
Abstract
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited, lethal ventricular arrhythmia triggered by catecholamines. Mutations in genes that encode cardiac ryanodine receptor (RyR2) and proteins that regulate RyR2 activity cause enhanced diastolic Ca2+ release (leak) through the RyR2 channels, resulting in CPVT. Current therapies for CPVT are limited. We found that Z16b, a meroterpenoid isolated from Ganoderma cochlear, inhibited Ca2+ spark frequency (CaSF) in R2474S/ + cardiomyocytes in a dose-dependent manner, with an IC50 of 3.2 μM. Z16b also dose-dependently suppressed abnormal post-pacing Ca2+ release events. Intraperitoneal injection (i.p.) of epinephrine and caffeine stimulated sustained ventricular tachycardia in all R2474S/+ mice, while pretreatment with Z16b (0.5 mg/kg, i.p.) prevented ventricular arrhythmia in 9 of 10 mice, and Z16b administration immediately after the onset of VT abolished sVT in 9 of 12 mice. Of translational significance, Z16b significantly inhibited CaSF and abnormal Ca2+ release events in human CPVT iPS-CMs. Mechanistically, Z16b interacts with RyR2, enhancing the “zipping” state of the N-terminal and central domains of RyR2. A molecular docking simulation and point mutation and pulldown assays identified Z16b forms hydrogen bonds with Arg626, His1670, and Gln2126 in RyR2 as a triangle shape that anchors the NTD and CD interaction and thus stabilizes RyR2 in a tight “zipping” conformation. Our findings support that Z16b is a novel RyR2 stabilizer that can prevent CPVT. It may also serve as a lead compound with a new scaffold for the design of safer and more efficient drugs for treating CPVT.
Similar content being viewed by others

Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a potentially lethal, heritable, cardiac channelopathy. Polymorphic ventricular arrhythmias, manifesting as bursts of monomorphic, polymorphic, or bidirectional ventricular tachycardia, are triggered by catecholamines released during exercise, stress, or sudden emotion in affected individuals with structurally normal hearts. Although the morbidity associated with CPVT is low, with an estimated prevalence of 1 in 5000 to 1 in 10,000, the mortality is high, with an estimated 30%–50% of untreated patients dying by the age of 40 [1].
Mutations in six genes have been identified as the genetic cause of CPVT. These genes encode proteins of the sarcoplasmic reticulum (SR) calcium release complex and comprise RYR2, CASQ2, and TRDN, which encode the cardiac ryanodine receptor calcium release channel (RyR2), calsequestrin 2, and triadin, respectively, and CALM1, CALM2, and CALM3, which encode identical calmodulin. Among these, dominant mutations in RYR2 are found in more than 60% of patients with CPVT. The current understanding of the cellular pathogenesis of CPVT is that these mutations cause enhanced diastolic Ca2+ release (leak) via the RyR2 channel upon catecholamine release. The abnormally enhanced SR Ca2+ leak drives the activity of the Na+/Ca2+ exchanger, creating a depolarizing Na+ current (INa) and triggering abnormal action potentials, either early after depolarization (EAD) or delayed after depolarization (DAD) [2].
Current therapies for CPVT include adrenergic receptor blockers, the Na+ channel blocker flecainide, and left cardiac sympathetic denervation. Beta-blockers are recommended as first-line therapy. However, in one study, more than 30% of patients with CPVT who received treatment experienced events during an 8-year follow-up period [3]. Flecainide has proved to be effective in most patients experiencing excitation-induced ventricular arrhythmia even with β-blocker treatment, but there are still patients who are not or incompletely protected [4, 5]. Therefore, more effective therapies are urgently needed.
The use of natural products as medicines has the benefits of multiple molecular architectures and immediate accessibility in quantities that are sufficient to explore their initial biological activity. We, therefore, performed a screen of the compounds in our natural product libraries, including more than 200 molecular entities isolated from fungi, insects, and plants, to find new, active compounds that have therapeutic potential for CPVT.
Spontaneous Ca2+ sparks indicate RyR2 function during diastole in living cardiomyocytes (CMs). Monitoring Ca2+ sparks in resting CMs, therefore, represents an ideal cellular platform for the rapid and convenient screening of chemicals that can inhibit SR Ca2+ leak and thus have potential efficacy in treating CPVT. During a pilot study, we discovered Z16b, a novel compound isolated from the fungus Ganoderma cochlear, inhibits spontaneous Ca2+ sparks in adult mouse ventricular myocytes (Fig. 1a, b).

a Protocol for screening active compounds effective at suppressing Ca2+ sparks, leading to the finding of Z16b. b Representative line-scan images of Ca2+ sparks in intact R2474S/ + CMs following ISO stimulation upon treatment with vehicle or Z16b. c A concentration–CaSF curve showing that the IC50 of Z16b on CaSF inhibition in R2474S/ + CMs is 3.2 μM; ncell = 25–36 cells/group, from at least three individual hearts. d Average data for CaSF, FDHM, and FWHM in the presence of vehicle and different concentrations of Z16b (nsparks = 102–327 sparks/group). e Representative traces of caffeine-elicited Ca2+ transients in WT and R2474S/ + CMs with or without addition of Z16b; n = 7–9 cells/group. f Average data for caffeine-elicited Ca2+ transients (ΔF/F0) in WT and R2474S/ + CMs. g Representative images of Ca2+ release events in R2474S/ + CMs treated with vehicle or indicated concentrations of Z16b following 1-Hz pacing for 1 min. Two forms of abnormal Ca2+ release events were observed in vehicle-treated R2474S/ + CMs (transients and waves). h Z16b reduced the frequency of post-pacing Ca2+ release events in a dose-dependent manner; n = 21–31 cells/group, from at least three individual hearts. *P < 0.05, **P < 0.01, and ***P < 0.001, versus vehicle.
Based on this finding, our present study investigated the possible therapeutic effects of Z16b in a gene-targeted mouse model of Ca2+ leak-induced arrhythmias (R2474S/+ mice). We further examined the potential therapeutic effect of Z16b on human CPVT, using induced pluripotent stem cell-derived cardiomyocytes (iPS-CMs) derived from a CPVT patient with a dominant mutation on RyR2 (R2401H). Furthermore, we sought the Z16b binding sites on RyR2 and analyzed the molecular mechanisms underlying the inhibition by Z16b of spontaneous Ca2+ leaks.
Materials and methods
Animals
RyR2R2474S/+ knock-in mice were kindly provided by Dr. Andrew Marks’s laboratory at Columbia University, New York, NY, USA. All animal studies were approved by the Animal Care and Use Committee of Shenzhen University, China, and conducted in accordance with NIH guidelines. Male and female RyR2R2474S/+ mice (2–4 months old) were used as the CPVT animal model, and their littermates not carrying mutant RyR2 were used as the WT control.
Scanning library and chemicals
The library of natural products used in this study includes more than 200 small molecular entities. The library was provided by Dr. Yong-xian Cheng’s laboratory, at Shenzhen University, China. All chemical compounds, including Z16b, were diluted with DMSO in stock solution to a concentration of 20 mM, stored at −20 °C, and protected from light. The purity of the chemicals was >95%, guaranteed by HPLC analysis. JTV-519 was purchased from Sigma Aldrich (USA). The CMs were pre-treated with the various natural chemicals for 10 min at a final concentration of 20 μM before stimulation with ISO, and the effects on Ca2+ sparks were measured.
Isolation of ventricular cardiomyocytes
Ventricular CMs were isolated from mice aged 2–4 months, as we have described previously [6].
Surface electrocardiography (ECG)
Mice were lightly anesthetized with isoflurane (2%) and placed on a heating pad to maintain a normal body temperature. A multifunctional physiological record system (TECHMAN BL-520N, China) was used for signal collection and analysis, and recordings were taken from a Lead II configuration, as we have described previously [7].
Measurement of SR Ca2+ content
SR Ca2+ content was detected as previous described [6]. Briefly, short bursts of caffeine (20 mM) were applied to completely empty the SR Ca2+ load, following a train of 1-Hz field stimulation to achieve steady-state SR Ca2+ loading in the ventricular myocytes. SR Ca2+ content was assessed by determining the amplitude of caffeine-elicited Ca2+ transients.
Detection of Ca2+ sparks
Mouse ventricular CMs or iPS-CMs were loaded with Flou-4-AM dye (Thermo Fisher Scientific, Waltham, MA, USA) for 15 min at room temperature (23 ± 1 °C). The Ca2+ sparks were imaged using a Carl Zeiss (Carl Zeiss Microscopy, White Plains, NY, USA) confocal microscope in the line-scan mode (see Supplementary Information for a more detailed description) at 37 °C by placing the cells on a stage top incubator on the confocal microscope (Zeiss Tokai Hit STXG-WSKMX -SET). To detect Ca2+ sparks, freshly prepared isoprenaline (ISO) was applied at a final concentration of 10 nM or 200 nM for mouse R2474S/ + or WT CMs, respectively, while 1 μM ISO was applied for Ca2+ imaging of human iPS-CMs. To measure the inhibitory effect of Z16b on Ca2+ sparks, the IC50 was calculated via a non-linear regression of the dose-response effect, using GraphPad Prism v8.0 (GraphPad Software, San Diego, CA, USA).
Molecular docking
The three-dimensional structure of Z16b was created using the CORINA online tool (http://www.molecular-networks.com) and converted to a PDBQT file with Open Babel [8]. The three-dimensional crystal structure of RyR2 (PDB code: 5GO9), obtained from the Protein Data Bank (https://www.rcsb.org/), was selected for the docking simulation study [9]. The docking was conducted using AutoDock Vina v.1.0.2 (https://vina.scripps.edu/). The docking parameters for AutoDock Vina were kept at their default values. The grid box encompassed the entire interface between the N-terminal domain (NTD) and the central domain (CD) of RyR2. The binding modes were clustered using the root mean square deviation among the Cartesian coordinates of the ligand atoms. The docking results were ranked by the binding free energy. The binding mode was demonstrated using PyMOL Molecular Graphics System v1.3 (Schrödinger, New York, NY, USA).
Molecular dynamic simulation
We used a previously developed molecular dynamics simulation to facilitate the simulation. The simulation region is restricted in the interface between NTD and CD. The initial protein–ligand complex is loaded from the docking output. The selected region of the protein was defined as 1.5 nm within the binding ligand, and other settings were as previously described [10]. GROMACS software (www.gromacs.org) was applied to run the MD simulation with AMBER-99SB force field [11, 12]. ACPYPE (https://pypi.org/) was used to produce the topology and partial charges of ligand [13] which relies on Antechamber [14]. The structure of the complex and the 2D protein–ligand interactions was visualized by Discovery Studio Visualizer 2019 (Dassault Systèmes, Vélizy-Villacoublay, France).
Expression of recombinant RyR2 fragments
The pcDNA3.1 plasmids containing RyR2 fragments of interest were constructed by HanyiBio Co. (Shanghai, China) and transfected into HEK293T cells using a JetOptimus DNA transfection kit (PolyPlus, New York, NY, USA), according to the manufacturer’s instructions. Cell lysates containing expressed RyR2 fragments were harvested for further assays, 48 h following transfection.
Generation of iPS-CMs
Monocytes were isolated from a blood sample of a patient with clinically diagnosed CPVT who carried a heterozygous RyR2-R2401H mutation, a previously confirmed causal mutation of CPVT [15]. These monocytes were then used for iPSC reprogramming and iPS-CM differentiation. The patient provided informed consent to participate in this study, under protocols approved by the Medical Research Ethics Committee of Guangdong Provincial Hospital, China. Commercially obtained iPS-derived cardiomyocytes (Cellapy Biotech, Beijing, China) were cultured according to the manufacturer’s instructions and used as the control in this study.
Probing the “zipping and unzipping” conformation of RyR2
We used a well-established method to investigate the conformational changes of RyR2 [16]. DPc10 is a 36-residue peptide that corresponds to the Gly2460–Pro2495 region of RyR2 (2460-GFCPDHKAAMVLFLDRVYGIEVQDFLLHLLEVGFLP-2495). A fluorescent label, 5-TAMRA (Invitrogen, Carlsbad, CA, USA), was added to the N-terminal of DPc10 (F-DPc10). The F-DPc10 used in this study was synthesized at JL Biochem Co. (Shanghai, China). Freshly isolated adult WT mouse CMs were permeabilized, and 5 μM F-DPc10 was applied in the presence of vehicle or 10 μM Z16b or 10 μM JTV-519 (Sigma, St. Louis, MO, USA). Fluorescent images were recorded at various time points, using a confocal microscope at 561-nm excitation and 582-nm collection. The fluorescence intensities were measured using ImageJ software (https://imagej.net/). The difference between the fluorescence intensities of the Z-line and the M-line represented the relative amount of F-DPc10 binding to RyR2. The binding kinetics of DPc10 fit into the nonlinear regression model: B = Bmax (1 − e−T/τwash-in), where Bmax indicates the maximal binding capacity of F-DPc10 and τwash-in reflects the velocity of F-DPc10 binding.
Statistical analysis
All values are presented as means ± SEM unless indicated otherwise. Student’s t-test was used for comparisons between two groups. One-way ANOVA, followed by the Tukey–Kramer correction, was performed for comparisons among multiple groups. The chi-square test was applied for rate comparisons between groups. Statistical analyses were performed using SPSS v17.0 software (IBM Corporation, Chicago, IL, USA). For the violin plots and box plots, the box and whiskers define the quartile ranges, with the center line as the median. A P value < 0.05 was considered to be statistically significant.
Results
Z16b decreases diastolic SR Ca2+ leak and abnormal Ca2+ handling
Diastolic Ca2+ sparks are elementary Ca2+ release events that reflect RyR2 activity and diastolic SR Ca2+ leak in intact CMs. We used CMs isolated from a validated mouse model of human CPVT (R2474S/+) that harbor a known human RyR2 mutation (RyR2-R2474S) to screen for chemical compounds that decrease the rate of spontaneous Ca2+ sparks. The Ca2+ spark frequency (CaSF) at resting state was very low in R2474S/ + mutant CMs but increased dramatically upon application of 10 nM isoproterenol (ISO) and simulated the characteristics of CPVT. The R2474S/ + mutant CMs were pre-treated with the various natural chemicals, and we identified that Z16b, a compound isolated from Ganoderma cochlear decreased the occurrence of Ca2+ sparks. Z16b was identified by high-performance liquid chromatography–mass spectrometry as a meroterpenoid (Fig. 1a). We found Z16b decreased CaSF in cardiomyocytes in a dose-dependent manner following ISO stimulation. CaSF was significantly decreased, by 30.0%, 59.8%, and 71.6%, when using 2.5, 5, and 10 μM Z16b, respectively, compared with vehicle treatment (Fig. 1b). The half-maximal inhibitory concentration (IC50) of Z16b on CaSF was 3.2 μM (Fig. 1c). Z16b also decreased the mass (full width at half maximum, FWHM) and duration (full duration at half maximum, FDHM) of Ca2+ sparks (Fig. 1d). The data indicate that Z16b suppresses diastolic SR Ca2+ leak. We also examined the effect of Z16b on Ca2+ spark in WT cardiomyocytes. ISO at 200 nM stimulated remarkable SR Ca2+ release in WT cells. Z16b at the concentrations of 5 μM and 10 μM significantly decreased CaSF, whereas 2.5 μM Z16b had no effect on CaSF (Supplementary Fig. S1).
Consistent with previous studies [17] the SR Ca2+ content was reduced in R2474S/ + mutant CMs due to increased Ca2+ leak, which was restored by treatment with 10 μM Z16b. The SR Ca2+ content in WT CMs was unaltered by treatment with Z16b (Fig. 1e, f). This is rational because the rate of Ca2+ sparks in normal cells is very low, which has no significant effect on SR Ca2+ content.
Enhanced SR Ca2+ leak triggers abnormal electrical activity in the heart, such as DAD, which consequently induces abnormal Ca2+ handling that can be monitored by Ca2+ fluorescence at the cellular level. We thus examined the effect of Z16b on abnormal post-pacing calcium release events (PPEs) in R2474S/ + cardiomyocytes, as suggested by Bezzerides et al. [18]. Strains of PPEs, including spontaneous transients and waves, were triggered in R2474S/ + CMs (Fig. 1g), while PPEs were rare in WT CMs (Supplementary Fig. S2). Z16b significantly decreased the incidence of PPEs in R2474S/ + CMs at a dose of 2.5 μM and almost completely abolished PPEs when the dose was increased to 10 μM (Fig. 1h). These results indicate that Z16b can prevent pro-arrhythmic activity in CPVT CMs.
Z16b suppresses arrhythmias in an R2474S/ + murine model of CPVT
Our findings in CMs suggested that Z16b may have a therapeutic effect on CPVT in vivo. To test this potential, we assessed the effect of Z16b on electrophysiological characteristics recorded with an electrocardiogram (ECG) in RyR2-R2474S/ + mice, a well-established CPVT model that recapitulates many of the clinical features of human CPVT, at rest and under arrhythmia-provoking conditions. R2474S/ + mice had normal electrophysiological characteristics during resting state, similar to their WT littermates (Supplementary Fig. S3). Following arrhythmic stimulation using epinephrine (Epi, 1 mg/kg) and caffeine (Caf, 60 mg/kg; via intraperitoneal injection, i.p.) [19, 20] all R2474S/ + mice readily generated long-sustained ventricular tachycardia (sVT), manifested as typical bidirectional VT and polymorphic VT. In contrast, in their WT littermates, Epi plus Caf stimulation triggered sinus tachycardia alone.
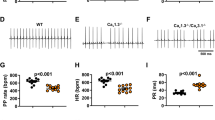
Prior to the experiment for detecting Z16b efficacy for treating CPVT, we determined the safe dose of Z16b by using echocardiography to examine the effect of Z16b on ECG and cardiac function. When Z16b was administered via i.p. at a dose of 0.5 mg/kg, it had no effect on basal heart rate (HR) or other major electrophysiological parameters in WT mice, including PR interval, QRS duration, and QT interval (Figs. 2a, b). In particular, the cardiac contractile function, as indexed by the left ventricular ejection fraction and fractional shortening, was unaltered (Supplementary Fig. S4). However, when the dose was increased to 2 mg/kg, a prolonged PR interval and slowed HR were observed (Supplementary Fig. S5). We thus used 0.5 mg/kg as the therapeutic dose in this study.

a Typical ECG traces in WT mice without (baseline) or with Z16b treatment (0.5 mg/kg body weight, i.p.). b Average heart rate, QRS duration, PR interval, and QT interval in WT mice with or without Z16b treatment; n = 6 mice/group. c Representative ECG traces in R2474S/ + mice receiving vehicle or Z16b pretreatment. Black ECG traces were recorded under resting conditions, while red ECG traces were recorded under Epi+Caf challenge. Average incidence of sVT (d) and death rate (e) in vehicle, Z16b, and JTV-519 pretreatment groups; n = 8–10 mice/group. f Representative ECG traces of R2474S/ + mice receiving vehicle or Z16b treatment, immediately after the onset of sVT. Pooled data of the rate of sVT termination (g) and death (h) in the vehicle and Z16b treatment groups; n = 8 or 12 mice/group. *P < 0.05, **P < 0.01, ***P < 0.001 versus vehicle group.
First, we investigated the potential of Z16b for preventing CPVT. Z16b was administered via i.p. to R2474S/ + mice 15 min prior to arrhythmic stimulation, and ECG was monitored for 30 min following stimulation. We found that preventive administration of Z16b significantly decreased the incidence of sVT compared with its incidence following administration of vehicle. Ventricular arrhythmia was completely prevented in 9 of 10 mice in the Z16b group. In contrast, sustained ventricular arrhythmia occurred in all 8 mice in the vehicle group (Fig. 2d). No mice died following Z16b treatment during the observation time of 24 h post-Epi+Caf stimulation, while two of eight mice died in the vehicle group (Fig. 2e).
Next, the therapeutic effect of Z16b on CPVT was compared with that of JTV-519, an anti-SR Ca2+ leak drug, characterized by its ability to stabilize RyR2. Administration of JTV-519 prior to Epi+Caf stimulation (0.5 mg/kg, i.p., 15 min before arrhythmic stimulation [21]) abolished sVT in seven of nine mice. The therapeutic efficacy of JTV-519 was comparable with that of Z16b. However, in contrast to Z16b, which had no effect on any ECG parameters, JTV-519 prolonged the PR interval, which might be due to the off-target effect as previously reported [22]. These results indicate that as an anti-CPVT drug, Z16b is superior to JTV-519 when they are applied at the same dose.
We next examined the rescue potential of Z16b on CPVT. The drug was administered immediately after the onset of sVT. During the 30 min observation time following Z16b treatment, we found that sVT was terminated in 9 of 12 mice. It took ~15 min (14.9 ± 7.0 min) to recover sinus rhythm in the sVT-terminated mice. However, sVT was not terminated in any of the mice in the vehicle group (Fig. 2g). Still, no mouse died after treatment with Z16b, but three of eight mice in the vehicle group died (Fig. 2h).
Z16b binds to RyR2 and stabilizes the RyR2 interdomain “zipping” state
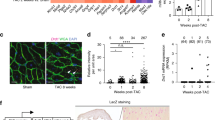
We addressed the possible mechanisms underlying how Z16b prevents abnormal RyR2 leaks. First, we wanted to know whether this effect was induced by a direct interaction between Z16b and RyR2. For this purpose, we designed and synthesized biotin-conjugated Z16b (biot-Z16b), in which the hydrogen on the 2′-hydroxyl in Z16b was substituted with biotin (Fig. 3a). This manipulation did not interfere with the activity of Z16b on SR Ca2+ release, as biot-Z16b (5 μM) significantly reduced CaSF in both intact and saponin-permeabilized R2474S/ + CMs (Supplementary Fig. S6), which was comparable to the effect of Z16b. Biot-Z16b was incubated with mouse-heart lysate overnight and its interaction with RyR2 was examined by a pull-down assay using streptavidin-coated magnetic beads. The results showed that R2474S/ + RyR2 and WT RyR2 in heart lysate could be pulled down by streptavidin-coated magnetic beads, suggesting biot-Z16b can bind with RyR2 (Fig. 3b).

a Molecular structure of biot-Z16b (detailed description of the synthesis procedures of biot-Z16b are described in the Supplementary Methods). b Immunoblotting assay of biot-Z16b or biotin incubated with heart lysates from R2474S/ + mice and WT mice, further enriched using streptavidin-coated magnetic beads. A Western blot of heart lysates was performed as an input control. c Representative images of F-DPc10 bound to WT RyR2 after treatment with vehicle, Z16b, or JTV-519 for 210 min. d Kinetics of F-DPc10 bound to WT RyR2 in saponin-permeabilized CMs. e Bmax and τwash-in of each group were calculated. f Representative line-scan images of Ca2+ sparks in saponin-permeabilized R2474S/ + CMs; g The CaSF in groups treated with vehicle or different concentrations of Z16b was calculated; n = 31–37 cells/group. **P < 0.01; ***P < 0.001, versus vehicle group.
Previous studies reported the disruption of the interaction between NTD and CD of RyR2 results in RyR2 channel “unzipping”, which accounts for the enhanced SR Ca2+ leak induced by CPVT-related mutations or RyR2 hyperphosphorylation (PKA and Ca2+/calmodulin-dependent protein kinase II-mediated) and oxidative stress modification [23,24,25,26,27]. To test whether the interdomain zipping model accounted for the Z16b-mediated anti-SR leak effect, we used a well-documented method to probe the interdomain “zipping” or “unzipping” state by applying a synergetic RyR2 CD peptide, DPc10 (Gly2460-Pro2495), which competitively binds to the NTD, inducing interdomain unzipping and aberrant channel opening [16]. The fluorescently-labeled DPc10 (F-DPc10) associated with RyR2 was measured by determining the difference in fluorescence between the Z-lines (Fz-line) and M-lines (FM-line) in CMs. The binding kinetics of F-DPc10, which represents the binding capability of F-DPc10 to RyR2 and reflects the stability of the interdomain zipping of RyR2 was analyzed as previously reported [28]. We found that pretreatment with Z16b (10 μM) induced a downward-shift of the time-course curve of F-DPc10 bound to Z-lines (Figs. 3c, d; time-lapse images are shown in Supplementary Fig. S7). The maximal binding of F-DPc10 (Bmax) in the Z16b group was significantly lower than that in the vehicle group, but comparable with the effect seen with 10 μM JTV-519 (vehicle 23.1 ± 0.6 a.u., Z16b 13.9 ± 0.8 a.u., and JTV-519 11.0 ± 0.8 a.u). The τwash-in, reflecting the binding rate of F-DPc10 to RyR2, was also decreased by the addition of Z16b (vehicle 84.7 ± 2.7 min, Z16b 140.5 ± 6.2 min, and JTV-519 131.7 ± 5.8 min) (Fig. 3). Collectively, these results indicate that Z16b enhances the interdomain interaction between the NTD and the CD, as does JTV-519 [28]. To explore the direct effect of Z16b in stabilizing RyR2, we examined the occurrence of spontaneous Ca2+ sparks in saponin-permeabilized R2474S/ + CMs following treatment with Z16b. As shown in Fig. 3f and g, Z16b decreased CaSF in a dose-dependent manner. The effect was quite similar to that seen in intact cells, where the minimum effective dose of Z16b on CaSF was 2.5 µM, and CaSF was decreased by 58.0%, 72.9%, and 91.9% by 2.5, 5, and 10 µM Z16b, respectively. These data suggest that Z16b inhibits RyR2 activity through direct Z16b-RyR2 interaction.
Since PKA and CaMKII-mediated RyR2 phosphorylation (at the site of Ser2808 and Ser2814, respectively) can induce interdomain “unzipping” and increase the open probability of RyR2 [23, 29], we examined the effect of Z16b on PKA and CaMKII activity. The results showed that Z16b had no effect on the phosphorylation levels of RyR2 at Ser2808 and Ser2814 in CMs following ISO stimulation, indicating that Z16b stabilizes RyR2 in a way that is unrelated to PKA and CaMKII regulation (Supplementary Fig. S8a, b). Previous studies have suggested that JTV-519 enhances FKBP12.6 binding to RyR2, which is critical for the stabilization of RyR2. In contrast, we observed no obvious change in FKBP12.6 binding to RyR2 in Z16b-treated cells compared with those cells treated with vehicle (Supplementary Fig. S8c, d). Taken together, our data indicate that Z16b stabilizes the RyR2 channel by directly enhancing RyR2 interdomain zipping without the participation of FKBP12.6.
It has been shown that blockers for L-type calcium channel (LCC), i.e., verapamil, and voltage-dependent Na+ channel, flecanide, has anti-CPVT effect [4, 30]. We thus examined the effect of Z16b on LCC current (ICa,L) and Na+ current (INa). The results showed that Z16b had a relative weak inhibition on ICa,L, with IC50 at 19.5 μM (Supplementary Fig. S9a, b). Z16b had no significant effect on INa at the concentration of 10 μM compared with vehicle treatment (Supplementary Fig. S9c). The results are consistent with our in vivo findings that the low effective dose of Z16b (0.5 mg/kg, i.p.) that was efficient for treating CPVT had no effect on ECG parameters, including QRS duration, PR and QT intervals due to no change in ICa,L and INa, but high dose of Z16b (2 mg/kg) prolonged PR interval most possibly by decreasing ICa,L (Supplementary Fig. S5) [31].
Probing the Z16b binding sites on RyR2
We applied a molecular docking simulation (Autodock Vina) to explore the possible interaction sites between RyR2 and Z16b. Given the critical role of the interaction between NTD and CD of RyR2 in the effect of Z16b, we selected a grid box that covered the interface of the NTD and CD. The docking model showed that Z16b exhibited relatively high binding affinity for RyR2 (-7.2 kcal/mol), further supporting the view that there is a direct interaction between Z16b and RyR2. The oxolane of Z16b comes into contact with His1670 in RyR2, and the two hydroxyl groups on the benzene ring of Z16b form two hydrogen bonds with Arg626 and Gln2126, in RyR2. Using a high performance computing-based molecular dynamic simulation (MD simulation) which mimics the physical movements of atoms and molecules by Newton’s equations of motion [11] we also found that Z16b interacted with Arg626, His1670, and Gln2126 via hydrogen bonds (Supplementary Fig. S10). We mapped the key residues on the RyR2 crystal structure and found that Arg626 is adjacent to the NTD (1–600 aa), Gln2126 is located at the CD (2000–2500 aa), and His1670 sits between the NTD and CD (Fig. 4a). Since the structure analysis of RyR2 is performed on pigs (Sus scrofa), we performed an alignment analysis of RyR2 amino acid sequences in humans, mice, and pigs. The result showed that the three residues are conserved in these species (Supplementary Fig. S11), highlighting the importance of these residues.

a Binding residues for Z16b on the crystal structure of RyR2, predicted by molecular docking simulation. b Full-length constructs of the RyR2 (upper image) and recombinant RyR2 fragments, constituting 600 to 2160 aa of RyR2 and a FLAG-tag fused on its C-terminal (lower image). Red bands represent predicted residues in RyR2 that bind with Z16b. c Pull-down assay results detecting the binding of Z16b with the RyR2 fragment. Biot-Z16b directly interacted with the RyR2 fragment, and the addition of Z16b competitively inhibited the binding effect of biot-Z16b. d Biot-Z16b lost its binding affinity for the fragment of RyR2 when the key residues Arg626, His1670, or Gln2126 were mutated to alanine.
To verify the docking model for Z16b binding with RyR2, we constructed a recombinant RyR2 fragment containing 600–2160 amino acids that covered all three predicted key residues (Arg626, His1670, and Gln2126), with a FLAG-tag fused to the C-terminal (Fig. 4b). We then tested the fragment’s interaction with biot-Z16b using a pull-down assay. The results showed that biot-Z16b, but not biotin, interacted with the fragment, and the addition of Z16b competitively weakened the binding affinity of biot-Z16b (Fig. 4c). These data support the in silico model of the Z16b–RyR2 complex.
To prove the role played by the three predicted residues in Z16b–RyR2 binding, we generated a point mutation in the 600–2160 aa recombinant fragments by replacing each of the residues with alanine (Arg626Ala, His1670Ala, and Gln2126Ala). Notably, the pull-down assay showed that each mutation significantly reduced the binding affinity of biot-Z16b. These results suggest that the residues Arg626, His1670, and Gln2126 are necessary for the interaction between Z16b and RyR2 (Fig. 4d).
Z16b prevents arrhythmogenic Ca2+ events in iPS-CMs derived from a patient with CPVT
The translational potential of Z16b for the treatment of CPVT was further examined in iPS-CMs obtained from a patient with CPVT who carries a reported causal mutation, R2401H, in RyR2 [32]. Monocytes isolated from the patient’s peripheral blood sample were collected and reprogrammed to become iPSCs, according to a previously reported method [33]. The successful reprogramming of iPSCs was confirmed by the positive expression of SSEA4 and NANOG, determined by immunofluorescent staining and a normal karyotype (Supplementary Fig. S12). After 20 passages, the iPSCs had differentiated into functional iPS-CMs, and the expression of the cardiomyocyte markers α-actinin (α-ACT) and cTNT was determined by immunofluorescent staining (Fig. 5a).

a Immunostaining images of the expression of cardiomyocyte markers α-Act and cTNT in iPS-CMs. Representative line-scan images (b) of Ca2+ sparks in control and R2401H/ + iPS-CMs and average data (c) for CaSF; n = 30–32 cells/group. Representative traces (d) of Ca2+ signals recorded by confocal line-scan in spontaneously beating iPS-CMs. e Quantification of the number of abnormal Ca2+ release events in each group; n = 33–36 cells/group. *P < 0.05, ***P < 0.001, versus baseline.
Consistent with previous studies, the control and R2401H mutant iPS-CMs exhibited periods of spontaneous rhythmic beating interspersed with periods of quiescence. Ca2+ sparks were observed in quiescent control and mutant iPS-CMs. CaSF in mutant cells was much higher than that in control cells. ISO stimulation (1 μM) significantly increased the occurrence of Ca2+ sparks in quiescent control and mutant cells, while Z16b (5 μM) significantly reduced CaSF in both types of cells (Fig. 5b, c). In spontaneously beating cells, abnormal Ca2+ release events in mutant iPS-CMs were excited by ISO stimulation, which was largely suppressed by 5 μM of Z16b (an 80.1% reduction). WT iPS-CMs had no significant abnormal Ca2+ release events, either with or without ISO stimulation (Fig. 5d, e).
Discussion
Here, we reported the discovery that a natural product, Z16b, isolated from Ganoderma cochlear, has a potent therapeutic effect on CPVT. It reduces CPVT episodes not only in a mouse model of CPVT but also in human iPS-CMs derived from a patient with CPVT. Functional analyses and molecular assays demonstrated that Z16b serves as an RyR2 stabilizer by enhancing the interaction between the CD and NTD of RyR2. Z16b has the features of a meroterpenoid whose structure is completely distinguishable from other reported RyR2 stabilizers and inhibitors. Furthermore, Z16b exerts a potent anti-CPVT effect, irrespective of whether it is applied before or after the onset of CPVT. These findings strongly suggest this compound has translational significance for the treatment of CPVT.
We had compared the effect of Z16b with that of JTV-519, and the results showed that Z16b had a similar efficacy for preventing ventricular arrhythmia in CPVT mice compared with that of JTV-519. However, Z16b had no effect on any ECG parameters (Fig. 2a), while JTV-519 increased the PR interval, which may have been caused by its off-target effect on electrical conductance [34]. Taking this into consideration, Z16b is safer than JTV-519. Nevertheless, the efficacy of Z16b at stabilizing RyR2 appears to be not so potent, with IC50 at 3.2 μM, despite the effective dose of Z16b (0.5 mg/kg, i.p.) for CPVT treatment is not cardiotoxic in mice. The study was also limited by not examining the efficiency of Z16b on suppressing DAD, a convincing cellular mechanism for CPVT. However, Z16b inhibited abnormal Ca2+ release events in CPVT mouse CMs and CPVT iPS-CMs (Figs. 1 and 5), the results of abnormal electrical activity which have been widely used to value the efficiency of a therapy for treating CPVT [18, 35]. Therefore, Z16b may serve as a novel lead compound and a new scaffold for the future design and refinement of more potent and clinical applicable new drugs for the treatment of CPVT, and other heart disease associated with SR Ca2+ leak, such as atrial fibrillation and heart failure [36,37,38].
In this study, we demonstrated that Z16b suppresses SR Ca2+ leak via direct interaction with RyR2. Functional evidence demonstrated that Z16b inhibited spontaneous Ca2+ release not only in intact but also in saponin-permeabilized CMs, suggesting Z16b has a direct effect on RyR2. Pull-down assays confirmed that Z16b binds with RyR2. One leading hypothesis suggests that the interaction between the NTD and CD plays a switch-like role in RyR2 gating, where the tight interdomain interaction (or zipping) facilitates the RyR2 ion pore to remain in a closed state, while disruption of interdomain interactions (or unzipping) induces enhanced RyR2 sensitivity and Ca2+ leakiness [39, 24]. It has been suggested that CPVT-related RyR2 point-mutations induce SR Ca2+ leak through disrupting the zipping state, eventually triggering DADs, EADs, and VT incidence [26, 40]. Here, we found that the binding of Z16b with RyR2 enhanced NTD and CD interaction, as demonstrated by the increased steric hindrance of F-DPc10 in association with RyR2 upon application of Z16b (Fig. 3). Our findings confirm the notion that the interaction between NTD and CD controls RyR2 gating and serves as a key target for modulating RyR2 function. Whether the association of FKBP12.6 with RyR2 is essential for RyR2 stabilization remains controversial [41,42,43]. Our data demonstrate that Z16b does not affect the interaction between FKBP12.6 with RyR2, supporting the argument that FKBP12.6 interaction is not essential for RyR2 stabilization.
An exciting recent study reported the crystal structures of RyR2 [9]. Our molecular docking and MD simulation analysis demonstrated that Z16b has a high binding affinity (−7.2 kcal/mol) for RyR2 and that Arg626, His1670, and Gln2126 in RyR2 are key residues for Z16b binding. Point mutation and pull-down assays further confirmed the in silico simulation, suggesting that molecular docking and MD simulation can be used as powerful tools for predicting detailed information about channel protein–drug interactions. Here, we proposed a model for Z16b stabilizing RyR2 (Fig. 6). Z16b binds with residues at Arg626 (adjacent to the NTD), His1670 (between the NTD and the CD), and Gln2126 (in the CD) in RyR2 to form a triangle-shape that anchors the NTD and CD interaction and thus stabilizes RyR2 in a tight, zipped conformation. As these three residues in RyR2 are conserved among species (Supplementary Fig. S11), Z16b should have similar effects on RyR2 in human CMs, which has indeed been confirmed in human iPS-CMs.

Z16b binds with Arg626, adjacent to the NCD; His1670, located between the NTD and the CD; and Gln2126, in the CD, in a triangle-shaped form, providing forces that enhance the interaction between the NTD and the CD and thus stabilizing RyR2.
A previous study used a quartz-crystal microbalance assay to demonstrate that JTV-519 specifically binds to the 2114–2149 aa fragment of RyR2. Interestingly, using our molecular docking model, we identified Gln2126 in this fragment as a critical residue for JTV-519 binding. This is in agreement with a previous study[28], although the docking simulation based on the reported crystal structure of RyR2 is thought to give more precise information. Notably, Gln2126 is a common residue for both Z16b and JTV-519 binding, suggesting it may be an important binding site for drugs that stabilize the interaction between NTD and CD. This will be verified in future studies and may provide a new strategy for screening anti-SR-leak drugs.
In summary, our study identified a novel RyR2 stabilizer, Z16b, from a series of natural products, and we showed that Z16b has a potent therapeutic effect on CPVT. The anti-SR leak potency of Z16b may be extended for the treatment of atrial fibrillation and heart failure, the development of which is closely associated with enhanced SR Ca2+ leak. The new meroterpenoid scaffold underscores the potential of Z16b as a lead compound for the design and refinement of safer and more efficient drugs in the future.
References
Wleklinski MJ, Kannankeril PJ, Knollmann BC. Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. J Physiol. 2020;598:2817–34.
Wehrens XH, Lehnart SE, Marks AR. Ryanodine receptor-targeted anti-arrhythmic therapy. Ann NY Acad Sci. 2005;1047:366–75.
van der Werf C, Zwinderman AH, Wilde AA. Therapeutic approach for patients with catecholaminergic polymorphic ventricular tachycardia: state of the art and future developments. Europace. 2012;14:175–83.
Kannankeril PJ, Moore JP, Cerrone M, Priori SG, Kertesz NJ, Ro PS, et al. Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: a randomized clinical trial. JAMA Cardiol. 2017;2:759–66.
Watanabe H, van der Werf C, Roses-Noguer F, Adler A, Sumitomo N, Veltmann C, et al. Effects of flecainide on exercise-induced ventricular arrhythmias and recurrences in genotype-negative patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2013;10:542–7.
Yang J, Zhang R, Jiang X, Lv J, Li Y, Ye H, et al. Toll-like receptor 4-induced ryanodine receptor 2 oxidation and sarcoplasmic reticulum Ca2+ leakage promote cardiac contractile dysfunction in sepsis. J Biol Chem. 2018;293:794–807.
Liu W, Wang G, Zhang C, Ding W, Cheng W, Luo Y, et al. MG53, a novel regulator of KChIP2 and I(to,f), plays a critical role in electrophysiological remodeling in cardiac hypertrophy. Circulation. 2019;139:2142–56.
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform. 2011;3:33.
Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, et al. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science. 2016;354:aah5324.
Zhang H, Yang Y, Li J, Wang M, Saravanan KM, Wei J, et al. A novel virtual screening procedure identifies pralatrexate as inhibitor of SARS-CoV-2 RdRp and it reduces viral replication in vitro. PLoS Comput Biol. 2020;16:e1008489.
Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–47.
Hornak V, Simmerling C. Generation of accurate protein loop conformations through low-barrier molecular dynamics. Proteins. 2003;51:577–90.
Sousa da Silva AW, Vranken WF. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes. 2012;5:367.
Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–60.
Aizawa Y, Ueda K, Komura S, Washizuka T, Chinushi M, Inagaki N, et al. A novel mutation in FKBP12.6 binding region of the human cardiac ryanodine receptor gene (R2401H) in a Japanese patient with catecholaminergic polymorphic ventricular tachycardia. Int J Cardiol. 2005;99:343–5.
Oda T, Yang Y, Nitu FR, Svensson B, Lu X, Fruen BR, et al. In cardiomyocytes, binding of unzipping peptide activates ryanodine receptor 2 and reciprocally inhibits calmodulin binding. Circ Res. 2013;112:487–97.
Nakamura Y, Yamamoto T, Kobayashi S, Tamitani M, Hamada Y, Fukui G, et al. Ryanodine receptor-bound calmodulin is essential to protect against catecholaminergic polymorphic ventricular tachycardia. JCI Insight. 2019;4:e126112.
Bezzerides VJ, Caballero A, Wang S, Ai Y, Hylind RJ, Lu F, et al. Gene therapy for catecholaminergic polymorphic ventricular tachycardia by inhibition of Ca2+/calmodulin-dependent kinase II. Circulation. 2019;140:405–19.
Wang Y, Li C, Shi L, Chen X, Cui C, Huang J, et al. Integrin β1D deficiency-mediated RyR2 dysfunction contributes to catecholamine-sensitive ventricular tachycardia in arrhythmogenic right ventricular cardiomyopathy. Circulation. 2020;141:1477–93.
Xiong J, Liu X, Gong Y, Zhang P, Qiang S, Zhao Q, et al. Pathogenic mechanism of a catecholaminergic polymorphic ventricular tachycardia causing-mutation in cardiac calcium release channel RyR2. J Mol Cell Cardiol. 2018;117:26–35.
Li N, Wang Q, Sibrian-Vazquez M, Klipp RC, Reynolds JO, Word TA, et al. Treatment of catecholaminergic polymorphic ventricular tachycardia in mice using novel RyR2-modifying drugs. Int J Cardiol. 2017;227:668–73.
Stams TR, Oros A, der Nagel R, Beekman JD, Chamberlin P, Dittrich HC, et al. Effects of K201 on repolarization and arrhythmogenesis in anesthetized chronic atrioventricular block dogs susceptible to dofetilide-induced torsade de pointes. Eur J Pharmacol. 2011;672:126–34.
Uchinoumi H, Yang Y, Oda T, Li N, Alsina KM, Puglisi JL, et al. CaMKII-dependent phosphorylation of RyR2 promotes targetable pathological RyR2 conformational shift. J Mol Cell Cardiol. 2016;98:62–72.
Uchinoumi H, Yano M, Suetomi T, Ono M, Xu X, Tateishi H, et al. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ Res. 2010;106:1413–24.
Oda T, Yang Y, Uchinoumi H, Thomas DD, Chen-Izu Y, Kato T, et al. Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J Mol Cell Cardiol. 2015;85:240–8.
Suetomi T, Yano M, Uchinoumi H, Fukuda M, Hino A, Ono M, et al. Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca²+ release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation. 2011;124:682–94.
Yano M, Okuda S, Oda T, Tokuhisa T, Tateishi H, Mochizuki M, et al. Correction of defective interdomain interaction within ryanodine receptor by antioxidant is a new therapeutic strategy against heart failure. Circulation. 2005;112:3633–43.
Yamamoto T, Yano M, Xu X, Uchinoumi H, Tateishi H, Mochizuki M, et al. Identification of target domains of the cardiac ryanodine receptor to correct channel disorder in failing hearts. Circulation. 2008;117:762–72.
Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–76.
Rosso R, Kalman JM, Rogowski O, Diamant S, Birger A, Biner S, et al. Calcium channel blockers and beta-blockers versus beta-blockers alone for preventing exercise-induced arrhythmias in catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:1149–54.
Bergenholm L, Parkinson J, Mettetal J, Evans ND, Chappell MJ, Collins T. Predicting QRS and PR interval prolongations in humans using nonclinical data. Br J Pharmacol. 2017;174:3268–83.
Itzhaki I, Maizels L, Huber I, Gepstein A, Arbel G, Caspi O, et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J Am Coll Cardiol. 2012;60:990–1000.
Ross S, Holliday M, Lim S, Semsarian C. Characterization of the first induced pluripotent stem cell line generated from a patient with autosomal dominant catecholaminergic polymorphic ventricular tachycardia due to a heterozygous mutation in cardiac calsequestrin-2. Stem Cell Res. 2019;37:101450.
Chen YJ, Chen YC, Wongcharoen W, Lin CI, Chen SA. Effect of K201, a novel antiarrhythmic drug on calcium handling and arrhythmogenic activity of pulmonary vein cardiomyocytes. Br J Pharmacol. 2008;153:915–25.
Kashimura T, Briston SJ, Trafford AW, Napolitano C, Priori SG, Eisner DA, et al. In the RyR2(R4496C) mouse model of CPVT, β-adrenergic stimulation induces Ca waves by increasing SR Ca content and not by decreasing the threshold for Ca waves. Circ Res. 2010;107:1483–9.
Dridi H, Kushnir A, Zalk R, Yuan Q, Melville Z, Marks AR. Intracellular calcium leak in heart failure and atrial fibrillation: a unifying mechanism and therapeutic target. Nat Rev Cardiol. 2020;17:732–47.
Yano M, Yamamoto T, Kobayashi S, Matsuzaki M. Role of ryanodine receptor as a Ca2+ regulatory center in normal and failing hearts. J Cardiol. 2009;53:1–7.
Yano M, Kobayashi S, Kohno M, Doi M, Tokuhisa T, Okuda S, et al. FKBP12.6-mediated stabilization of calcium-release channel (ryanodine receptor) as a novel therapeutic strategy against heart failure. Circulation. 2003;107:477–84.
Oda T, Yano M, Yamamoto T, Tokuhisa T, Okuda S, Doi M, et al. Defective regulation of interdomain interactions within the ryanodine receptor plays a key role in the pathogenesis of heart failure. Circulation. 2005;111:3400–10.
Seidel M, Thomas NL, Williams AJ, Lai FA, Zissimopoulos S. Dantrolene rescues aberrant N-terminus intersubunit interactions in mutant pro-arrhythmic cardiac ryanodine receptors. Cardiovasc Res. 2015;105:118–28.
Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113:829–40.
Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99:292–8.
Kushnir A, Wajsberg B, Marks AR. Ryanodine receptor dysfunction in human disorders. Biochim Biophys Acta Mol Cell Res. 2018;1865:1687–97.
Acknowledgements
We appreciate Dr. Long-sheng Song for critical comments. This study was supported by National Natural Science Foundation of China (Grant No. 81970250, 31671179), Natural Science Foundation of Guangdong, China (Grant No. 2021A1515010787), Shenzhen Key Laboratory of Metabolism and Cardiovascular Homeostasis (Grant No. ZDSYS20190902092903 237), Basic Research Foundation of Shenzhen (Grant No. JCYJ20180508152222104, JCYJ20190808123018993, JCYJ20190808121819024, JCYJ20210324094006018), and Shenzhen Science and Technology Innovation Committee Project (Grant No. JCYJ20180507182239272).
Author information
Authors and Affiliations
Contributions
JL generated the hypotheses and designed the experiments, and YXC provided natural product libraries for drug screening including Z16b. JFW performed most experiments and created the figures. GW participated in the key experiments and created the figures. FYQ and DLH prepared and purified Z16b for this study. YW and ALS carried out the molecular docking simulation. HPZ and CLW performed the MD simulation. SYZ and YL provided the clinical sample.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Wan, Jf., Wang, G., Qin, Fy. et al. Z16b, a natural compound from Ganoderma cochlear is a novel RyR2 stabilizer preventing catecholaminergic polymorphic ventricular tachycardia. Acta Pharmacol Sin 43, 2340–2350 (2022). https://doi.org/10.1038/s41401-022-00870-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-00870-1
