
Abstract
Poly(ethylene glycol) (PEG)-based hydrogels are promising materials for biomedical applications because of their excellent hydrophilicity and biocompatibility. However, conventional chemically cross-linked PEG hydrogels are brittle under mechanical loading. The mechanical resilience and rapid recovery abilities of hydrogel implants are critical in load-bearing tissues, such as articular cartilage, which are routinely subjected to cyclic loadings of high magnitude and frequency. Here, we report the fabrication of novel supramolecular PEG hydrogels by polymerizing N,N-dimethylacrylamide with supramolecular cross-linkers self-assembled from adamantane-grafted PEG and mono-acrylated β-cyclodextrin. The resultant PEG–ADA supramolecular hydrogels exhibit substantial deformability, excellent capacity to dissipate massive amounts of loading energy, and have a rapid, full recovery during excessive, ultrafast, and non-resting cyclic compression. Furthermore, the energy dissipation capacity of the PEG–ADA (adamantane-grafted Poly(ethylene glycol)) hydrogels can be regulated by changing the concentration, molecular weight and cross-linking density of PEG. According to in vitro cell metabolism and viability tests, the PEG–ADA hydrogels are non-cytotoxic. When placed over a monolayer of myoblasts that were subjected to instantaneous compressive loading, the PEG–ADA hydrogel cushion significantly enhanced cell survival under this deleterious mechanical insult compared with the effects of the conventional PEG hydrogel. Therefore, PEG–ADA hydrogels are promising prosthetic biomaterials for the repair and regeneration of load-bearing tissues.
Similar content being viewed by others

Introduction
Hydrogels are widely thought to be promising scaffold materials for tissue engineering and regenerative medicine1,2,3,4. Particularly, poly(ethylene glycol) (PEG) hydrogels are promising materials for many biomedical applications, including wound dressing, drug delivery, and tissue engineering, due to their excellent biocompatibility, non-immunogenicity, and resistance to protein adsorption. However, the brittle mechanical property and lack of fatigue resistance of conventional covalently cross-linked PEG hydrogels restrict the wide-spread application, especially where biomechanical loading is significant. To overcome these limitations of conventional chemical hydrogels, researchers have capitalized on a wide array of physical interactions to develop physical hydrogels with unique properties, including self-healing and shear-thinning5,6,7. However, conventional physical hydrogels are usually too weak to be implanted in load-bearing sites of clinical interest, such as the knee joint5,8,9,10. Thus, the development of strategies to improve the mechanical properties of hydrogel scaffolds, such as mechanical resilience and fatigue resistance, has become crucial to the research field of hydrogels. Extensive pioneering studies have been conducted to improve the stability and robustness of physical hydrogels. Liu et al. employed H-bond and dipole–dipole interactions to enhance the mechanical strength and fatigue resistance of physical hydrogels11,12. Meijer et al. fabricated tough supramolecular hydrogels cross-linked by the H-bonds between self-complementary UPy units13,14. Scherman et al. demonstrated the fabrication of supramolecular cross-linked hydrogels by the host–guest interactions of cucurbit [n] uril complexes15,16,17. In addition to their pioneering study on double-network hydrogels, Gong et al. developed ionic bond-based polyampholyte hydrogels that possess excellent robustness and viscoelasticity6,18,19,20,21. Aida et al. developed mechanically robust hydrogels by incorporating various nanocomposites22,23,24. The aforementioned research demonstrates the great potential of supramolecular chemistry to improve the mechanical properties and fatigue resistance of hydrogels. However, none of these studies have investigated the energy dissipation capacity of hydrogels under high-frequency mechanical loadings. In addition, the fatigue resistance of these hydrogels is achieved either under small deformations11,25 or after a long waiting time (>30 min)6,26. These limitations significantly compromise the fatigue resistance of hydrogels under high-frequency cyclic loading of large deformations.
The β-cyclodextrin and adamantane (βCD–ADA) host–guest complex is stable under aqueous conditions with a high-binding constant (K = ~105 m−1)27 and rapid binding kinetics (ka = ~108 m−1 s−1, kd = ~103 s−1)28,29,30. Therefore, the βCD–ADA complex is an ideal non-covalent system for mediating energy dissipation during loading and network recovery during unloading in antifatigue hydrogels. However, researchers have paid little attention to the energy dissipation capacity of the supramolecular hydrogels cross-linked by the βCD–ADA complexes, mainly because of the mechanical weakness of such hydrogels. Most of the such weak host–guest hydrogels were formed by mixing βCD-modified polymers with ADA-modified polymers and verified with the “tube-inversion” test10,31,32. Recently, Harada et al. fabricated highly elastic polyacrylamide hydrogels cross-linked by host–guest cross-linkers that are preassembled from βCD monomers and ADA monomers33,34. This method significantly improved the stability and elasticity of the hydrogels and demonstrated the important role of energy dissipation by host–guest complexations in enhancing the reversible elastic deformation and toughness of the host–guest hydrogels. Furthermore, Suo et al. also reported highly stretchable and tough hydrogels by introducing energy dissipation structures35. In contrast, the lack of energy dissipation in covalently cross-linked hydrogels leads to stress concentration, premature rupture and permanent damage of hydrogel networks under excessive loadings. This leads to a diminished capacity of covalently cross-linked hydrogels to resist cyclic loading with an increasing number of loading cycles. The introduction of energy-dissipating host–guest cross-links will potentially improve the mechanical properties of the hydrogels, such as deformability and toughness, under excessive cyclic loadings.
Because of its excellent hydrophilicity and biocompatibility, PEG was selected as the platform material for testing the energy-dissipating mechanism of the novel hydrogels fabricated in this study. The brittle mechanical property and lack of fatigue resistance of conventional covalently cross-linked PEG hydrogels (e.g., PEGDA hydrogels) restrict their wide-spread applications, especially where biomechanical loading is significant. Herein, we utilized the energy dissipation capacity of the βCD–ADA complex as the cross-linker to fabricate mechanically stable supramolecular poly(N,N-dimethylacrylamide) (PDMA) PEG hydrogels, which are prepared by cross-linking the N,N-dimethylacrylamide monomer with the preassembled adamantane-grafted poly(ethylene glycol) (PEG–ADA) and βCD supramolecular cross-linkers (Ac-βCD–ADA–PEG–ADA–Ac-βCD). The supramolecular cross-linkers were formed by the self-assembly of ADA-grafted PEG polymers and mono-acrylated β-cyclodextrin monomers (mono-Ac-βCD) under aqueous conditions. The excellent water solubility of this supramolecular cross-linker eliminates the additional organic solvent typically required by conventional methods of host–guest hydrogel fabrication to dissolve the guest molecule, thereby facilitating the biomedical applications of such hydrogels.
The focus of this study is to investigate the mechanical properties of these supramolecular PEG hydrogels, including deformability, energy dissipation, and fatigue resistance, in comparison with those of the chemical hydrogels cross-linked by conventional PEG diacrylate (PEGDA) and a co-monomer of identical concentrations, and cell behaviors responding to such unique mechanical properties. Our strategy successfully optimizes the mechanical performance of PEG hydrogels under stringent mechanical loading, and our findings provide critical insights into the translation of host–guest hydrogels to biomedical applications. The supramolecular cross-links based on the host–guest complexation significantly enhance the energy dissipation, fatigue resistance, and stress-relaxation of supramolecular PEG hydrogels and protect cells from deleterious mechanical forces. To the best of our knowledge, our work is the first to capitalize on host–guest supramolecular chemistry to develop energy-dissipating PEG hydrogels and to systemically study the effects of design parameters on the energy dissipation of PEG hydrogels. Our findings provide valuable guidance on the design of prosthetic hydrogels for loading bearing implantation sites with surrounding mechanosensitive cells and tissues.
Materials and methods
Hydrogel fabrication
Details about the synthesis of ADA–PEG–ADA and host monomer (mono-Ac-βCD) are provided in the supporting information. The precursor solution of supramolecular PEG–ADA hydrogels was prepared by dissolving and mixing ADA–PEG–ADA and mono-Ac-βCD in phosphate-buffered saline (PBS). After thorough stirring of the mixture for 1 h, the co-monomer (N,N-dimethylacrylamide (DMA), aqueous solution), initiator (ammonium persulfate (APS)), and catalyst (1,2-dis(dimethylamino)ethane (TEMED)) were added to the mixture. The final solution was quickly transferred to a cylindrical mold (4.7-mm diameter and 3-mm height). The hydrogels were removed from the mold after 2 h and washed with PBS. The PEG–ADA hydrogels were fabricated using four different concentrations of ADA–PEG–ADA (3, 6, 9, and 12% (w/v)) with equivalent molar amounts of mono-Ac-βCD to the molar amount of ADA. The control PEGDA hydrogels were fabricated using PEGDA instead of the Ac-βCD–ADA–PEG–ADA–Ac-βCD as cross-linkers under the same conditions. For all comparative assessments between the PEG–ADA and PEGDA hydrogels, both hydrogels were fabricated using a DMA concentration of 1 m and APS/TEMED concentrations of 20 mM each. In the PEG–ADA hydrogels, the DMA concentration was later adjusted from 0.5 to 2 min, and the APS/TEMED concentrations were later adjusted from 10 mM each to 100 mM each to study the effect of DMA and APS/TEMED concentrations on the properties of the PEG–ADA hydrogel.
Loading–unloading compression test
Before mechanical testing, all as-prepared hydrogels were immersed in PBS for 12 h to ensure complete swelling. The dimensions of the hydrogels were measured by digital Vernier calipers. The compression tests were conducted on a Mach-1TM micromechanical system (Biomomentum Inc., Canada). A continuous loading speed was set at the strain rate of 5% per second, and the maximum loading strain was set at 90% to prevent damage to the testing instrument. After determining the maximum strain in the unconfined compression test, the peak strain in the continuous loading–unloading compression test was set to be slightly lower than the maximum strain (supramolecular PEG-ADA hydrogel: 90%, PEGDA hydrogel: 60%).
Loading–unloading compression test with varying loading speeds
The dependence of the hydrogel mechanical properties on the strain rate was explored by conducting a continuous loading–unloading compression test with a series of accelerating loading speeds: 2% per second, 5% per second, 10% per second, 12.5% per second, 16% per second, and 25% per second. Two loading–unloading cycles were applied for each loading speed. There was no resting between the two cycles or cycles with different loading speeds. In this test, the peak strain of each cycle was 90%.
Stress-relaxation test
Stress-relaxation tests were conducted to compare the relaxation modes between the PEG–ADA and PEGDA hydrogels. Both hydrogels were compressed to the peak strains (in 0.5 s of loading time) by applying a series of 10% strain rampings with 5 min of relaxation between successive rampings. The peak strains of the PEGDA and PEG–ADA hydrogels were 60 and 90%, respectively.
Rheological test
Frequency sweep and strain sweep were performed on a rheometer (Anton Paar MCR301) with 8-mm-diameter plates at a 0.2-mm gap (plate-to-plate distance). All measurements were conducted at 25 °C, unless otherwise stated. Strain sweep was performed at a set frequency of 1 Hz. Frequency sweep was performed at a set strain of 5%.
In vitro cytotoxicity assay
Human mesenchymal stem cells (hMSCs, Lonza) were expanded to fourth passages in growth media consisting of α-minimum essential medium supplemented with 16.7% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Pen/Strep). The cells were cultured at 37 °C with 5% CO2.
The conditioned cell culture medium obtained by soaking the PEG–ADA hydrogels in the cell culture medium for varying durations (2, 14, and 28 days) were used to assay the impact of the hydrogels on cultured cells. The hydrogels used in this assay were fabricated following the same scheme described earlier in the text. After the hydrogels were fabricated and washed with PBS, they were exposed to UV light for 30 min for sterilization. Three hydrogels were placed in the cell culture medium (25 mL) for varying durations. The hydrogels were removed from the media at the time points of interest (2, 14, and 28 days), and the media were then frozen at −80 °C. After removal of the hydrogels at all-time points, the frozen media were thawed and used as cell culture media.
Passage-four hMSCs were seeded at a density of 5000 cells cm−2 in 24-well plates with 1 mL of conditioned medium per well, and the cells were cultured for varying durations (24 h, 48 h, 72 h, and 7 days). Alamar Blue (50 μL) was added to each well at the time points of interest. After 4 h of culture, the culture medium (100 μL) containing the Alamar Blue dye was added to 96-well plates. The absorbances of the media at 562 nm were measured using a microplate reader. After 7 days of culture, the viability of the cultured cells was tested using calcein-AM (0.5 μL/mL) or propidium iodide. Fluorescence images were taken at excitation wavelengths of 488 and 514 nm.
In vitro mechanical loading
C2C12 mouse skeletal myoblasts (ATCC, Manassas, USA) expanded to fourth passages were grown to 95% confluency in a growth medium consisting of Dulbecco’s Modified Eagle Medium supplemented with 4.5 g L−1 glucose, 10% FBS, and 1% penicillin/streptomycin (Gibco®). We conducted uniaxial compression tests on 9% (w/v) PEGDA and PEG–ADA hydrogels covering a monolayer of myoblasts. The hydrogels were compressed by a flat platen at a strain rate of 25% per second to reach the peak compressive stress of 500 Pa, followed by 5 min of stress-relaxation while maintaining a constant compressive strain on the hydrogels. Subsequently, calcein-AM (0.5 μL mL−1) and PI (3 mM) were used to assess the percentage of cell death occurring below the hydrogel cushion. Fluorescence images of cell viability staining were taken at ×100 magnification on a motorized stage (ECLIPSE Ti, Nikon). The images were combined to generate one full-field image of the C2C12 monolayer.
Results and discussion
Fabrication of PEG–ADA supramolecular hydrogels
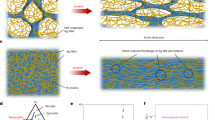
The grafting degree of ADA on both ends of the HO–PEG–OH of three different molecular weights (2 K, 4 K, and 10 K) was estimated to be 100%, as demonstrated by the 1H NMR spectra (Figure S1, S2, and S3). The preparation of mono-functionalization of βCD (mono-Ac-βCD) was also confirmed by the 1H NMR (Figure S4). Mixing of ADA–PEG–ADA and mono-Ac-βCD yielded the supramolecular cross-linker of ac-βCD–ADA–PEG–ADA–Ac-βCD, which was self-assembled through efficient complexation between ADA and the βCD monomer, confirmed by the 2D-NOESY NMR analysis (Figure S5)36. Subsequent UV exposure induced radical polymerization of the supramolecular cross-linker in the presence of co-monomer (DMA), generating the free-standing supramolecular PEG–ADA hydrogels (Fig. 1).

a ADA-grafted PEG polymers. b βCD grafted with a single-acrylate group as the host monomer. c The self-assembly of ADA–PEG–ADA and mono-Ac-βCD in PBS yielded the supramolecular Ac-βCD–ADA–PEG–ADA–Ac-βCD cross-linker. d The PEG–ADA supramolecular hydrogels were obtained by the radical polymerization of the supramolecular cross-linker in the presence of co-monomer (DMA) with APS/TEMED as the initiator and catalyst. e PEG–ADA hydrogel network structure. f The representation of PEGDA polymers. g The PEGDA hydrogels were obtained by radical polymerization in the presence of a co-monomer (DMA, 1 M) with APS/TEMED (20/20 mM) as the initiator and catalyst
PEG–ADA hydrogels can withstand excessive mechanical loadings and effectively dissipate massive loading energy
The supramolecular PEG–ADA hydrogels demonstrate significantly improved deformability compared with the covalently cross-linked PEGDA hydrogels. The compression test revealed no fractures in any of the PEG-ADA hydrogel groups at compressive strains of up to 90% (Figure S6b), which is significantly higher than the fracture strain of the corresponding PEGDA hydrogels fabricated using an identical method (Figure S6a). The rheological strain sweep analysis also revealed that the fracture shear strain of the PEG–ADA hydrogels (1261.5%) (Figure S6d) was much larger than that of the PEGDA hydrogels (79.5%) (Figure S6c). In addition, the structural integrity or surface smoothness of PEG–ADA hydrogels were maintained when the hydrogels were sliced by a knife or abraded by abrasive paper under torsion (Fig. 2a, c, d and f). By contrast, the PEGDA hydrogels ruptured under either type of these mechanical challenges (Fig. 2b, e).

Cutting (a) and torsion (d) tests of the PEGDA hydrogels and PEG–ADA hydrogels. The PEGDA hydrogels easily ruptured when sliced by a knife (b) or abraded by abrasive paper under torsion (e). The structural integrity of the PEG–ADA hydrogels was maintained when sliced by a knife (c), and their surface smoothness was maintained when abraded by abrasive paper under torsion (f). The PEG cross-linker concentration of both PEGDA and PEG–ADA hydrogels was 9%, and the mono-Ac-βCD concentration of PEG–ADA hydrogels was 6%. Both hydrogels were fabricated using the DMA concentration of 1 M and the APS/TEMED concentrations of 20 mM each
These results indicate that replacing the covalent cross-linking with the physical cross-linking mediated by the βCD–ADA host–guest complexation effectively enhances the deformability of the PEG hydrogels. The loading and unloading test revealed the significant energy dissipation capacity of the PEG–ADA hydrogels during loading, as demonstrated by the gap between the loading and unloading curves (Fig. 3a). By contrast, no such hysteresis was observed in the loading–unloading curves of the PEGDA hydrogels (Fig. 3a), thereby indicating little dissipation of the applied loading energy by PEGDA hydrogels. Furthermore, quantitative analysis of the energy dissipation capacity, which was conducted by integrating the area between the loading and unloading curves of the force versus displacement, confirmed the significantly enhanced energy dissipation capacity of the PEG–ADA hydrogels compared with that of the PEGDA hydrogels (Fig. 3b). We believe that the dissipation of loading energy is mediated by the dissociation of βCD–ADA complexes in the PEG–ADA hydrogels, and this phenomenon may help reduce the fracturing of the hydrogel network structures, thereby leading to superior deformability.

a Loading–unloading curves of the PEG-ADA hydrogels (red) and the PEGDA hydrogels (black) at the maximum of the safe strain. b Quantitative analysis of the energy dissipation capacity (at the maximum of the safe strain) of the PEGDA hydrogels (black) and PEG–ADA hydrogels (red) with different PEG cross-linker concentrations. The PEG–ADA hydrogels dissipated 8.31, 12.62, 12.10, and 13.86 times more energy than the PEGDA hydrogels with identical PEG cross-linker content (3%, 6%, 9%, and 12%, respectively). c Stress-relaxation curves of the PEGDA hydrogels (solid lines) and the PEG–ADA hydrogels (dotted lines) with different peak compressive strains. d The relaxation rate constants of PEGDA hydrogels (red triangle) and the PEG–ADA hydrogels (black and blue dot) were obtained by exponential fitting of the curves in c. Statistical significance was determined based on the method described in Supporting Information (*p < 0.05, **p < 0.01, ***p < 0.001). The PEG cross-linker concentration of both PEGDA and PEG–ADA hydrogels was 9%, and the mono-Ac-βCD concentration of the PEG–ADA hydrogels was 6%. Both hydrogels were fabricated using the DMA concentration of 1M and the concentrations of APS/TEMED of 20 mM each
PEG–ADA hydrogels exhibit rapid stress-relaxation because of dissociation of host–guest cross-links
We also performed the stress-relaxation test on the PEG–ADA hydrogels, because energy dissipation in the hydrogels can also influence stress-relaxation kinetics. In the stress-relaxation test, the two types of hydrogels were compressed to the peak strains by applying a series of 10% strain rampings with 5 min of relaxation between successive rampings (Fig. 3c). The peak strain of the PEGDA hydrogels was set at 60% to avoid hydrogel rupture, and the peak strain of the PEG–ADA hydrogels was set at 90% to prevent damage to the testing instrument. For clarity, all stresses obtained during the stress-relaxation test were normalized to the corresponding peak stresses.
Curve fitting of the stress-relaxation profiles demonstrated that the PEG–ADA hydrogels had one additional relaxation mechanism compared with that of the PEGDA hydrogel (Fig. 3d). The relaxation curves of the PEGDA hydrogels were well fitted to the exponential equation with a single-decay factor (k1, Equation (1)); whereas, the relaxation curves of the PEG–ADA hydrogels could be fitted to the exponential equation with two decay factors (k1′ and k2, Equation (2)).
Fitting equations:
σt is the stress at a certain time t; σ0 is the peak stress; A, B, and C are constants; k1 is relaxation rate constant of the PEGDA hydrogels; k1′ and k2 are the slow and rapid relaxation rate constants of the PEG–ADA hydrogels, respectively.
The slow relaxation rate constants (k1′) of the PEG-–ADA hydrogels under varying compressive strains (60: 0.0188, 70: 0.0128, 80: 0.0136, and 90%: 0.0147) were comparable to the relaxation rate constants (k1) of the PEGDA hydrogels under slightly lower compressive strains (30: 0.0056, 40: 0.0050, 50: 0.0061, and 60%: 0.0064). This finding suggests the existence of a common stress-relaxation mechanism for both hydrogels. The stress-relaxation of covalently cross-linked hydrogels such as PEGDA is known to be dominated by water exudation37,38. Therefore, these data indicate that water migration also contributes to the stress-relaxation mechanism of the supramolecular PEG–ADA hydrogels. Furthermore, the rapid relaxation rate constants (k2) of the PEG–ADA hydrogels (60: 0.1183, 70: 0.1141, 80: 0.1961, and 90%: 0.2345) are approximately one order of magnitude higher than the relaxation rate constants (k1) of the PEGDA hydrogels. This result suggests the presence of a more effective stress-relaxation mechanism for the PEG–ADA hydrogels. In addition, the k2 value increased with increasing compressive peak strain, indicating that stress-relaxation mediated by the rapid relaxation mode of the PEG–ADA hydrogels is more rapid at a higher strain level. We postulate that the rapid stress-relaxation of the PEG–ADA hydrogels is achieved by the effective dissipation of a large amount of energy through the rapid disassociation of the βCD–ADA complexes, because this is the only structural difference between the PEGDA and PEG–ADA hydrogels.
PEG–ADA hydrogels maintain consistent mechanical properties under excessive cyclic compression without rest
Continuous cyclic loading–unloading compression tests (peak strain of 90%) on the PEG–ADA hydrogels (positioned in a dry test chamber) without rest revealed significant hysteresis during each loading cycle, as demonstrated by the gap between the loading and unloading curves (hysteresis loop) (Fig. 4a). Furthermore, complete overlap was observed for the hysteresis loops of different loading cycles. This finding indicates that the PEG-ADA hydrogels can rapidly recover the network structure upon unloading.

a Loading–unloading curves of the PEG–ADA hydrogels (positioned in a dry test chamber with exposure to air) recorded during 10 cycles of non-resting cyclic compression with a loading speed of 2% strain per second. b Loading–unloading curves of PEG–ADA hydrogels subjected to a series of compression cycles with accelerating loading speed from 2% per second to 25% per second, and two continuous cycles were performed at each loading speed. The tested hydrogel was immersed in PBS solution to mimic the physiological environment. The PEG cross-linker concentration of both PEGDA and PEG–ADA hydrogels was 9%, the mono-Ac-βCD concentration of the PEG–ADA hydrogels was 6%. Both hydrogels were fabricated using the DMA concentration of 1M and concentrations of APS/TEMED of 20 mM each
More importantly, the PEG–ADA hydrogels maintained the effective energy dissipation when immersed in PBS solution and could completely recover the original mechanical properties, even under ultrafast loading frequency (Fig. 4b). This study found that the PEG–ADA hydrogels maintained a consistent hysteresis loop during continuous cyclic compression with a series of accelerating loading speeds (2% per second, 5% per second, 10% per second, 12.5% per second, 16% per second, and 25% per second). Moreover, complete overlap was observed for the hysteresis loops of different loading–unloading cycles despite the increasing loading speed.
The rapid recovery of the PEG–ADA hydrogels may result from two structural components in these hydrogels. One structural component is the βCD–ADA complexes, which exhibit rapid binding kinetics (ka = ~108 m−1 s−1, kd = ~103 s−1)28,29,30,39, and their instant dissociation and re-association favors high-fidelity re-association between specific pairs of βCD and ADA, with minimal mismatching. The other structural component is the elastic chains (PDMA) formed by polymerized DMA. The PDMA chains help keep the host–guest complexes close to their original locations. Therefore, the βCD and ADA molecules that are dissociated during loading are more likely to rapidly re-associate with their original partners or those close by upon unloading. Such preservation of the spatial organization of the host–guest cross-links limits the plastic deformation and improves the fatigue resistance of the PEG–ADA hydrogels.
Energy dissipation capacity of PEG–ADA hydrogels is dependent on several key parameters
We examined the dependence of the energy dissipation capacity of the PEG–ADA hydrogels on different gelation parameters. The amount of loading energy dissipated by the PEG–ADA hydrogels increased with increasing PEG cross-linker concentration (3%: 0.25 × 10−3 ± 1.32 × 10−8 J, 6%: 0.65 × 10−3 ± 5.25 × 10−8 J, and 9%: 1.10 × 10−3 ± 5.02 × 10−8 J, Fig. 5a). However, the amount of energy dissipated by the PEG–ADA hydrogels became constant at a PEG cross-linker concentration of >9% (12%: 1.09 × 10−3 ± 5.31 × 10−8 J, Fig. 5a). The amount of energy dissipated by the PEG–ADA hydrogels decreased with increasing PEG molecular weight (2 K: 0.66 × 10−3 ± 1.71 × 10−5 J, 4 K: 0.61 × 10−3 ± 2.30 × 10−5J, and 10 K: 0.28 × 10−3 ± 4.52 × 10−5 J, Fig. 5b). The same trend was observed for the influence of the PEG cross-linker concentration and molecular weight on the percentage of dissociated βCD–ADA complexes and the fraction of dissipated energy (Figure S7a, S7b, S8a, and S8b). The amount of energy dissipated by the PEG–ADA hydrogels increased with increasing DMA concentration (0.5 M: 0.04 × 10−3 ± 2.18 × 10−6 J, 1 M: 0.61 × 10−3 ± 2.30 × 10−5 J, and 2 M, 2.44 × 10−3 ± 1.62 × 10−4 J, Fig. 5c). The percentage of dissociated βCD–ADA complexes and the fraction of dissipated energy also increased with increasing DMA concentration (Figure S7c, S8c). The initiator concentration exerted no significant effect on the energy dissipation capacity (20 mM: 0.61 × 10−3 ± 2.30 × 10−5 J, 50 mM: 0.56 × 10−3 ± 7.17 × 10−5 J, and 100 mM: 0.53 × 10−3 ± 4.35 × 10−4 J, Fig. 5d), the percentage of dissociated βCD–ADA complexes, and the fraction of dissipated energy of the PEG–ADA hydrogels. Table S1 summarizes the effect of the gelation parameters on the energy dissipation-related parameters. Table S1 also lists the relationship between the gelation parameters and the swelling ratio to illustrate the relationship between the hydrogel network structure and the energy dissipation capacity of the PEG–ADA hydrogels.

The amount of energy dissipated by the PEG–ADA hydrogels with varying a PEG cross-linker concentration (3%, 6%, 9%, and 12%; the DMA concentration was 1M, and the APS/TEMED concentrations were 20 mM each), b PEG molecular weight (the PEG cross-linker concentration was controlled to 9% for different polymer weights, the concentration of mono-Ac-βCD was 6% according to the PEG cross-linker concentration, the DMA concentration was 1M, and the APS/TEMED concentrations were 20 mM each), c co-monomer concentration (the PEG cross-linker concentration was 9%, the mono-AcβCD concentration was 6% and the APS/TEMED concentrations were 20 mM each), or d initiator (APS/TEMED) concentrations. Statistical significance was determined based on the method described in Supporting Information (*p-value <0.05, **p-value <0.01, ***p-value <0.001, the PEG cross-linker concentration was 9%, the mono-Ac-βCD concentration was 6% and the DMA concentration was 1M)
The PEG–ADA hydrogels dissipated energy through the dissociation of the βCD–ADA complexes. Despite variation in the gelation parameters, only a small fraction of the βCD–ADA complexes was dissociated during compression (Figure S7). This finding indicates that the βCD–ADA complex can dissipate energy with high efficiency. This study also demonstrated that the PEG cross-linker concentration and molecular weight affected the energy dissipation capacity of the hydrogels by changing the hydrogel network structure. As confirmed by the swelling ratio data (Figure S9), a more compact hydrogel network structure can be achieved by increasing the PEG cross-linker concentration or decreasing the PEG molecular weight. A previous study showed that the hydrogels with a more compact structure can dissipate an increasing amount of energy40. This previous finding is consistent with our finding that less swollen PEG–ADA hydrogels dissipate a larger absolute amount and fraction of applied loading energy. The DMA concentration exerted little influence on the swelling ratio of the hydrogels, and the amount of dissipated energy significantly increased with increasing DMA concentration. This result indicates that the PDMA chains may contribute to energy dissipation because of their elasticity.
PEG–ADA hydrogels are non-cytotoxic
In the in vitro cytotoxicity test, the conditioned cell culture media obtained by immersing the PEG–ADA hydrogels in the growth medium for varying durations (2, 14, and 28 days) were used for cell culture. No significant difference was observed in the metabolic activity of the hMSCs between the experimental groups, in which the cells were cultured in conditioned media, and the control group, in which the cells were cultured in regular growth medium (Fig. 6a). No dead cells were observed in all the experimental groups as confirmed by the cell viability test using calcein/PI staining after seven days of culture (Fig. 6b). This result shows that the PEG–ADA hydrogels are cytocompatible. Most of the DMA monomers were polymerized during hydrogel fabrication. The level of un-polymerized DMA and the initiator in the PEG–ADA hydrogels is compatible with cell viability. The appropriate biocompatibility of the PEG–ADA hydrogels shows the potential application of the PEG–ADA hydrogels as implant materials for tissue repair.

Cell metabolic activity (a) and viability (b) in growth medium and conditioned medium obtained after incubation for varying durations with PEG–ADA hydrogels. Cell viability was checked as long as 7 days of culture
PEG–ADA hydrogel cushion promotes the survival of underlying cells subjected to deleterious mechanical loading
A 500-Pa instantaneous compressive stress (25% per second strain rate) was applied to a monolayer of myoblasts through a PEG-ADA or PEGDA hydrogel cushion (Fig. 7a, b). The peak strain induced by the 500-Pa stress was maintained for 5 min of stress-relaxation. Upon completion of the mechanical stress, cell viability staining revealed that the cells below the PEGDA hydrogel cushion showed an extensive area of cell death that matched the size of the hydrogel cushion (Fig. 7d). By contrast, the cells below the PEG–ADA hydrogel cushion showed a much-reduced area of cell death. Quantitative analysis revealed that the percentage of cell death below the PEG–ADA hydrogels was significantly lower than that below the PEGDA hydrogels (50.50 ± 3.05% vs. 82.47 ± 15.22%, p < 0.05, Fig. 7c, d). The substantial dissipation of the loading energy and rapid relaxation by the PEG–ADA hydrogel cushion may have contributed to the enhanced survival of myoblasts under such deleterious mechanical stresses. Many cell types have been shown to be able to sense the mechanical cues in their microenvironment, and the associated mechanotransduction signaling has been implicated in many major diseases, such as cancers, pressure ulcers, fibrosis, and osteoporosis41. This finding revealed the acute sensitivity of cells to mechanical properties, especially the dynamic mechanical properties of cell-contacting materials, which mediate the mechanical signals perceived by the cells.

Cartoon illustration of cell protection from compression by (a) PEG–ADA hydrogel and (b) PEGDA hydrogel. c Percentage of cell death under deleterious compression in monolayers of cells cushioned by PEG–ADA and PEGDA hydrogels. Hydrogels were compressed instantaneously to a peak stress of 500 Pa, followed by 5 min of stress-relaxation. d Viable cells were stained by calcein-AM (green), and non-viable cells were stained by PI (red)
Conclusion
In summary, we fabricated novel PEG-based supramolecular hydrogels by cross-linking supramolecular cross-linkersthat were self-assembled from adamantane-grafted PEG polymer (ADA–PEG–ADA) and mono-acrylated β-cyclodextrin monomer (mono-Ac–βCD). Compared with conventional covalently cross-linked PEGDA hydrogels, these PEG–ADA hydrogels exhibit high deformability, excellent capacity to dissipate massive amounts of loading energy, and superior antifatigue properties under excessive, ultrafast, and non-resting cyclic compression. The improvement of these mechanical properties results from the employment of the βCD–ADA host–guest complex as the cross-linker, which has a high binding constant and rapid binding kinetics. PEG–ADA hydrogels can rapidly recover completely, even under a high compression rate of up to 25% strain increase per second. The parametric analysis of the PEG–ADA hydrogels suggests that the gelation parameters can be modified to customize the hydrogel bulk properties to address the specific demands of different applications. The excellent water solubility of this supramolecular cross-linker eliminates the additional organic solvent typically required by conventional methods for host–guest hydrogel fabrication to dissolve the hydrophobic guest molecules. This is important for the biomedical applications of such hydrogels. The in vitro cell metabolism and viability tests showed that our PEG–ADA hydrogels are non-toxic to cells. By effectively dissipating loading energy and relaxing applied stress, the PEG–ADA hydrogel cushions significantly promoted the survival of the underlying myoblasts compared with the effect of the PEGDA hydrogels. Therefore, we believe that the PEG–ADA hydrogels are promising biomaterials for tissue repair and regeneration at load-bearing sites. Our findings will provide valuable guidance on the design of prosthetic hydrogels for load-bearing implantation sites with surrounding mechanosensitive cells and tissues.
References
Wichterle, O. & Lim, D. Hydrophilic gels for biological use. Nature 185, 117–118 (1960).
Lee, K. Y. & Mooney, D. J. Hydrogels for tissue engineering. Chem. Rev. 101, 1869–1879 (2001).
Hoffman, A. S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 54, 3–12 (2002).
Drury, J. L. & Mooney, D. J. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials 24, 4337–4351 (2003).
Appel, E. A., del Barrio, J., Loh, X. J. & Scherman, O. A. Supramolecular polymeric hydrogels. Chem. Soc. Rev. 41, 6195–6214 (2012).
Sun, T. L. et al. Physical hydrogels composed of polyampholytes demonstrate high toughness and viscoelasticity. Nat. Mater. 12, 932–937 (2013).
Koopmans, C. & Ritter, H. Formation of physical hydrogels via host− guest interactions of β-cyclodextrin polymers and copolymers bearing adamantyl groups. Macromolecules 41, 7418–7422 (2008).
Cheung, H. Y., Lau, K. T., Lu, T. P. & Hui, D. A critical review on polymer-based bio-engineered materials for scaffold development. Compos Part B-Eng. 38, 291–300 (2007).
Zhao, X. Multi-scale multi-mechanism design of tough hydrogels: building dissipation into stretchy networks. Soft Matter 10, 672–687 (2014).
Rodell, C. B., Kaminski, A. L. & Burdick, J. A. Rational design of network properties in guest-host assembled and shear-thinning hyaluronic acid hydrogels. Biomacromolecules 14, 4125–4134 (2013).
Bai, T. et al. Construction of an ultrahigh strength hydrogel with excellent fatigue resistance based on strong dipole–dipole interaction. Soft Matter 7, 2825–2831 (2011).
Zhang, Y., Li, Y. & Liu, W. Dipole–dipole and h‐bonding interactions significantly enhance the multifaceted mechanical properties of thermoresponsive shape memory hydrogels. Adv. Funct. Mater. 25, 471–480 (2015).
Bastings, M. M. C. et al. A fast pH-switchable and self-healing supramolecular hydrogel carrier for guided, local catheter injection in the infarcted myocardium. Adv. Healthc. Mater. 3, 70–78 (2014).
Guo, M. et al. Tough stimuli-responsive supramolecular hydrogels with hydrogen-bonding network junctions. J. Am. Chem. Soc. 136, 6969–6977 (2014).
Appel, E. A. et al. Supramolecular cross-linked networks via host−guest complexation with cucurbit[8]uril. J. Am. Chem. Soc. 132, 14251–14260 (2010).
Barrow, S. J., Kasera, S., Rowland, M. J., del Barrio, J. & Scherman, O. A. Cucurbituril-based molecular recognition. Chem. Rev. 115, 12320–12406 (2015).
McKee, J. R. et al. Healable, stable and stiff hydrogels: combining conflicting properties using dynamic and selective three-component recognition with reinforcing cellulose nanorods. Adv. Funct. Mater. 24, 2706–2713 (2014).
Tanaka, Y., Gong, J. P. & Osada, Y. Novel hydrogels with excellent mechanical performance. Progress Polym. Sci. 30, 1–9 (2005).
Webber, R. E., Creton, C., Brown, H. R. & Gong, J. P. Large strain hysteresis and mullins effect of tough double-network hydrogels. Macromolecules 40, 2919–2927 (2007).
Nakajima, T. et al. True chemical structure of double network hydrogels. Macromolecules 42, 2184–2189 (2009).
Gong, J. P. Why are double network hydrogels so tough? Soft Matter 6, 2583–2590 (2010).
Kim, Y. S. et al. Thermoresponsive actuation enabled by permittivity switching in an electrostatically anisotropic hydrogel. Nat. Mater. 14, 1002 (2015).
Sano, K., Ishida, Y. & Aida, T. Synthesis of anisotropic hydrogels and their applications. Angew. Chem. Int. Ed. 57, 2532–2543 (2018).
Liu, M. et al. An anisotropic hydrogel with electrostatic repulsion between cofacially aligned nanosheets. Nature 517, 68 (2014).
Zhang, J., Wang, N., Liu, W., Zhao, X. & Lu, W. Intermolecular hydrogen bonding strategy to fabricate mechanically strong hydrogels with high elasticity and fatigue resistance. Soft Matter 9, 6331–6337 (2013).
Haque, M. A., Kurokawa, T., Kamita, G. & Gong, J. P. Lamellar bilayers as reversible sacrificial bonds to toughen hydrogel: hysteresis, self-recovery, fatigue resistance, and crack blunting. Macromolecules 44, 8916–8924 (2011).
Chen, G. & Jiang, M. Cyclodextrin-based inclusion complexation bridging supramolecular chemistry and macromolecular self-assembly. Chem. Soc. Rev. 40, 2254–2266 (2011).
Novo, M., Granadero, D., Bordello, J. & Al-Soufi, W. Host-guest association studied by fluorescence correlation spectroscopy. J. Incl. Phenom. Macro 70, 259–268 (2011).
Blass, J., Albrecht, M., Bozna, B. L., Wenz, G. & Bennewitz, R. Dynamic effects in friction and adhesion through cooperative rupture and formation of supramolecular bonds. Nanoscale 7, 7674–7681 (2015).
Bohne, C. Supramolecular dynamics. Chem. Soc. Rev. 43, 4037–4050 (2014).
Wang, L. L. et al. Injectable, guest–host assembled polyethylenimine hydrogel for siRNA delivery. Biomacromolecules 18, 77–86 (2017).
Park, K. M., Roh, J. H., Sung, G., Murray, J. & Kim, K. Self-healable supramolecular hydrogel formed by Nor-Seco-Cucurbit[10]uril as a supramolecular crosslinker. Chem. – Asian J. 12, 1461–1464 (2017).
Tomatsu, I., Hashidzume, A. & Harada, A. Redox‐responsive hydrogel system using the molecular recognition of β‐cyclodextrin. Macromol. Rapid Commun. 27, 238–241 (2006).
Kakuta, T. et al. Preorganized hydrogel: self-healing properties of supramolecular hydrogels formed by polymerization of host–guest-monomers that contain cyclodextrins and hydrophobic guest groups. Adv. Mater. 25, 2849–2853 (2013).
Sun, J.-Y. et al. Highly stretchable and tough hydrogels. Nature 489, 133 (2012).
Wei, K. et al. Robust biopolymeric supramolecular “host−guest macromer” hydrogels reinforced byin situformed multivalent nanoclusters for cartilage regeneration. Macromolecules 49, 866–875 (2016).
Zhao, X. H., Huebsch, N., Mooney, D. J. & Suo, Z. G. Stress-relaxation behavior in gels with ionic and covalent crosslinks. J. Appl. Phys. 107, https://doi.org/10.1063/1.3343265 (2010).
Peleg, M. & Pollak, N. The problem of equilibrium conditions in stress-relaxation analyses of solid foods. J. Texture Stud. 13, 1–11 (1982).
Gomez-Casado, A. et al. Probing multivalent interactions in a synthetic host-guest complex by dynamic force spectroscopy. J. Am. Chem. Soc. 133, 10849–10857 (2011).
Haque, M. A., Kurokawa, T. & Gong, J. P. Super tough double network hydrogels and their application as biomaterials. Polymer 53, 1805–1822 (2012).
Pattabhi, S. R. Polyelectrolyte multilayers and extracellular matrix effects on mesenchymal stem cell growth and differentiation, The FSU Digital Library (2014).
Acknowledgements
Project 31570979, 31530024, and 81672213 are supported by the National Natural Science Foundation of China. This project is supported by National Key R&D Program of China (2016YFC1100203). The work described in this paper is supported by a General Research Fund grant from the Research Grants Council of Hong Kong (Project No. 14205817 & 14202215). This work is supported by the Health and Medical Research Fund, Food and Health Bureau, Government of the Hong Kong Special Administrative Region (reference no: 03140056). This research is supported by the Chow Yuk Ho Technology Centre for Innovative Medicine, The Chinese University of Hong Kong. This research is also supported by project BME-p3-15 of the Shun Hing Institute of Advanced Engineering, The Chinese University of Hong Kong.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, X., Dong, C., Wei, K. et al. Supramolecular hydrogels cross-linked by preassembled host–guest PEG cross-linkers resist excessive, ultrafast, and non-resting cyclic compression. NPG Asia Mater 10, 788–799 (2018). https://doi.org/10.1038/s41427-018-0071-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41427-018-0071-0
This article is cited by
-
Sustained release delivery of favipiravir through statistically optimized, chemically cross-linked, pH-sensitive, swellable hydrogel
BMC Pharmacology and Toxicology (2024)
-
Effect of xanthan gum on the physical and chemical features of microfibrillated cellulose films plasticized by glycerol
Biomass Conversion and Biorefinery (2023)
-
Convergent synthesis of diversified reversible network leads to liquid metal-containing conductive hydrogel adhesives
Nature Communications (2021)
