2) optic atrophy, bilateral optic disc pallor on fundus examination, (3) optic atrophy unexplained by any other diseases including infectious, ischaemic, toxic, compression, metabolic etiology, and congenital abnormalities, (4) OPA1 or OPA3 mutations identified by using gene testing and (5) absence of other ocular disorders that could severely affect visual acuity. The inclusion criteria for young patients with NTG were as follows [13]: (1) glaucomatous optic disc damage and glaucomatous visual field defects, (2) open anterior chamber angle, (3) IOP ≤ 21 mmHg, (4) age at diagnosis ≤30 years, (5) absence of any secondary glaucoma, absence of prior history of long-term use of steroid drugs, ocular trauma, uveitis or any kinds of diseases with IOP elevation, (6) absence of other ocular disorders that could severely affect visual acuity, (7) ancillary laboratory tests with no evidence of infectious, ischaemic, toxic, and metabolic etiology, (8) brain/orbit MRI showing no compression lesions and (9) absence of congenital abnormalities of optic disc development. All the NTG patients in our cohort were evaluated by glaucoma specialists and neuro-ophthalmologists together and were followed up for an average of 5 years to exclude other possible diseases.
The detailed medical and family histories of the participants, including age at onset and diagnosis, symptoms at onset, and ocular manifestations, were recorded. All patients underwent a complete ophthalmologic examination, including measurement of best-corrected visual acuity (BCVA), slit-lamp biomicroscopy, ophthalmoscopy, gonioscopy, colour fundus photography, B-mode ultrasonography, and IOP measurement (Goldmann applanation tonometer). Perimetry was performed for cooperative children and adults using Octopus 101 (Haag-Streit, Inc., Köniz, Switzerland) or Humphrey Visual Field Analyzer 750 (Zeiss Humphrey Systems, Dublin, CA, USA).
Optical coherence tomography examination
RNFL and GCC were analysed using spectral domain OCT equipment (RTVue-XR Avanti; Optovue, Fremont, CA, USA). RNFL and GCC thickness and cup/disc ratio (C/D ratio) parameters were calculated using the system’s built-in software. The measured parameters included average RNFL thickness, average RNFL thickness in each hemisphere (inferior and superior), RNFL thickness in each regional sector (superotemporal, inferotemporal, temporal upper, temporal lower, superonasal, inferonasal, nasal upper, and nasal lower), average GCC thickness, average GCC thickness in each hemisphere (inferior and superior), and C/D ratio.
Genetic analysis
Whole blood samples of all the patients in both ADOA and NTG groups and their available first-degree relatives were collected for genomic DNA extraction. Genetic testing was performed using next-generation sequencing. A panel of 194 genes associated with hereditary neuro-ophthalmic diseases was sequenced using the Illumina NovaSeq 6000 (Illumina, Inc., San Diego, CA, USA) sequencing system with 200× average depth. Segregation in available relatives was verified using Sanger sequencing.
The reference sequences NM_015560.2 (OPA1) and NM_001017989.3 (OPA3) were used for mutation analysis. The detected mutations were annotated using ANNOVAR [14] and the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/), and 1000 Genomes Project (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp) databases. All detected variants were verified using Sanger sequencing. The novel mutations were classified as pathogenic, likely pathogenic, variant of uncertain significance, likely benign, and benign according to the American College of Medical Genetics and Genomics guidelines [15]. The pathogenicity of detected missense mutations was assessed using Polymorphism Phenotyping 2 (PolyPhen2) [16] and Sorting Intolerant from Tolerant (SIFT) [17].
Statistical analysis
Statistical analysis was performed using SPSS statistical software (version 20; SPSS Inc, Chicago, IL). Data were presented as mean ± standard deviation. The differences between the clinical parameters of the two groups were examined using the Mann–Whitney U test or one-way analysis of variance according to the results of the Kolmogorov–Smirnov test for normality. A mixed model was established using the generalised estimating equation to compare the OCT parameters between the two groups (corrected for measurements of two eyes). The chi-square test was used to compare the sex distributions between the two groups. P < 0.05 was considered statistically significant.
Results
Twenty-four unrelated patients with a genetic diagnosis of ADOA and 21 unrelated young patients with NTG were enroled in this study. The demographic and clinical features of all the patients are summarised in Supplementary Table 1. The ADOA group had a significantly younger mean age at onset (13.92 ± 10.73 years) than the NTG group (23.67 ± 4.98 years, P = 0.002). There were no significant differences in sex distribution between the two groups. Nine patients with ADOA (9/24, 37.5%) had family histories of poor vision or dyschromatopsia, whereas one patient with NTG (1/21, 4.8%) had a family history of glaucoma.
Ophthalmological characteristics
In the ADOA group, patients presented with reduced visual acuity between the ages of 3 and 36 years; 58.3% (14/24) of the patients in this group had an age at onset younger than 10 years old. The BCVA of the ADOA group (0.75 ± 0.32) was significantly poorer than that of the NTG group (0.16 ± 0.19, P = 0.002). There were no significant differences in IOP between the two groups. Temporal or diffused optic disc pallor was observed in the fundus of patients with ADOA, whereas pathological cupping of the ONH, thinning of the neuroretinal rim, and nerve fibre layer defects were observed in the fundus of patients with NTG. The perimetry results of the patients with ADOA mainly showed central scotoma, paracentral scotoma, and mild diffuse decreases in visual sensitivity. In the NTG group, the observed visual field defects had various patterns, including paracentral scotoma, nasal step, arcuate scotoma, and temporal wedge (Fig. 1).

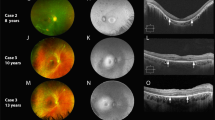
Patients with autosomal dominant optic atrophy (A, B) show temporal optic disc pallor in optic disc photographs, central scotoma, mild diffuse decreases in visual sensitivity, and reduction in papillomacular bundle thickness in optical coherence tomography (OCT) images. Patients with normal tension glaucoma (C, D) show optic nerve head cupping and nerve fibre layer defects in optic disc photographs, nasal visual field defects, and diffuse reduction in retinal nerve fibre layer (RNFL) thickness or asymmetric reduction in RNFL thickness in OCT images. The different colour in OCT images reflects different thickness of RNFL and ganglion cell complex. Red colour means the thickness is outside normal, yellow colour means borderline, and green colour means within normal. SN superonasal, NU nasal upper, NL nasal lower, IN inferonasal, IT inferotemporal, TL temporal lower, TU temporal upper, ST superotemporal.
Optical coherence tomography characteristics
The patterns of the loss of RGCs and their axons were measured in 16 patients with ADOA (31 eyes) and 21 patients with NTG (42 eyes) using OCT (Table 1, Figs. 1, 2). Eight patients with ADOA did not undergo OCT because of the lack of cooperation. RNFL analysis showed that the thicknesses in the nasal lower, temporal lower, and temporal upper sectors were significantly thinner in the ADOA group (51.94 ± 11.95, 35.65 ± 9.51, and 45.16 ± 21.87 μm) than in the NTG group (59.55 ± 14.32, 58.33 ± 21.20, and 66.57 ± 18.62 μm; P = 0.042, P < 0.001, and P < 0.001, respectively; Fig. 3). Furthermore, the average total RNFL thickness of the ADOA group (69.39 ± 11.35 μm) was thinner than that of the NTG group (78.96 ± 13.49 μm; P = 0.013). The RNFL thicknesses in the superotemporal, inferotemporal, inferonasal, nasal upper, and superonasal sectors were not significantly different between the two groups. GCC analysis showed that the ADOA group had significantly lower GCC thickness in all sectors of the foveal region than did the NTG group (all P < 0.001). ONH analysis revealed that the C/D ratio of the NTG group was significantly larger than that of the ADOA group (P = 0.005).

A Optical coherence tomography (OCT) images of three patients with autosomal dominant optic atrophy (ADOA) showing reduced papillomacular bundle thickness. B OCT images of three patients with normal tension glaucoma (NTG) showing reduced inferior RNFL and GCC thickness. The different colour in OCT images reflects different thickness of RNFL and GCC. Red colour means the thickness is outside normal, yellow colour means borderline, and green colour means within normal. SN superonasal, NU nasal upper, NL nasal lower, IN inferonasal, IT inferotemporal, TL temporal lower, TU temporal upper, ST superotemporal.

The average RNFL thickness and sectoral (superonasal, nasal upper, nasal lower, inferonasal, inferotemporal, temporal lower, temporal upper, and superotemporal) RNFL thickness in the autosomal dominant optic atrophy group (ADOA) (shown in grey) and the normal tension glaucoma group (NTG) (shown in red). Error bars indicate the standard deviation of the mean. Asterisks indicate significance. SN superonasal, NU nasal upper, NL nasal lower, IN inferonasal, IT inferotemporal, TL temporal lower, TU temporal upper, ST superotemporal, AVG average. *P < 0.5; ***P < 0.001.
Genetic analysis
All patients underwent genetic analysis using next-generation sequencing. No mutations in OPA1/OPA3 were detected in the NTG group. In the ADOA group, 23 pathogenic variants of OPA1 were detected in 23 patients, and 1 pathogenic variant of OPA3 was identified in 1 patient (Table 2). Apart from one patient with two heterozygous OPA1 mutations (c.1516+1 G>T and c.1498G>T), all the other patients had one heterozygous mutation. No mitochondrial DNA mutations associated with Leber hereditary optic neuropathy were detected in the ADOA group. Among the 24 patients who showed genetic mutations, 2 carried de novo mutations, 9 carried heterozygous mutations inherited from their parents, and 13 patients had no available genetic samples from parents.
Among the 23 detected OPA1 mutations, there were 8 missense (8/23, 34.8%), 6 frameshift (6/23, 26.1%), 6 nonsense (6/23, 26.1%), and 3 splicing mutations (3/23, 13.0%). The frameshift mutation c.2708_2711delTTAG (p.Val903Glyfs*3) was detected in two patients; however, all other mutations were detected only in one patient. Eleven of the mutations were novel, including p.Ser107X, p.Asp273Metfs*35, p.291delVal, p.Ala500Ser, p.Leu546Pro, p.Glu558Valfs*3, p.Glu680X, p.Gly647Glufs*24, c.2496+1G>A, p.His844Tyr, and p.Asp950Metfs*18, whereas the other twelve have been reported previously [12, 18,19,20,21,22,23,24,25]. An OPA3 mutation (c.123C>G, p.Ile41Met) that has been previously detected in three Chinese patients with ADOA was detected in one patient in the present study [26, 27]. The PolyPhen2 and/or SIFT prediction showed that except for the p.His844Tyr OPA1 variant, most detected missense variants were highly deleterious to OPA1 or OPA3.
Discussion
The present study compared the clinical manifestations and OCT characteristics of ADOA and NTG to help differentiate these conditions. In particular, patients with ADOA can be misdiagnosed with NTG because of the similarities in the presentations between the two diseases [28, 29].
The age at onset for the ADOA group in the present study was 13.92 ± 10.73 years and 58.3% of the patients presented with reduced visual acuity in the first decade of life. This finding is consistent with a previous meta-analysis, which showed that ADOA occurred before 10 years of age in more than two-thirds of analysed cases [9]. NTG is prevalent among adults older than 40 years; thus, young age at onset may differentiate most patients with ADOA from those with NTG. However, the diagnosis of ADOA may be difficult when it presents in adults. Likewise, NTG may be difficult to diagnose when it presents in young people. Besides, some literatures showed that the incidence of NTG was increasing in young Asian patients [30,31,32,33]. Therefore, we enroled patients with NTG who were younger than 30 years to determine the differences between the features of NTG and ADOA more accurately. Notably, although we enroled young patients with NTG (23.67 ± 4.98 years), the average age at onset of the ADOA group (13.92 ± 10.73 years) was still significantly younger than that of the NTG group (P = 0.002).
Most patients with ADOA presented to our clinic with poor BCVA (0.75 ± 0.32) and complained of vision loss. However, most patients with NTG visited our clinic for suspected glaucomatous ONH damage noted in regular health examinations. The BCVA of the NTG group (0.16 ± 0.19) was significantly better than that of the ADOA group (0.75 ± 0.32) (P = 0.002). The poor BCVA observed in patients with ADOA is caused by the impairment of the papillomacular bundle in the early stage of the disease [34].
ADOA and NTG have distinct characteristic fundus manifestations. In the present study, the patients with ADOA often presented with temporal or total optic disc pallor, whereas those with NTG frequently showed pathological ONH cupping, thinning of the neuroretinal rim, and nerve fibre layer defects. However, the disc morphology of some patients with ADOA also showed a C/D ratio > 0.5 and/or deep excavation, which might confuse the clinical diagnosis of the disease [8]. Thus, we evaluated the OCT features of the patients to reveal the differences between the two diseases.
OCT is useful for the quantitative assessment of RNFL and GCC thickness in all sectors. However, the differences in OCT features between ADOA and NTG have not been investigated. ADOA predominantly involves damage to the macular RGCs and papillomacular bundle [35]. Therefore, patients with ADOA often present with diffuse GCC thinning and reduction in RNFL thickness in the temporal quadrant during OCT examination [36,37,38,39]. Meanwhile, glaucomatous damage of the arcuate bundles and macular often occurs early in NTG [40, 41]. Reduction in RNFL thickness in the inferotemporal segment or the inferior quadrant has been reported to be the best indicator for early detection of glaucoma [42, 43]; moreover, early changes in macular thickness can be observed in NTG using OCT [40]. These differential patterns of RNFL and GCC thinning in ADOA and NTG result from the different mechanisms of damage to the RGC in the two diseases. The pathogenesis of ADOA includes the mitochondrial dysfunction of RGCs [2, 3], and the main mechanisms of glaucomatous neurodegeneration in NTG include a lower tolerance of normal IOP, vascular dysregulation, and high translaminar pressure gradient [44]. In the present study, a significant reduction in the average RNFL thickness and RNFL thicknesses in the nasal lower, temporal lower, and temporal upper sectors was observed in patients with ADOA compared with patients with NTG. The diffuse thinning of the macular GCC can also differentiate ADOA from NTG. Therefore, observation of RNFL thinning in the temporal lower, temporal upper, and nasal lower sectors along with GCC thinning on OCT might indicate ADOA.
Regarding perimetry, the patients with ADOA in the present study often presented with central or paracentral scotoma but rarely presented with peripheral visual field defects. Mild diffuse decrease in visual sensitivity was also common among the patients with ADOA, even in those with severe RNFL defects observed on OCT images. The mismatch between visual field defects and OCT features is an important indicator of ADOA. In the NTG group, the visual field defects were often consistent with RNFL thinning seen on OCT images. The patients often presented with peripheral visual field defects in the early stage of the disease. Therefore, the combined analysis of visual field and OCT examination results is essential for the differentiation of ADOA from NTG.
To date, 13 genes or loci have been identified to cause ADOA (https://www.omim.org/). OPA1 and OPA3 are the two most common causative genes. OPA1 is responsible for approximately 60%–80% of all ADOA cases [4]. In the present study, 23 OPA1 variants and 1 OPA3 variant were detected in 24 patients with ADOA. The OPA1 gene, located on chromosome 3q29, comprises 32 coding exons and has 8 isoforms (https://www.ncbi.nlm.nih.gov/gene/4976) [20]. As a dynamin-related GTPase protein, OPA1 plays a key role in maintaining the mitochondrial structure and function [2, 3]. To date, more than 400 OPA1 variants have been identified [34]. Haploinsufficiency is the main pathogenic mechanism of OPA1 variants. OPA1 variants can impact mitochondrial fusion, cristae structure remodelling, mitochondrial respiration activity, and control of apoptosis [2, 3, 34]. Among the 23 OPA1 mutations detected in the present study, 52.2% (12/23, 6 frameshift mutations and 6 nonsense mutations) induced premature translation termination and resulted in haploinsufficiency. These variants could be harmful to OPA1 transcription and translation and ultimately damage mitochondrial function. The hot spot mutation c.2708_2711delTTAG, which was detected in approximately 17% of cases in the eOPA1 database [2], was also detected in two of the patients in the present study. Except for c.2708_2711delTTAG, all other variants were detected only in one patient each, consistent with previous studies, indicating that OPA1 mutations are mostly family-specific [2, 34]. Although OPA1 polymorphisms are reported to be associated with NTG in Caucasians [45,46,47], the relationship could not be consistently replicated in other studies [48, 49]. The contribution of OPA1 to the pathogenesis of NTG is still unclear. OPA3, which is an outer mitochondrial membrane lipid metabolism regulator, is another gene that causes ADOA. To date, five OPA3 variants have been detected in six Chinese patients [26, 27]. Interestingly, the OPA3 variant (c.123C>G) detected in one of the patients in the present study has been previously detected in three Chinese patients [26, 27]. The OPA3 variant could harm the normal function of OPA3, including maintenance of energy metabolism and balancing of apoptosis [3]. Genetic testing is essential for the differentiation of ADOA from NTG when the clinical diagnosis is difficult.
In conclusion, this study demonstrated that the average RNFL thickness and RNFL thicknesses in the temporal lower, temporal upper, and nasal lower sectors were thinner in patients with ADOA than in those with NTG. Additionally, GCC thickness was more likely to be reduced in ADOA than in NTG. The differences in OCT features between the two diseases may assist in their differential diagnoses. This study also presented the genetic spectrum of Chinese patients with ADOA. Due to the overlapping clinical features of ADOA and NTG, genetic testing and complete ophthalmic examinations should be performed in patients suspected to have ADOA or young patients suspected to have NTG.
Summary
What was known before
-
ADOA is an important differential diagnosis of NTG.
-
They have many overlapping clinical manifestations and are often misdiagnosed in clinical practice.
What this study adds
-
Our study revealed a distinct difference between the patterns of RNFL and GCC loss in ADOA and NTG, which will help to differentiate ADOA from NTG in young patients.
-
Additionally, this study expanded the genetic spectrum of ADOA.
Change history
12 April 2022
The figures were changed to color figures in pdf.
References
Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies—disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114.
Amati-Bonneau P, Milea D, Bonneau D, Chevrollier A, Ferre M, Guillet V, et al. OPA1-associated disorders: phenotypes and pathophysiology. Int J Biochem Cell Biol. 2009;41:1855–65.
Lenaers G, Hamel C, Delettre C, Amati-Bonneau P, Procaccio V, Bonneau D, et al. Dominant optic atrophy. Orphanet J Rare Dis. 2012;7:46.
Le Roux B, Lenaers G, Zanlonghi X, Amati-Bonneau P, Chabrun F, Foulonneau T, et al. OPA1: 516 unique variants and 831 patients registered in an updated centralized Variome database. Orphanet J Rare Dis. 2019;14:214.
Kim KE, Park KH. Update on the prevalence, etiology, diagnosis, and monitoring of normal-tension glaucoma. Asia Pac J Ophthalmol. 2016;5:23–31.
Zhao J, Solano MM, Oldenburg CE, Liu T, Wang Y, Wang N, et al. Prevalence of normal-tension glaucoma in the Chinese population: a systematic review and meta-analysis. Am J Ophthalmol. 2019;199:101–10.
Fournier AV, Damji KF, Epstein DL, Pollock SC. Disc excavation in dominant optic atrophy: differentiation from normal tension glaucoma. Ophthalmology. 2001;108:1595–602.
Votruba M, Thiselton D, Bhattacharya SS. Optic disc morphology of patients with OPA1 autosomal dominant optic atrophy. Br J Ophthalmol. 2003;87:48–53.
Ham M, Han J, Osann K, Smith M, Kimonis V. Meta-analysis of genotype-phenotype analysis of OPA1 mutations in autosomal dominant optic atrophy. Mitochondrion. 2019;46:262–9.
Katz BJ. Disc excavation in dominant optic atrophy. Ophthalmology. 2002;109:1947.
Kim GN, Kim JA, Kim MJ, Lee EJ, Hwang JM, Kim TW. Comparison of lamina cribrosa morphology in normal tension glaucoma and autosomal-dominant optic atrophy. Investig Ophthalmol Vis Sci. 2020;61:9.
Toomes C, Marchbank NJ, Mackey DA, Craig JE, Newbury-Ecob RA, Bennett CP, et al. Spectrum, frequency and penetrance of OPA1 mutations in dominant optic atrophy. Hum Mol Genet. 2001;10:1369–78.
Chen Y, Hughes G, Chen X, Qian S, Cao W, Wang L, et al. Genetic variants associated with different risks for high tension glaucoma and normal tension glaucoma in a Chinese population. Investig Ophthalmol Vis Sci. 2015;56:2595–600.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Baris O, Delettre C, Amati-Bonneau P, Surget MO, Charlin JF, Catier A, et al. Fourteen novel OPA1 mutations in autosomal dominant optic atrophy including two de novo mutations in sporadic optic atrophy. Hum Mutat. 2003;21:656.
Liskova P, Tesarova M, Dudakova L, Svecova S, Kolarova H, Honzik T, et al. OPA1 analysis in an international series of probands with bilateral optic atrophy. Acta Ophthalmol. 2017;95:363–9.
Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet. 2001;109:584–91.
Zhang J, Yuan Y, Lin B, Feng H, Li Y, Dai X, et al. A novel OPA1 mutation in a Chinese family with autosomal dominant optic atrophy. Biochem Biophys Res Commun. 2012;419:670–5.
Shimizu S, Mori N, Kishi M, Sugata H, Tsuda A, Kubota N. A novel mutation in the OPA1 gene in a Japanese patient with optic atrophy. Am J Ophthalmol. 2003;135:256–7.
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, et al. Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet. 2000;26:207–10.
Yu-Wai-Man P, Shankar SP, Biousse V, Miller NR, Bean LJ, Coffee B, et al. Genetic screening for OPA1 and OPA3 mutations in patients with suspected inherited optic neuropathies. Ophthalmology. 2011;118:558–63.
Iida K, Ohkuma Y, Hayashi T, Katagiri S, Fujita T, Tsunoda K, et al. A novel heterozygous splice site OPA1 mutation causes exon 10 skipping in Japanese patients with dominant optic atrophy. Ophthalmic Genet. 2016;37:354–6.
Chen J, Xu K, Zhang X, Jiang F, Liu L, Dong B, et al. Mutation screening of mitochondrial DNA as well as OPA1 and OPA3 in a Chinese cohort with suspected hereditary optic atrophy. Investig Ophthalmol Vis Sci. 2014;55:6987–95.
Li Y, Li J, Jia X, Xiao X, Li S, Guo X. Genetic and clinical analyses of DOA and LHON in 304 Chinese patients with suspected childhood-onset hereditary optic neuropathy. PLoS ONE. 2017;12:e0170090.
Craig JE, Hewitt AW, Dimasi DP, Howell N, Toomes C, Cohn AC, et al. The role of the Met98Lys optineurin variant in inherited optic nerve diseases. Br J Ophthalmol. 2006;90:1420–4.
Newman NJ, Biousse V. Hereditary optic neuropathies. Eye. 2004;18:1144–60.
Moon Y, Lee JY, Jeong DW, Kim S, Han S, Kook MS. Relationship between nocturnal intraocular pressure elevation and diurnal intraocular pressure level in normal-tension glaucoma patients. Investig Ophth Vis Sci. 2015;56:5271–9.
Park JH, Yoo C, Kim YY. Peripapillary vessel density in young patients with open-angle glaucoma: comparison between high-tension and normal-tension glaucoma. Sci Rep. 2019;9:19160.
Lim JH, Park JS, Lee SY, Hong YJ. Incidence of and risk factors for glaucoma in lost-to-follow-up normal-tension glaucoma suspect patients. BMC Ophthalmol. 2016;16:62.
Jeong DW, Kook MS, Lee KS, Lee JR, Han S. Circadian pattern of intraocular pressure fluctuations in young myopic eyes with open-angle glaucoma. Investig Ophthalmol Vis Sci. 2014;55:2148–56.
Lenaers G, Neutzner A, Le Dantec Y, Juschke C, Xiao T, Decembrini S, et al. Dominant optic atrophy: Culprit mitochondria in the optic nerve. Prog Retin Eye Res. 2021;83:100935.
Chun BY, Rizzo JF 3rd. Dominant optic atrophy: updates on the pathophysiology and clinical manifestations of the optic atrophy 1 mutation. Curr Opin Ophthalmol. 2016;27:475–80.
Yu-Wai-Man P, Bailie M, Atawan A, Chinnery PF, Griffiths PG. Pattern of retinal ganglion cell loss in dominant optic atrophy due to OPA1 mutations. Eye. 2011;25:597–601.
Milea D, Sander B, Wegener M, Jensen H, Kjer B, Jorgensen TM, et al. Axonal loss occurs early in dominant optic atrophy. Acta Ophthalmol. 2010;88:342–6.
Pretegiani E, Rosini F, Rufa A, Gallus GN, Cardaioli E, Da Pozzo P, et al. Genotype-phenotype and OCT correlations in autosomal dominant optic atrophy related to OPA1 gene mutations: report of 13 Italian families. J Neurol Sci. 2017;382:29–35.
Barboni P, Savini G, Cascavilla ML, Caporali L, Milesi J, Borrelli E, et al. Early macular retinal ganglion cell loss in dominant optic atrophy: genotype-phenotype correlation. Am J Ophthalmol. 2014;158:628–36.
Hou H, Moghimi S, Zangwill LM, Shoji T, Ghahari E, Penteado RC, et al. Macula vessel density and thickness in early primary open-angle glaucoma. Am J Ophthalmol. 2019;199:120–32.
Zhang X, Dastiridou A, Francis BA, Tan O, Varma R, Greenfield DS, et al. Comparison of glaucoma progression detection by optical coherence tomography and visual field. Am J Ophthalmol. 2017;184:63–74.
Kanamori A, Nakamura M, Escano MF, Seya R, Maeda H, Negi A. Evaluation of the glaucomatous damage on retinal nerve fiber layer thickness measured by optical coherence tomography. Am J Ophthalmol. 2003;135:513–20.
Bowd C, Zangwill LM, Berry CC, Blumenthal EZ, Vasile C, Sanchez-Galeana C, et al. Detecting early glaucoma by assessment of retinal nerve fiber layer thickness and visual function. Investig Ophthalmol Vis Sci. 2001;42:1993–2003.
Killer HE, Pircher A. Normal tension glaucoma: review of current understanding and mechanisms of the pathogenesis. Eye. 2018;32:924–30.
Mabuchi F, Tang S, Kashiwagi K, Yamagata Z, Iijima H, Tsukahara S. The OPA1 gene polymorphism is associated with normal tension and high tension glaucoma. Am J Ophthalmol. 2007;143:125–30.
Aung T, Ocaka L, Ebenezer ND, Morris AG, Krawczak M, Thiselton DL, et al. A major marker for normal tension glaucoma: association with polymorphisms in the OPA1 gene. Hum Genet. 2002;110:52–6.
Woo SJ, Kim DM, Kim JY, Park SS, Ko HS, Yoo T. Investigation of the association between OPA1 polymorphisms and normal-tension glaucoma in Korea. J Glaucoma. 2004;13:492–5.
Liu Y, Schmidt S, Qin X, Gibson J, Munro D, Wiggs JL, et al. No association between OPA1 polymorphisms and primary open-angle glaucoma in three different populations. Mol Vis. 2007;13:2137–41.
Yao W, Jiao X, Hejtmancik JF, Leske MC, Hennis A, Nemesure B, et al. Evaluation of the association between OPA1 polymorphisms and primary open-angle glaucoma in Barbados families. Mol Vis. 2006;12:649–54.
Acknowledgements
The authors are grateful to the biobank of the Eye & ENT Hospital of Fudan University. The authors would also like to thank all the patients and their families.
Funding
This work was supported by funds from the National Natural Science Foundation of China (Grant no. 81870692), the Shanghai Committee of Science and Technology, China (Grant no. 20S31905800), and the Clinical Research Plan of SHDC (Grant no. SHDC2020CR6029). The authors were supported by grants from the National Key Research and Development Programme of China (Grant no. 2020YFA0112700), from the State Key Programme of National Natural Science Foundation of China (Grant no. 82030027), and the subject of major projects of the National Natural Science Foundation of China (Grant no. 81790641). The sponsor or funding organisations had no role in the design or conduct of this research.
Author information
Authors and Affiliations
Contributions
Concept and design, YC and GT; literature search, YZ and YC; clinical studies, YZ, YC, GT and XS; data acquisition, YZ, YC, GT and XS; data analysis, YZ, GT and YC; statistical analysis, YZ, GT and YC; paper preparation, YZ; paper editing and paper review, YZ, GT and YC.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhang, Y., Sun, X., Tian, G. et al. Comparison of the clinical and genetic features of autosomal dominant optic atrophy and normal tension glaucoma in young Chinese adults. Eye 37, 624–630 (2023). https://doi.org/10.1038/s41433-022-01990-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-022-01990-y
