
Abstract
Cryomicroneedles (cryoMNs) offer a convenient and minimally invasive way to precisely deliver therapeutic cells intradermally for treating local and systemic diseases. cryoMNs are manufactured by shaping and freezing the cell-containing cryogenic media in a microneedle template, which allows cells to be packaged in advance for direct usage in the clinic. However, the current cryoMNs require cold-chain transportation and storage and do not permit the loading of autologous cells in situ. This article introduces the second generation of cryoMNs (S-cryoMNs) that address these limitations. Specifically, S-cryoMNs are made by dipping a porous MN scaffold in the cell suspension before cryopreservation. The porous scaffold can be transported at room temperature, and researchers can load any cells with the optimized cryogenic medium. As a proof-of-concept, we examined the loading and intradermal delivery of three cell types in clinically relevant in vitro and in vivo models, including mesenchymal stem cells for wound healing, melanocytes for vitiligo treatment, and antigen-pulsed dendritic cells for cancer vaccination.
Similar content being viewed by others
Introduction
The intradermal delivery of therapeutic cells has great potential for treating both local and systemic diseases (e.g. mesenchymal stem cells (MSCs) for wound healing, melanocytes for vitiligo treatment, and dendritic cells (DCs) and chimeric antigen receptor (CAR) T cells for immunotherapy). The key to successful treatment is the precise delivery of cells at the desired location and depth for an optimal response and outcome. Microneedles (MNs) are minimized hypodermic needle arrays that were recently suggested to be powerful platforms for the intradermal delivery of therapeutic cells. For example, Lee et al. developed a hydrogel MN with an outer poly(lactic-co-glycolic) protective shell to deliver MSCs for wound regeneration1. Chen et al. seeded human keratinocytes and human follicle dermal papilla cells on the surface of solid polymethyl methacrylate or metal MN patches2. Subsequently, the coated cells could be transplanted into a hydrogel or the targeted tissues within 3 days. Li et al. seeded CAR-T cells on the surface of porous MNs and implanted them via direct intratumoral injection3. However, all these cell-loaded MN platforms require fresh preparation and timely application of the cell products to preserve cell viability and functionality.
To overcome these challenges, we recently introduced cryomicroneedle (cryoMN) technology, which allows cells to be packaged into MNs in advance for direct usage in the clinic4,5. These cryoMNs are manufactured by shaping and freezing the cell-containing cryogenic media in the MN template. The prepared devices can be stored for months in a freezer and are easily transported and deployed. This approach circumvents complex and redundant procedures during each cell administration. As a proof-of-concept, we explored cancer immunotherapy through the intradermal delivery of ovalbumin-pulsed DCs4. The experimental data showed that DC vaccination via cryoMNs elicited robust antigen-specific immune responses and provided strong protection against tumors in mice. cryoMNs are prefabricated through stepwise cryogenic molding of the cell suspension before they are demolded and stored in liquid nitrogen/a −80 °C freezer. This reinforces the need for an ultralow temperature freezer for the transportation and storage of cryoMNs. Moreover, this approach does not allow the loading of autologous cells by the user in situ.
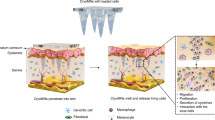
This article describes the development of the second generation of cryoMNs (S-cryoMNs) that addresses these limitations (Fig. 1). Specifically, we propose the use of a porous sponge-like MN that allows the loading of cells via one-step dipping. These MN scaffolds are made through lyophilization of crosslinked hydrogels. They can be packed, transported, and stored at room temperature. Cells suspended in the optimized cryopreservation formula can then be loaded into the porous MNs by the user through the dipping process, which is driven by capillary force6. The loaded MNs can be directly used in situ after freezing or stored in liquid nitrogen/a −80 °C freezer in the end-use clinical facility for later usage. S-cryoMNs are relatively simple to fabricate and use. This process also minimizes the potential contamination and cell damage that might occur during the storage and distribution of 1st generation cryoMN products from central manufacturers. As a proof-of-concept, we developed and applied S-cryoMNs for the intradermal delivery of both allogeneic and autologous cells, including MSCs for wound healing, melanocytes for vitiligo treatment, and antigen-pulsed DCs for cancer vaccination.

Second-generation cryomicroneedles (S-cryoMNs) were fabricated through the dipping method with a porous MN scaffold for intradermal cell delivery.
Results
Fabrication and characterization of porous MN scaffolds
The porous scaffolds used to create S-cryoMNs were made by lyophilizing hydrogels in the MN template. As a proof-of-concept, we used methacrylated hyaluronic acid (MeHA) to demonstrate this principle. Hyaluronic acid (HA) with different molecular weights (48 kDa and 300 kDa) was first modified with methacrylic anhydride to obtain photocrosslinkable 48-MeHA and 300-MeHA. The degrees of substitution, evaluated by proton nuclear magnetic resonance (1H NMR), were 93.6% and 70.3% for 48-MeHA and 300-MeHA, respectively (Fig. S1). Later, an aqueous solution of MeHA was used to fill the negative polydimethylsiloxane (PDMS) MN mold, which was photocrosslinked before freeze-drying (Fig. 2A). Here, the ice was the porogen that introduced the micropores in the MN structure7,8,9. The morphology of the derived MN scaffold was similar to that of the original MN master template. While the stainless steel master MN template had a height of 1200 μm and a base width of 300 μm (Fig. S2), the height and base width were 700 μm and 270 μm, respectively, for the porous MNs made of 48-MeHA, and 620 μm and 320 μm, respectively, for the MNs made of 300-MeHA. Changes in the MN scaffold dimensions, which are due to the shrinkage of PDMS and the polymeric matrix, have been observed in many previous studies10,11,12.

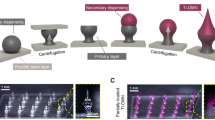
A Illustration of porous MN scaffold fabrication using the MeHA hydrogel. Scale bar: 2 mm. Optimization of (B) 300-MeHA concentration (2, 4, 5, and 6 wt%) and (C) 48-MeHA concentration (2, 4, 6, and 8 wt%) during MN scaffold fabrication. Optimization of the crosslinking time for (D) 4 wt% 300-MeHA and (E) 6 wt% 48-MeHA during MN scaffold fabrication.
The porous structure of the MNs was tunable by adjusting the MeHA concentration and photocrosslinking time. We examined the changes in morphology of the MN scaffolds made with different MeHA concentrations (2, 4, 5, and 6 wt% 300-MeHA and 2, 4, 6, and 8 wt% 48-MeHA) with the same crosslinking time (5 min). As shown in Fig. 2B, C, 300-MeHA MNs provided a porous structure when the concentration of the polymer was 4%. Similar results were obtained for 48-MeHA MNs at 4% and 6% wt% 48-MeHA. Lower concentrations of 48-MeHA and 300-MeHA did not provide a stable MN structure, while higher concentrations significantly decreased the porosity6,13. The crosslinking time is another important parameter during fabrication. With the optimized MeHA concentrations (4% for 300-MeHA and 6% for 48-MeHA), we examined UV crosslinking times of 3 min (CL3), 5 min (CL5), 10 min (CL10), and 20 min (CL20) (Fig. 2D, E). In general, the longer the UV exposure time was, the lower the porosity was14. Specifically, when the crosslinking time was 10 min or longer, we obtained a shell structure instead of a porous structure. This shell structure is not preferrable for loading cells.
Therefore, CL3 and CL5 of 48-MeHA (CL3-48-MeHA and CL5-48-MeHA) and CL3 of 300-MeHA (CL3-300-MeHA) were identified as the optimal formulations due to their intact MN structure and observable porosity. We further analyzed the porosity and dimensions of the samples via the SEM images using ImageJ. The CL3-48-MeHA and CL5-48-MeHA MNs had average MN heights of 697.1 ± 21.0 μm and 682.9 ± 10.7 μm, respectively. The CL3-300-MeHA MNs were slightly shorter at 616.4 ± 32.8 μm. The average pore sizes for CL3-48-MeHA, CL5-48-MeHA, and CL3-300-MeHA were 81.0 ± 36.8 μm, 56.6 ± 22.8 μm, and 54.2 ± 23.9 μm, respectively. These pore sizes match well with that of mammalian cells, which are usually 10–30 μm in suspension. Of these three hits, CL3-300-MeHA was used as a representative for the following studies.
While the rest of the work was done with the MeHA porous MN scaffold, it should be noted that other hydrogel-derived scaffolds can also be used. As shown in Fig. S3, porous MN scaffolds may be prepared from other hydrogel formulations, including but not limited to cryogelation of polyvinyl alcohol (PVA), cryogelation of gelatin (gelatin), EDC/NHS (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide)-crosslinked gelatin (EDC/NHS-Gel), 1,4-butanediol diglycidyl ether crosslinked hyaluronic acid (BDDE-HA), photocrosslinked gelatin methacryloyl (GelMA), and photocrosslinked polyethylene glycol diacrylate (PEGDA).
Cell loading and release from S-cryoMNs
S-cryoMNs are made by dipping the porous MN scaffold in a cell suspension and freezing. Like in the first generation of cryoMNs, the cryogenic medium is the key factor for maintaining cell viability and function in the device. Dimethyl sulfoxide (DMSO) is the most popular cryoprotective agent because it binds water molecules to prevent ice crystallization and cell damage15,16. However, the commonly used concentration (10%, v/v) can be toxic to cells and induce cell membrane damage and gene expression alterations17. During the intradermal delivery of therapeutic cells, a high dose of DMSO might cause unwanted side effects on both the therapeutic cells and surrounding skin cells.
Macromolecular cryoprotectants, in which hydrophilic macromolecules bind with water and increase solution viscosity to inhibit extracellular ice growth, have been reported to be a partial substitute for DMSO to reduce toxicity18. Hydrogel-forming polymers are macromolecules characterized by good biocompatibility and good affinity for water molecules. Some of these materials, such as polyethylene glycol (PEG) and hydroxyethyl starch (HES), have been applied in the cryopreservation of red blood cells19,20, stem cells21,22,23, and other mammalian cells24,25,26. Therefore, hydrogel polymers are considered potential candidates for replacing some of the DMSO to develop cryoprotective media with low toxicity.
We screened eight hydrogel-forming polymers that have been used to cryopreserve cells: PEG, HES, methylcellulose (MC), sodium carboxymethyl cellulose (CMC), HA, chitosan, gelatin, and alginate. These polymers were mixed with low doses of DMSO (1% and 2%) as cryogenic media for the cryopreservation of human dermal fibroblasts. Among the eight candidates, cryogenic media containing PEG, HES, MC, and CMC ensured cell survival (Fig. S4). Later, the concentrations of PEG, HES, MC and CMC were further evaluated from 0, 0.25, 0.5, 1.0 to 2.5 wt% (Fig. S5). When the concentration of DMSO was lower than 1%, the cryogenic solution did not sufficiently protect the cells during cryopreservation. With 2% DMSO, all cryogenic solutions of the four polymers provided similar or even greater cell viability than 10% DMSO. We further confirmed the preservation ability of these four best formulations (2.5 wt% PEG, 2.5 wt% MC, 2.5 wt% CMC, and 0.25 wt% HES with 2.0% DMSO) on three other cell types, namely, human keratinocytes (HaCaT cells), a human malignant melanoma cell line (A375), and human bone marrow-derived MSCs. As shown in Fig. 3A, the fibroblasts, A375 cells, and HaCaT cells maintained comparable, if not greater, viabilities in these cryogenic formulations compared to those in classic cryogenic media supplemented with 10% DMSO (except 2% DMSO + 0.25% HES for HaCaT cells). However, no advantage was observed for hMSCs except for 2% DMSO + 2.5% CMC. However, compared with media supplemented with 10% DMSO, cryogenic solutions containing PEG, HES, MC, or CMC did not induce any significant toxicity (Fig. S6). Finally, the formulation containing 2.5% CMC and 2% DMSO was ultimately selected as the representative cryogenic solution for subsequent cell loading.

A Screening of cryogenic formulations for S-cryoMNs with low toxicity. The relative cell viability in each cryoprotective medium was calculated by normalization to that of the positive control 10% DMSO (v/v) in DPBS. B Digital images of S-cryoMNs before and after thawing. Scale bar: 2 mm. C Live/dead staining of A375 cells after their release from S-cryoMNs. Scale bar: 200 μm. D Confocal images of S-cryoMNs loaded with A375 labeled with CellTracker™ (green). Scale bar: 200 μm. E Top view confocal images and (F) quantification of the cells released from S-cryoMNs loaded with different cell densities ranging from 2.5 × 106, 5.0 × 106, and 7.5 × 106 to 1.0 × 107 cells/ml. G Top view and (H) 3D reconstruction of the confocal images showing cell release from S-cryoMNs into the agarose gel phantom. Scale bar: 100 μm. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001; ns: p > 0.05, no significant difference (ns).
The representative porous MN scaffold (CL3-300-MeHA) was immersed in cryogenic medium containing cells (A375 cells as an example) for 1 min. The loading of cells into the porous MN scaffold was driven by capillary force during wetting. After the scaffold was prewetted, the cells could not reach the matrix (Fig. S7A). The polymeric component (CMC) increased the viscosity of the cryogenic solution and thus reduced the climbing viscosity of the solution inside the scaffold27, which was beneficial for uniformly distributing cells in the porous scaffold (Fig. S7B).
Later, the cell-loaded S-cryoMN patches were frozen in a cryopreservation box. After overnight freezing, the patches were stored at −80 °C for the short-term or in liquid nitrogen for longer storage (>7 days). When the frozen S-cryoMNs were placed at room temperature (24 °C), they melted and completely thawed within 2 min (Figs. 3B and S8). The encapsulated A375 cells had an average viability of 76.3 ± 7.7% (Fig. 3C). Cells labeled with green dye were observed inside the tips of the S-cryoMNs (Fig. 3D), and the number of loaded cells inside the S-cryoMNs was tuned according to the initial cell density in the cell suspension. The release of cells from S-cryoMNs was tested on an agarose skin phantom. When the cell concentration was increased from 2.5 × 106 to 5.0 × 106, 7.5 × 106, and 1.0 × 107 cells/ml in cryogenic medium, the number of cells released from each needle increased accordingly from ~10 cells/needle to 240 cells/needle (Fig. 3E, F). The loaded cells were released from the S-cryoMNs upon melting and degradation (Fig. S9A). The degradation behavior of the porous MN scaffold could be efficiently tuned by adjusting the second crosslinking degree to achieve a desirable release profile (Fig. S9A, B). In the following studies, a second crosslinking cycle of 8 min was adopted to achieve relatively rapid release within 8 h.
As shown in Fig. 3G, H, the S-cryoMNs easily penetrated the agarose gel and delivered the stained A375 cells into the agarose gel at a depth of ~200 μm. The mechanical strength of the S-cryoMNs was tested via a compressive mechanical test in which the S-cryoMNs could bear 0.058 N of force with a single needle, which indicated that the strength of the S-cryoMNs was sufficient for skin penetration28 (Fig. S10). Skin penetration was also validated on ex vivo porcine skin, where the S-cryoMNs were easily inserted into the skin, and an approximate penetration depth of 450 µm was determined from the histological images (Fig. S11). Note that the agarose gel is much more elastic than porcine skin is; thus, the S-cryoMNs had less penetration into the gel phantom.
Using S-cryoMNs for the delivery of allogeneic and autologous cells
In cell therapies, the cells to be delivered might originate from the patient (autologous cells) or a donor (allogeneic cells). These therapeutic cells are collected, expanded, and engineered ex vivo before administration to the patient. The S-cryoMNs utilize a dip-loading procedure that allows their on-site preparation for subsequent cell delivery as well as long-term storage for later application (Fig. 4A). Here, we used hMSCs and melanocytes as model cells to demonstrate the potential applications of S-cryoMNs for delivering both allogenic cells and autologous cells.

A Schematic of the application of S-cryoMNs for allogeneic and autologous cell therapies. The secretion effects of hMSCs delivered using S-cryoMNs on promoting (B) in vitro wound closure and (C) vascularization. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; ns no significant difference. n = 3.
MSCs are promising therapeutic cells because of their immunosuppressive and tissue repair properties29. Autologous MSCs can suffer from insufficient quantities, decreased biological activity (elderly individuals as donors), or impaired functions (donors with systemic diseases). Using allogeneic MSCs from young healthy donors is a reasonable approach to resolving these issues. Here, we loaded 1 × 105 hMSCs into S-cryoMNs and examined their potential application in wound healing through scratch and tube formation assays30. A scratch assay was used to examine the migration capability of dermal fibroblasts in response to hMSCs released from the S-cryoMNs. We first examined the migration and proliferation of fibroblasts into the wounded area at 24 and 48 h after treatment under four conditions: low serum concentration (1% FBS instead of 5% FBS in the other groups), positive control (20 ng/ml TGF-β1), blank S-cryoMNs, and hMSC-loaded S-cryoMNs. As shown in Figs. 4B and S12, treatment with S-cryoMNs significantly promoted the migration of fibroblasts. Closure was 50.8 ± 6.1% at 24 h and 86.5 ± 5.0% at 48 h in the cell-loaded S-cryoMN group, while closure was 25.5 ± 11.4% at 24 h and 59.1 ± 7.6% at 48 h for the group treated with blank S-cryoMNs. This result is comparable to that of the positive control. We also carried out a tube formation assay under four different conditions: low serum concentration (1.5% FBS instead of 5% FBS in the other groups), a positive control (40 ng/ml VEGF), blank S-cryoMNs, and hMSC-loaded S-cryoMNs. Eight hours after treatment, a tube-like network was clearly observed in the cell-loaded S-cryoMN group (Fig. S13). In the group treated with hMSC-loaded S-cryoMNs, there was a 2.3-fold increase in the total tube length compared to that of the group treated with blank S-cryoMNs (Fig. 4C). Treatment with hMSC-loaded S-cryoMNs increased the numbers of tubes and branching nodes by 2.7 times and 2.0 times, respectively, compared to those in the blank S-cryoMN group.
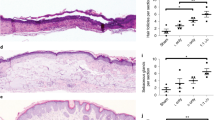
Vitiligo is a skin depigmentation disorder caused by the autoimmune destruction of melanocytes, which results in a loss of melanin expression31,32. Autologous melanocyte transplantation is a surgical strategy for regenerating melanocytes and repairing vitiliginous areas33,34. As melanocytes typically reside in the basal epidermis and dermal junctions35, recipient sites are commonly exposed to the papillary dermis by invasive abrasion methods (laser abrasion36, dermabrasion37,38, and the suction blister method39) to ensure the survival of melanocytes after transplantation. Here, we used B16-loaded and melanocyte (Mela)-loaded S-cryoMNs to restore melanin expression in model mice. The B16-loaded S-cryoMNs (characterization details are shown in Fig. S14) were applied to the backs of the mice via a thumb press to intradermally deliver B16 cells, and the results from this method were compared with those from the dermabrasion technique (Fig. 5A). As shown in Fig. 5B, C, 1 day after cell transplantation via B16-loaded S-cryoMNs, the delivered cells could be visualized at the base of the epidermis and dermis, ranging from ~50 μm to 200 μm. On day 3, the melanin production by the delivered B16 cells was comparable to that of the dermabrasion group.

A Timeline of the delivery of B16 melanoma cells and human primary melanocytes to the immunodeficient model mice. The dermabrasion method was compared with the S-cryoMNs delivery methods. B Melanin expression and (C) quantitative analysis of the transplanted B16 cells using dermabrasion, blank or B16-loaded S-cryoMNs 1 and 3 days after application. Melanin was stained with Masson-Fontana stain and is indicated by the red arrow. At each time point, three independent samples were analyzed. D Melanin expression and (E) quantitative analysis of the transplanted melanocytes obtained via dermabrasion, blank or Mela-loaded S-cryoMNs 1 and 3 days after application. Melanin was stained with Masson-Fontana stain and is indicated by the red arrow. The scab is indicated using a green arrow. At each time point, three independent samples were analyzed. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; ns no significant difference. n = 3.
We also applied melanocyte (Mela)-loaded S-cryoMNs (characterization details are shown in Fig. S14A, B) for intradermal transplantation of melanocytes. On day 0, each mouse received four treatments on four different dorsal sites: no treatment, Mela-loaded S-cryoMNs, blank S-cryoMNs, and dermabrasion transplantation (Fig. 5A). In the dermabrasion treatment, the epidermis was mechanically removed to expose the superficial dermis to obtain a melanocyte suspension with a density of ~250 cells/mm2, which was consistent with that of Mela-loaded S-cryoMNs (~220 cells/mm2)40,41. The Mela-loaded S-cryoMNs were pressed onto the mouse skin and not removed until the patches had melted and softened. On day 1 after treatment, the melanocytes released from the S-cryoMNs were visualized in the mouse skin from the epidermis to the dermis (depth up to ~300 μm), and these cells were found mostly in the basal layer of the epidermis and around the hair follicles (indicated by the red arrows in Fig. 5D). Three days after S-cryoMN transplantation, the delivered melanocytes remained viable and produced melanin. Moreover, there was no significant decrease in the number of cells according to semiquantitative analysis of the histological images (Fig. 5E). The efficacy of the Mela-loaded S-cryoMNs was similar to that of the dermabrasion technique in terms of intradermal transplantation of melanocytes. In contrast, the dermabrasion method was markedly more invasive, as the epidermis was destroyed on day 1 and a scab had formed on day 3, as shown in Fig. 5D.
OVA-DC S-cryoMNs for cancer vaccination in vivo
DCs are potent antigen-presenting cells (APCs) that capture and present antigens to T lymphocytes for long-lasting and specific immune memory. Additionally, autologous DCs have been applied for the development of vaccines in the clinic. We developed DC-loaded S-cryoMNs and tested their potential for use in vaccination in a mouse model. DCs were pulsed with ovalbumin (OVA) as a model antigen4, after which the surface markers of mature and active DCs were examined (CD11c, CD86, and MHCII). Lipopolysaccharide (LPS)-pulsed DCs served as the positive control (Fig. S15A). Specifically, OVA stimulation generated 63.5 ± 6.9% CD11c+CD86+ DCs and 68.8 ± 9.1% CD11c+MHCII+ DCs, which was comparable to the LPS-pulsed DCs (72.4 ± 12.0% CD11c+CD86+ DCs and 74.6 ± 8.2% CD11c+MHCII+ DCs). After being loaded into S-cryoMNs (OVA-DC S-cryoMNs), the OVA-DCs maintained high viability after short-term (83.1% after 1 week at −80 °C) and long-term storage (77.2% after 1 month of storage in liquid nitrogen and 63.4% after 3 months of storage in liquid nitrogen) (Fig. S15B, C).
We optimized the dosage and application frequency of OVA-DC S-cryoMN vaccination. Healthy mice were subcutaneously (s.c.) vaccinated twice a week with different dosages of OVA-DCs (0, 1, 2, or 8 × 105). The levels of OVA-specific antibodies in the blood significantly increased in the third week and became saturated after eight treatments (Fig. S16). The 2 × 105 OVA-DC dosage produced the best antigen-specific immune response. Therefore, we used S-cryoMNs with 2 × 105 OVA-DCs (two patches per dose with each patch containing 1 × 105 OVA-DCs) to vaccinate the mice over 4 weeks for a total of 8 treatments (Fig. 6A, B). OVA-DC S-cryoMNs penetrated mouse skin up to ~200 µm in the dermal area, as shown in Fig. 6C. After one treatment with OVA-DC S-cryoMNs, prelabeled OVA-DCs were observed in the draining lymph nodes (Fig. S17A, B). On day 28, the mice were inoculated with B16-OVA melanoma cells. The tumors were measured every 2 days and excised on day 24 post inoculation. As shown in Figs. 6D, E and S18, tumor growth was much slower and the tumors were much smaller in mice inoculated with OVA-DC S-cryoMNs (average size of 186.1 ± 144.3 mm3 and weight of 135.2 ± 114.7 mg) compared with mice treated with blank S-cryoMNs (average size of 905.8 ± 445.1 mm3 and weight of 1037.1 ± 662.2 mg).

A The timeline of mouse vaccination using OVA-DC S-cryoMNs. B Photos before and after the administration of two patches of OVA-DC S-cryoMNs to shaved mice. C Histological analysis of the depth of S-cryoMN penetration into mouse skin. Scale bar: 100 μm. D Photos of excised tumors from B16-OVA melanoma-bearing mice without or with vaccination with OVA-DC S-cryoMNs. E Tumor volume growth curves of B16-OVA melanoma-bearing mice without or with vaccination with OVA-DC S-cryoMNs. F Quantification of CD11c+CD86+ and CD11c+ MHCII+ DCs inside the excised draining lymph nodes without or with vaccination with OVA-DC S-cryoMNs. G Proliferation of and (H) secretion of interferon gamma (IFN-γ) by splenocytes from vaccinated mice after restimulation with 50 µg/ml OVA. I The activity of OVA-specific cytotoxic T lymphocytes (CTLs) without or with vaccination by OVA-DC S-cryoMNs. The cytotoxic effects of splenocytes (effector cells) on B16-OVA cells (target cells) were analyzed at different effector cell-to-target cell ratios (E:T ratios). Each group included four independent animals. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001; ns no significant difference. n = 4.
The vaccinated mice had a greater percentage of CD11c+CD86+ DCs (2.16 ± 0.23% vs. 1.22 ± 0.24% in the blank S-cryoMNs group) and more CD11c+MHCII+ DCs (1.04 ± 0.32% vs. 0.72 ± 0.26% of the blank S-cryoMNs group) in the excised draining lymph nodes (Figs. 6F and S19). After splenocytes were collected from vaccinated mice and restimulated with OVA, OVA-DC S-cryoMNs-vaccinated mice exhibited a significantly greater proliferation rate (~1.3-fold greater than that of the blank S-cryoMNs group; Fig. 6G) and greater secretion of IFN-γ (736.4 ± 145.1 pg/ml vs. 202.1 ± 33.0 pg/ml in the blank S-cryoMNs group; Fig. 6H). As shown in Fig. 6I, immunization with OVA-DC S-cryoMNs induced significantly greater OVA-specific toxicity to splenic T lymphocytes than vaccination with blank S-cryoMNs. The above results suggested that vaccination with OVA-DC S-cryoMNs could induce potent antigen-specific immune responses with strong antitumorigenic effects.
The S-cryoMNs were safe throughout the entire experimental period. All of the thumb-pressed S-cryoMNs left a clear micropattern on the mouse skin, which visually disappeared within 1 h (Fig. S20A); there was no obvious skin damage. After 8 applications of S-cryoMNs over 1 month, we excised the skin and performed H&E staining and did not observe any increase in the infiltration of inflammatory cells in the skin tissue (Fig. S20B). Such frequent administration of S-cryoMNs induced neither weight loss (Fig. S20C) nor pathological damage to major organs (Fig. S20D).
Discussion
The feature of S-cryoMNs is their porous spongy structure, which can rapidly absorb aqueous solutions while maintaining its original structure. This sponge-like structure allowed the cell suspension to be absorbed through capillary force during wetting and the MN morphology to be maintained (Fig. S7). This approach will facilitate the future large-scale fabrication and distribution of porous MN scaffolds that can be loaded with cells and frozen into S-cryoMNs in situ for the intradermal injection of cells. When the MeHA hydrogel was used to construct the porous MN scaffold, two-step UV crosslinking was carried out (Fig. 2). Both the UV lamp intensity and crosslinking duration could affect the degree of crosslinking. In this work, the crosslinking degree was tuned with a fixed UV light intensity to study the relationship between the photocrosslinking degree and the pore size in the MN scaffold (Fig. 2D, E). The first crosslinking procedure in the PDMS mold was to achieve a pore size that was appropriate to accommodate cells. The second crosslinking procedure was carried out on MeHA MNs after their removal from the mold. The additional crosslinking protocol minimized the swelling of the porous structure when the cell suspension was added and also served to adjust the degradation behavior of the scaffold to achieve a desirable release profile (Fig. S9).
This porous MN scaffold-assisted loading method also minimized the steps from cell harvesting to cell delivery. The porous MN scaffold rapidly absorbed the therapeutic cell suspension and was subsequently frozen to a solid state to form S-cryoMNs. The cryoprotective medium was formulated with a low concentration of DMSO and biocompatible polymers (PEG, HES, MC, and CMC) to have low toxicity while maintaining good cryoprotection for various types of cells (fibroblasts, HaCaT cells, A375 cells, hMSCs, melanocytes, B16 cells, and DCs) (Figs. 3A and S14, S15B). There are other types of cryoprotectant agents, such as sugars, polysaccharides, and proteins. We chose polymers because of their relatively low cost, batch-to-batch consistency, and easy accessibility.
The potential applications of S-cryoMNs were shown through three clinically relevant in vitro and in vivo models. The first is hMSC delivery for wound healing29,30. The delivered hMSCs promoted cell migration according to data from scratch and endothelial tube formation assays (Fig. 4B, C). The second example is the delivery of melanocytes for vitiligo treatment in vivo (Fig. 5). We examined the delivery of both mouse melanoma cells and human primary melanocytes and proved the potential ability of these cells to restore melanin levels in mouse skin. The third example is the delivery of antigen-pulsed DCs for cancer immunotherapy. After in vivo vaccination with antigen-pulsed DC-loaded S-cryoMNs, strong antigen-specific immune responses were induced, and the growth of melanoma was significantly delayed (Fig. 6). In addition to DCs, modified T cells42 and macrophages43 have also been utilized in cell-based cancer immunotherapy and can specifically attack cancer cells. This approach addresses the resistance issues that occur in traditional drug therapies44,45. Additionally, in comparison with the immediate therapeutic effects of traditional drugs and phototherapies46,47, cell therapy has potential for long-lasting effects that continue to eliminate cancer cells48. While S-cryoMNs have addressed the issues faced by the first generation of cryoMNs, they also face some specific challenges that need further improvement. The first is the speed of cell release. First-generation cryoMNs dissolve in the skin in a few seconds, which too fast for clinical application. S-cryoMNs release cells during melting and degradation (Fig. S9A, B), and the degradation behavior of the porous MN scaffold could be tuned by adjusting the second crosslinking parameters to achieve a desirable release profile. In the future, further optimization of the crosslinking conditions and identification of other potential scaffold materials are needed to achieve dissolution and drug release in the skin in 2–5 min. Second, S-cryoMNs are still applied through a thumb press. This approach would result in different penetration depths into the skin at different locations or after application by different operators (the results in Figs. 3G, H and S11 are very different). We hope that there will be an applicator that can achieve consistent penetration of MNs by automatically adjusting the pressing force based on real-time measurements of the mechanical properties of the skin.
Conclusion
This article reports S-cryoMNs that aimed to address the limitations of the first generation of cryoMNs. S-cryoMNs were generated by dipping a porous MN scaffold in a cell suspension before cryopreservation. The porous scaffold can be transported at room temperature, and researchers can load any type of cells with the optimized cryogenic medium. As a proof-of-concept, we examined the loading and intradermal delivery of three cell types that are clinically relevant in vitro and in vivo: hMSCs for wound healing, melanocytes for vitiligo treatment, and DCs for cancer vaccination. We believe that this new version of cryoMNs will facilitate the adoption of MN technology in the field of cell therapy and regenerative medicine.
Materials and methods
Sodium hyaluronic acid (300-HA, Mw 300 kDa) and sodium hyaluronic acid (48-HA, Mw 48 kDa) were purchased from Freda Biochem Co., Ltd. (China). N,N-Dimethylformamide (DMF, 227056), methacrylic anhydride (MAA, 276685), dimethyl sulfoxide (DMSO, 276855), sodium carboxymethyl cellulose (CMC, Mw 90 kDa, 419273), methylcellulose (MC, viscosity 15 cP, M7140), hydroxyethyl starch (HES, medium Mw, Y0001277), poly(ethylene glycol) (PEG, Mw 10 kDa, P6667), chitosan (low Mw, 448869), gelatin (from bovine skin, G9391), and alginic acid sodium salt (from brown algae, A0682) were purchased from Sigma-Aldrich (Singapore). The AlamarBlue cell viability reagent, LIVE/DEAD™ viability/cytotoxicity kit and CellTracker™ Green CMFDA were purchased from Thermo Fisher Scientific (USA). All other materials except those specifically mentioned were acquired from Sigma-Aldrich (Singapore).
Synthesis of MeHA
HA was methacrylated according to a published protocol11. Briefly, 1.0 g of HA (Mw of 300 kDa or 48 kDa) was dissolved in 50 ml of deionized water at 4 °C until complete dissolution. Then, 33 ml of DMF was added to the HA solution to achieve a water/DMF ratio of 3:2 (v/v). Then, 1.22 g of MAA was subsequently added dropwise to the solution while maintaining the pH at 8–9. The reaction mixture was left overnight with continuous stirring at 4 °C. Later, 2.46 g of NaCl and pure ethanol were added sequentially to precipitate the product MeHA. The crude product was collected by centrifugation and redissolved in deionized water. MeHA was purified by dialysis against deionized water for 7 days. The purified product was obtained by lyophilization and stored at 4 °C. MeHA was characterized by 1H NMR spectroscopy (Bruker Avance II 300 MHz NMR) to determine the modification degree.
Fabrication of porous MeHA MNs
The stainless steel MN mold (Micropoint Technologies Pte Ltd., Singapore) had a base diameter of 300 μm, a tip radius of 5 μm, and a tip height of 1200 μm. To fabricate the negative mold, polydimethylsiloxane (PDMS, 10 mm thick; Dow Corning 184 Sylgard) was poured over the master template to replicate the structure. After degassing in a vacuum oven, the PDMS was cured at 70 °C for 1 h and carefully peeled away from the template.
To generate a porous structure, MeHA (Mw of 48 kDa or 300 kDa) was mixed with the photoinitiator (Irgacure 2959) at a mass ratio of 100:1 and dissolved in deionized water. The mixture was cast in the PDMS mold and centrifuged at 4000 rpm for 5 min to fill the voids. Later, the patch was crosslinked by UV exposure. MNs with different crosslinking degrees were named CL3, CL5, CL10, and CL15, defined by their exposure time to UV light (3, 5, 10, and 15 min, respectively). Then, these MNs were frozen at −40 °C and lyophilized. The resulting porous MNs were carefully peeled off from the PDMS mold and crosslinked with UV light for 8 min to strengthen the scaffold matrix. The morphology and porous structures of the MeHA MNs were examined by a microlens-equipped digital camera and field-emission scanning electron microscope (FESEM, JEOL JSM-6700, Japan). The porosity of the MNs was analyzed using ImageJ software. For each type of porous MN, three independent images at ×500 magnification were analyzed. The scaffold area was determined by the software, and the porosity of each porous MN was calculated according to the following equation:
where Atotal and Ascaffold are the areas of the whole image and the scaffold part, respectively.
Compressive mechanical test
The mechanical properties of the S-cryoMNs were tested using an Instron 5942 micro tester. The S-cryoMNs were first placed flat on a precooled metal container with the tips facing upward, followed by cooling the detector and the flat metal container with liquid nitrogen to provide a low-temperature environment for testing. S-cryoMNs (CL3, CL5, CL10, and CL15) made of 300 kDa MeHA were examined at a constant speed (0.5 mm min−1) of vertically oriented force applied to the MN tips. Pure CMC cryoMNs made with only cryoprotective medium (2.5 wt% CMC and 2 v/v % DMSO) were used for comparison.
Cell culture
Human dermal fibroblasts (CellResearch Corporation Pte Ltd.), human immortalized keratinocytes (HaCaT cells, Lonza), human mesenchymal stem cells (hMSCs, Lonza), and a human malignant melanoma cell line (A375, ATCC) were cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. Human umbilical vein endothelial cells (HUVECs, Lonza) were cultured in EGMTM-2 endothelial cell growth medium (EGM-2, Lonza) supplemented with 5% FBS. B16 cells (mouse melanoma cell line) were cultured in high-glucose DMEM medium. Human epidermal melanocytes (ThermoFisher Scientific, USA) were cultured in Medium 254 supplemented with 1% Human Melanocyte Growth Supplement (HMGS-2). The cells were grown under 5% CO2 at 37 °C, and the medium was replaced every 2 or 3 days.
Optimization of the cryoprotective medium
Cryoprotective medium was prepared by dissolving the polymers (HA, gelatin, alginic acid, chitosan, MC, CMC, PEG, and HES) and DMSO at the predetermined concentrations (0, 0.25, 0.5, 1.0, 2.5 wt% for the polymers and 0, 1, 2 v/v % for DMSO) in Dulbecco’s phosphate-buffered saline (DPBS) (Gibco). Human dermal fibroblasts were used to evaluate the cryoprotective effect of the freezing medium. After the fibroblasts reached a confluency greater than 90%, the cells were trypsinized and suspended in freezing medium at a concentration of 2 × 105 cells/ml. The cells were cryopreserved by gradient freezing and finally stored in a −80 °C ultralow temperature freezer for 72 h. Later, the cells were thawed in a 37 °C water bath and seeded on 96-well plates and 48-well plates using fresh complete medium. After culturing for 24 h, the cells were imaged by phase contrast microscope and viability was assessed using the AlamarBlueTM cell viability assay. The cells cryopreserved in 10 v/v% DMSO under the same experimental conditions were used as the positive control. The relative viabilities of the cells from each cryoprotective medium were calculated by normalization to the positive control.
Fabrication of S-cryoMNs through the dipping method with porous MNs
The porous MN scaffolds were sterilized with UV light for 30 min before the operation. Cells (hMSCs, A375 cells, B16 cells, melanocytes, and DCs) were first suspended in cryoprotective solution at a density of 1 × 106 cells/ml or at other stated densities. The porous MNs were soaked in 100 μl of cell suspension with the tip facing downward for 1 min to allow full absorption. The cell-loaded S-cryoMNs were placed inside the cryopreservation box and frozen at −80 °C for 1 day. The prepared S-cryoMNs were finally stored in liquid nitrogen.
Wound/scratch healing assay
Cell migration toward the wounded area was assessed using a scratch assay. Dermal fibroblasts were seeded in the bottom layer of a 48-well transwell (Costar®) at 90% confluency and cultured overnight. A linear defect in the fibroblast monolayer was created by scratching with a 1000 μl pipette tip. After the cells were scratched, the damaged monolayer was washed twice with DPBS, and 1.5 ml of low-serum culture medium was added to each well (1% FBS in the negative control group and 5% FBS in the positive control, blank S-cryoMN, and hMSC-loaded S-cryoMN groups; n = 3). In the S-cryoMN treatment groups, blank or hMSC-loaded S-cryoMN patches were dissolved in 200 μl of culture medium and transferred to one transwell insert. In the positive control group, low-serum culture medium was supplemented with 20 ng/ml transforming growth factor (TGF)-β1 to accelerate fibroblast proliferation. Bright field images of the wounded area were taken at predetermined time points (0, 24, and 48 h after treatment) with an Olympus IX71 inverted microscope. The width of the linear defect was measured using ImageJ software. The extent of wound closure was calculated by the following equation:
Endothelial cell tube formation assay
The ability of hMSC-loaded MNs to promote angiogenesis was assessed using an endothelial cell tube formation assay. HUVECs were prestained with CellTracker™ one night before the tube formation assay. Matrigel® Matrix Basement Membrane (Corning) was applied to coat the wells. Briefly, 100 μl of Matrigel® Matrix was added to each well of a 48-well plate and allowed to solidify overnight at 37 °C. The tube formation assays included 40,000 stained HUVECs in 300 μl of EGM-2 medium (1.5% FBS for the negative control group and 5% FBS for the positive control, blank S-cryoMN, and hMSC-loaded S-cryoMN groups; n = 3) in each well. In the positive control group, 40 ng/ml of VEGF was added to the EGM-2 medium to promote the formation of tube-like structures. In the S-cryoMNs treatment groups, blank or hMSC-loaded S-cryoMN patches were dissolved in each well. After 24 h of incubation, bright field and fluorescence images of each group were recorded with an Olympus IX71 inverted microscope. The total length of each tube, number of tubes, and number of branch points were determined using ImageJ to quantify the angiogenetic potential of each treatment.
Intradermal cell transplantation using B16-loaded or Mela-loaded S-cryoMNs
The animal experiments were performed on B/C nude mice (female, 4–6 weeks) in accordance with a protocol approved by the Ethics Committee of Shanghai Ninth People’s Hospital (HKDL [2018] 234). The nude mice were obtained from the specific-pathogen-free (SPF) laboratory of Shanghai Ninth People’s Hospital. The animals were housed in ventilated cages under a 12:12 h light-dark (LD) cycle at constant temperature (22 ± 2 °C) and relative humidity (50 ± 10%). Twelve mice were randomly assigned to four different groups for treatment with B16-loaded or Mela-loaded S-cryoMNs (n = 3). All the mice received treatment on day 0, and the mice in each group were sacrificed at predetermined time points (day 1 and day 3) for histological analysis. After the mice were anesthetized, four treatments were administered to the back of each mouse: no treatment (upper left flank), dermabrasion (upper right flank), B16-loaded S-cryoMNs (lower left flank), and blank S-cryoMNs (lower right flank). For dermabrasion treatment, the skin was sterilized and then scratched using a surgical blade until very small bleeding spots appeared. Later, ~25 µl of B16 cell suspension at a concentration of 2 × 106 cells/ml was carefully applied to the wounded area. Five minutes after applying the cell suspension, the treatment site was protected using a Tegaderm™ film (3 M™, China). In the B16-loaded S-cryoMN groups, the application sites were disinfected, and two patches were applied to deliver ~4 × 103 B16 cells per mouse. Two patches of blank S-cryoMNs containing cryopreservation medium only were applied to the lower right flank of the back skin of each mouse. After the S-cryoMNs were applied, the treatment sites were covered with Tegaderm™ film for protection.
Mela-loaded S-cryoMNs were applied for melanocyte delivery using a similar protocol in which only the dose of the melanocytes was modified. For melanocyte delivery, 25 µl of melanocyte suspension (1 × 106 cells/ml) was used for dermabrasion treatment, and ~2 × 103 melanocytes were delivered to each mouse using Mela-loaded S-cryoMNs. The doses of melanocytes for dermabrasion and Mela-loaded cryoMN treatment were determined to achieve a melanocyte density of ~250 cells/mm2 after application. Three mice were sacrificed at each timepoint (day 1 and day 3), and their skin was harvested. The skin sections were stained using a Masson-Fontana staining kit (Beijing Solarbio Science & Technology Co., Ltd., China) to visualize melanin expression in the transplanted B16 cells and melanocytes.
Preparation and antigen stimulation of bone marrow-derived dendritic cells (BMDCs)
BMDCs were isolated from the bone marrow of C57BL/6 mice according to a previously reported protocol4. Briefly, the femur bones were dissected, and the bone marrow was collected. Later, the red blood cells were lysed using ACK lysis buffer. The cells were resuspended to a concentration of 1 × 106 cells/ml, cultured in complete RPMI 1640 medium supplemented with GM-CSF (PeproTech) and IL-4 (PeproTech); the medium was replaced with fresh medium every 2 days. On the 7th day, nonadherent and loosely adherent cells were collected and pulsed with 100 µg/ml LPS and 50 µg/ml OVA for 24 h to obtain LPS-pulsed and OVA-pulsed DCs, respectively.
Determination of OVA-specific immunoglobulin levels using ELISA
Tail vein blood was collected, and plasma was separated by centrifugation at 1000 × g for 15 min at 4 °C. Briefly, a NuncTM MaxiSorbTM ELISA plate (BioLegend, Inc.) was coated with 50 µl of 5 µg/ml OVA for 16–18 h and later blocked with 1.0% BSA for 1 h at room temperature. Fifty microliters of diluted plasma (1:100) was added to each well of a coated plate. After 1 h of incubation and washing, HRP-conjugated goat anti-mouse IgG (1:2000 dilution; BioLegend, Inc.) was added to the plate, which was incubated for 1 h. The TMB substrate was added, and the absorbance of color that developed was subsequently quantified at 450 nm (SpectraMAX® M5e microplate reader).
In vivo vaccination using OVA-DCs S-cryoMNs
This experiment was performed on C57BL/6 mice (male, 6–8 weeks) in accordance with ethical approval by the Animal Research Ethics Sub-Committee of City University of Hong Kong (reference no. A-0493). The animals were purchased from the Laboratory Animal Research Unit of City University of Hong Kong. The animals were housed in ventilated cage systems under a 12:12 h LD cycle at constant temperature and humidity. The mice were randomly allocated to two treatment groups: the blank group and the OVA-DC S-cryoMNs group (n = 4). Each mouse was vaccinated twice a week for a total of eight treatments, in which 2 × 105 OVA-DCs were administered in each treatment in the OVA-DC S-cryoMN group. After S-cryoMNs were applied, the treatment sites were covered with Tegaderm™ film for protection, which was removed together with the S-cryoMNs after 8 h. On day 28, the mice were sacrificed, and the lymph nodes, spleens, skin, and major organs were collected. Lymphocytes from excised lymph nodes were stained for surface markers of mature DCs (CD11c, MHCII, and CD86) to evaluate the homing effects of the delivered OVA-DC vaccines. Splenocytes were obtained from excised spleens and restimulated with OVA to evaluate specific proliferation and IFN-γ secretion in the presence of the model antigen. Proliferation was assessed using an AlamarBlueTM viability assay, and IFN-γ secretion was measured using a mouse IFN-γ ELISA kit (BioLegend, Inc., USA). OVA-specific lysis of cytotoxic T lymphocytes was subsequently determined. Splenocytes and B16-OVA cells were cocultured at ratios of 10:1, 33:1, and 100:1 in U-bottom 96-well plates for 2 h. The percentages of lysed B16-OVA cells were determined by a CyQUANTTM LDH cytotoxicity assay kit (Thermo Fisher Scientific). Treated and untreated skin and major organs were fixed with 4% paraformaldehyde and cryosectioned for histological analysis.
Statistical analysis
Each experiment was repeated at least three times in triplicate unless otherwise specified. Student’s t test was used to determine p values, with p < 0.05 considered to indicate statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
References
Lee, K. et al. A patch of detachable hybrid microneedle depot for localized delivery of mesenchymal stem cells in regeneration therapy. Adv. Funct. Mater. 30, 2000086 (2020).
Chen, Y.-H., Lin, D.-C., Chern, E. & Huang, Y.-Y. The use of micro-needle arrays to deliver cells for cellular therapies. Biomed. Microdevices 22, 63 (2020).
Li, H. et al. Scattered seeding of CAR T cells in solid tumors augments anticancer efficacy. Natl. Sci. Rev. 9, nwab172 (2021).
Chang, H. et al. Cryomicroneedles for transdermal cell delivery. Nat. Biomed. Eng. 5, 1008–1018 (2021).
Cui, M. et al. Ocular delivery of predatory bacteria with cryomicroneedles against eye infection. Adv. Sci. 8, e2102327 (2021).
Gao, Y. et al. Highly porous silk fibroin scaffold packed in PEGDA/sucrose microneedles for controllable transdermal drug delivery. Biomacromolecules 20, 1334–1345 (2019).
Liu, L., Kai, H., Nagamine, K., Ogawa, Y. & Nishizawa, M. Porous polymer microneedles with interconnecting microchannels for rapid fluid transport. RSC Adv. 6, 48630–48635 (2016).
Li, J. et al. Fabrication of a Ti porous microneedle array by metal injection molding for transdermal drug delivery. PLoS ONE 12, e0172043 (2017).
van der Maaden, K. et al. Microneedle-based drug and vaccine delivery via nanoporous microneedle arrays. Drug Deliv. Transl. Res. 5, 397–406 (2015).
Tekko, I. A. et al. Localised and sustained intradermal delivery of methotrexate using nanocrystal-loaded microneedle arrays: potential for enhanced treatment of psoriasis. Eur. J. Pharm. Sci. 152, 105469 (2020).
Hao, C. et al. A swellable microneedle patch to rapidly extract skin interstitial fluid for timely metabolic analysis. Adv. Mater. 29, 1702243 (2017).
Bok, M. et al. Microneedles integrated with a triboelectric nanogenerator: an electrically active drug delivery system. Nanoscale 10, 13502–13510 (2018).
Hou, Q., Grijpma, D. W. & Feijen, J. Porous polymeric structures for tissue engineering prepared by a coagulation, compression moulding and salt leaching technique. Biomaterials 24, 1937–1947 (2003).
Bai, Y.-X. & Li, Y.-F. Preparation and characterization of crosslinked porous cellulose beads. Carbohydr. Polym. 64, 402–407 (2006).
Hey, J. & MacFarlane, D. Crystallization of ice in aqueous solutions of glycerol and dimethyl sulfoxide. 1. A comparison of mechanisms. Cryobiology 33, 205–216 (1996).
Gao, D. & Critser, J. Mechanisms of cryoinjury in living cells. ILAR J. 41, 187–196 (2000).
Zhong, Y., McGrath, J. K. & Gong, B. Dipropinonates of sugar alcohols as water-soluble, nontoxic CPAs for DMSO-free cell cryopreservation. ACS Biomater. Sci. Eng. 7, 4757–4762 (2021).
Gore, M., Narvekar, A., Bhagwat, A., Jain, R. & Dandekar, P. Macromolecular cryoprotectants for the preservation of mammalian cell culture: lessons from crowding, overview and perspectives. J. Mater. Chem. B 10, 143–169 (2022).
Deller, R. C., Vatish, M., Mitchell, D. A. & Gibson, M. I. Synthetic polymers enable non-vitreous cellular cryopreservation by reducing ice crystal growth during thawing. Nat. Commun. 5, 3244 (2014).
Deller, R. C., Vatish, M., Mitchell, D. A. & Gibson, M. I. Glycerol-free cryopreservation of red blood cells enabled by ice-recrystallization-inhibiting polymers. ACS Biomater. Sci. Eng. 1, 789–794 (2015).
Naaldijk, Y., Staude, M., Fedorova, V. & Stolzing, A. Effect of different freezing rates during cryopreservation of rat mesenchymal stem cells using combinations of hydroxyethyl starch and dimethylsulfoxide. BMC Biotechnol. 12, 49 (2012).
Imaizumi, K. et al. A simple and highly effective method for slow-freezing human pluripotent stem cells using dimethyl sulfoxide, hydroxyethyl starch and ethylene glycol. PLoS ONE 9, e88696 (2014).
Wang, H.-Y., Lun, Z.-R. & Lu, S.-S. Cryopreservation of umbilical cord blood-derived mesenchymal stem cells without dimethyl sulfoxide. CryoLetters 32, 81–88 (2011).
Ashwood-Smith, M., Warby, C., Connor, K. & Becker, G. Low-temperature preservation of mammalian cells in tissue culture with polyvinylpyrrolidone (PVP), dextrans, and hydroxyethyl starch (HES). Cryobiology 9, 441–449 (1972).
Stolzing, A., Naaldijk, Y., Fedorova, V. & Sethe, S. Hydroxyethylstarch in cryopreservation—mechanisms, benefits and problems. Transfus. Apher. Sci. 46, 137–147 (2012).
Asada, M., Ishibashi, S., Ikumi, S. & Fukui, Y. Effect of polyvinyl alcohol (PVA) concentration during vitrification of in vitro matured bovine oocytes. Theriogenology 58, 1199–1208 (2002).
Bai, H. et al. Biomimetic gradient scaffold from ice-templating for self-seeding of cells with capillary effect. Acta Biomater. 20, 113–119 (2015).
Lee, C. et al. Evaluation of the anti‐wrinkle effect of an ascorbic acid‐loaded dissolving microneedle patch via a double‐blind, placebo‐controlled clinical study. Int. J. Cosmet. Sci. 38, 375–381 (2016).
Parekkadan, B. & Milwid, J. M. Mesenchymal stem cells as therapeutics. Annu. Rev. Biomed. Eng. 12, 87–117 (2010).
Yaojiong, W., Liwen, C., Scott, P. G. & Tredget, E. E. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells 25, 2648–2659 (2007).
Jin, Y. et al. Common variants in FOXP1 are associated with generalized vitiligo. Nat. Genet. 42, 576–578 (2010).
Yaghoobi, R., Omidian, M. & Bagherani, N. Vitiligo: a review of the published work. J. Dermatol. 38, 419–431 (2011).
Zokaei, S. et al. Cultured epidermal melanocyte transplantation in vitiligo: a review article. Iran J. Public Health 48, 388–399 (2019).
Dillon, A. B., Sideris, A., Hadi, A. & Elbuluk, N. Advances in vitiligo: an update on medical and surgical treatments. J. Clin. Aesthet. Dermatol. 10, 15–28 (2017).
Lin, J. Y. & Fisher, D. E. Melanocyte biology and skin pigmentation. Nature 445, 843–850 (2007).
Chen, Y.-F. et al. Treatment of vitiligo by transplantation of cultured pure melanocyte suspension: analysis of 120 cases. J. Am. Acad. Dermatol. 51, 68–74 (2004).
Quezada, N., Filho, C. A. M., De La Sotta, P. & Uribe, P. Melanocytes and keratinocytes transfer using sandpaper technique combined with dermabrasion for stable vitiligo. Dermatol. Surg. 37, 192–198 (2011).
Verma, R. et al. A comparative study of efficacy of cultured versus non cultured melanocyte transfer in the management of stable vitiligo. Med. J. Armed Forces India 70, 26–31 (2014).
Mutalik, S. & Ginzburg, A. Surgical management of stable vitiligo: a review with personal experience. Dermatol. Surg. 26, 248–254 (2000).
Fan, S. M.-Y. et al. Preclinical evaluation of melanocyte transplantation by chitosan-based melanocyte spheroid patch to skin prepared by controlled sunburn blistering. J. Biomed. Mater. Res. Part B Appl. Biomater. 106, 2535–2543 (2018).
Swope, V. B., Supp, A. P. & Boyce, S. T. Regulation of cutaneous pigmentation by titration of human melanocytes in cultured skin substitutes grafted to athymic mice. Wound Repair. Regen. 10, 378–386 (2002).
Foy, S. P. et al. Non-viral precision T cell receptor replacement for personalized cell therapy. Nature 615, 687–696 (2023).
Mantovani, A., Allavena, P., Marchesi, F. & Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 21, 799–820 (2022).
Dzobo, K., Senthebane, D. A. & Dandara, C. The tumor microenvironment in tumorigenesis and therapy resistance revisited. Cancers 15, 376 (2023).
Saleh, R. & Elkord, E. Acquired resistance to cancer immunotherapy: role of tumor-mediated immunosuppression. Semin. Cancer Biol. 65, 13–27 (2020).
Xu, Y. et al. Long wavelength–emissive Ru (II) metallacycle–based photosensitizer assisting in vivo bacterial diagnosis and antibacterial treatment. Proc. Natl Acad. Sci. 119, e2209904119 (2022).
Li, C. et al. Near-infrared metal agents assisting precision medicine: from strategic design to bioimaging and therapeutic applications. Chem. Soc. Rev. 52, 4392–4442 (2023).
Leidner, R. et al. Neoantigen T-cell receptor gene therapy in pancreatic cancer. N. Engl. J. Med. 386, 2112–2119 (2022).
Acknowledgements
C.X. acknowledges the support of CityU Strategic Interdisciplinary Research Grant (#7020029), the General Research Fund (GRF) from the Research Grants Council (RGC) of the Hong Kong Special Administrative Region, China (Project No. CityU11202222), and the Research Institute of Tsinghua at Pearl River Delta (9239094). W.L. acknowledges funding support from the National Natural Science Foundation of China (31870967, 81671921).
Author information
Authors and Affiliations
Contributions
C.X. and M.Z. conceived the original idea of the study. C.X., W.L. and K.Y. supervised the project. M.Z., T.H. and Y.Y. contributed to the design of the study. M.Z., T.H., Y.Y., X.Q., H.Y., Y.Z. and Q.Z. performed the experiments and analyzed the results. C.X. and M.Z. drafted the manuscript with input from all the authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zheng, M., Hu, T., Yang, Y. et al. In situ-formed cryomicroneedles for intradermal cell delivery. NPG Asia Mater 16, 11 (2024). https://doi.org/10.1038/s41427-024-00531-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41427-024-00531-1
