
Abstract
Connexin 43 (Cx43) is the most important protein in the gap junction channel between cardiomyocytes. Abnormalities of Cx43 change the conduction velocity and direction of cardiomyocytes, leading to reentry and conduction block of the myocardium, thereby causing arrhythmia. It has been shown that IL-1β reduces the expression of Cx43 in astrocytes and cardiomyocytes in vitro. However, whether caspase-1 and IL-1β affect connexin 43 after myocardial infarction (MI) is uncertain. In this study we investigated the effects of VX765, a caspase-1 inhibitor, on the expression of Cx43 and cell-to-cell communication after MI. Rats were treated with VX765 (16 mg/kg, i.v.) 1 h before the left anterior descending artery (LAD) ligation, and then once daily for 7 days. The ischemic heart was collected for histochemical analysis and Western blot analysis. We showed that VX765 treatment significantly decreased the infarct area, and alleviated cardiac dysfunction and remodeling by suppressing the NLRP3 inflammasome/caspase-1/IL-1β expression in the heart after MI. In addition, VX765 treatment markedly raised Cx43 levels in the heart after MI. In vitro experiments were conducted in rat cardiac myocytes (RCMs) stimulated with the supernatant from LPS/ATP-treated rat cardiac fibroblasts (RCFs). Pretreatment of the RCFs with VX765 (25 μM) reversed the downregulation of Cx43 expression in RCMs and significantly improved intercellular communication detected using a scrape-loading/dye transfer assay. We revealed that VX765 suppressed the activation of p38 MAPK signaling in the heart tissue after MI as well as in RCMs stimulated with the supernatant from LPS/ATP-treated RCFs. Taken together, these data show that the caspase-1 inhibitor VX765 upregulates Cx43 expression and improves cell-to-cell communication in rat heart after MI via suppressing the IL-1β/p38 MAPK pathway.
Similar content being viewed by others


Introduction
Globally, myocardial infarction (MI) is the leading cause of death. MI is mainly caused by coronary artery stenosis [1], which leads to myocardial ischemia, injury, and cardiac arrest. Ventricular tachycardia is the most common and potentially lethal complication of MI [2]. Primary arrhythmia is the major cause of sudden cardiac death in individuals with MI [3]. Although existing antiarrhythmic drugs can be used to treat the various stages of cardiac action potentials, many drugs have poor efficiency and a significant risk of adverse effects, especially drug-induced arrhythmia or organ damage [4, 5]. Therefore, seeking new therapeutic targets and exploring the mechanism of arrhythmia assume significance for reducing the mortality rate of MI.
Gap junctions (GJs) are transmembrane channels for electrical and chemical coupling and are the basis for maintaining normal cell-to-cell communication connections, electrical conduction, and normal rhythmic contraction between cardiomyocytes [6,7,8]. Alterations in electrical coupling via GJ channels have been found to lead to aberrant conduction and arrhythmogenesis [9]. Connexins (Cxs) in the myocardium are predominantly distributed in the intercalary discs and are mainly expressed as Cx45, Cx43, and Cx40 [10]. Cx43 is the most important protein in the gap junction channel between cardiomyocytes. Abnormalities in the quantity, distribution, and phosphorylation level of Cx43 can cause cardiomyocytes to change the conduction velocity and direction, leading to reentry and conduction block of the myocardium, thereby causing arrhythmia [11,12,13]. Identifying the molecular mechanism causing Cx43 dysfunction after MI is critical for reducing the occurrence of arrhythmias.
Sterile inflammation is a key element in tissue repair, but it can also contribute to severe cardiac damage and inappropriate ventricular remodeling after MI [14]. Interleukin-1 (IL-1) regulates immune cell recruitment, cytokine synthesis, and extracellular matrix degradation and contributes to the inflammatory response following MI [15]. An abrupt increase in the IL-1β expression level decreases the left ventricular ejection fraction (LVEF), impairs cardiac function, and causes cardiac hypertrophy and fibrosis [16,17,18]. NLR family pyrin domain-containing 3 (NLRP3) is a multiprotein complex consisting of an NLRP3 scaffold, which comprises nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3, an ASC (PYCARD) adaptor, and a pro-caspase-1 effector. The NLRP3 inflammasome interacts with ASC to recruit pro-caspase-1 and convert it into activated caspase-1 and then promotes transformation of the intracellular cytokine precursor pro-IL-1β into mature and biologically active IL-1β, followed by its release into the extracellular environment [19]. It has been demonstrated that IL-1β is closely associated with the occurrence of arrhythmia and with abnormal changes in myocardial ion channels and that IL-1β treatment can reduce the in vitro expression of Cx43 in astrocytes and cardiomyocytes [20,21,22,23]. However, whether and how IL-1β and caspase-1 affect Cx43 is uncertain.
Therefore, this study aimed to evaluate the effects of the caspase-1 inhibitor VX765 on Cx43 expression and cell–cell communication after MI and to clarify the molecular mechanism by which caspase-1/IL-1β regulate Cx43, which may provide a new therapeutic target for myocardial remodeling after myocardial injury.
Materials and methods
Animal experiments
Male Sprague–Dawley (SD) rats weighing 220–250 g were obtained from the Experimental Animal Center of Zhengzhou University (Zhengzhou, China). All animals were kept under standard laboratory conditions: room temperature 20 ± 2 °C, relative humidity 40%–60%, 12 h light-dark cycles, and chaw and water ad libitum. All animal experiments were approved by the Animal Experiments Committee of Zhengzhou University and conformed to the Guide for the Care and Use of Laboratory Animals (NIH Publication, No. 85–23, revised 1996).
Myocardial infarction (MI) model
By permanently ligating the left anterior descending artery (LAD) as previously reported, we established a rat model of MI [24, 25]. Briefly, rats were randomly divided into three groups. The sham group (only threaded at the same site without ligation), the MI group (infarcted rats treated with vehicle only), and the VX765 + MI group (VX765, a caspase-1 inhibitor, 16 mg/kg, HY-13205, Med Chem Express, Monmouth Junction, NJ, USA), which was administered intravenously 1 h before surgery and thereafter once every day for 7 consecutive days [26].
Electrocardiogram (ECG) detection
Before and after surgery, a lead II ECG (Nihon Kohden CardiofaxC, Japan) was recorded. The presence of an enhanced ST segment in the ECG following LAD ligation indicated that the rat MI model had been successfully established. As previously reported, the QT interval was measured and adjusted for heart rate (QTc) using Bazett’s equation [27].
Echocardiographic measurements
Cardiac function was evaluated 7 days after the operation via transthoracic echocardiography. Left ventricular fractional shortening (LVFS), left ventricular ejection fraction (LVEF), and left ventricular end-diastolic diameter (LVED.d) were recorded through M-mode tracing (Vevo 2100; Visual Sonics, Toronto, Canada).
Hematoxylin and eosin (H&E) staining
The heart samples were individually removed and immediately submerged in 4% paraformaldehyde solution for 24 h. Following fixation and paraffin embedding, 4 μm thick segments were cut and stained with H&E for overall morphological evaluation with an optical microscope (BX60; Olympus, Japan), image acquisition and analysis (ImageJ Launcher, National Institutes of Health, Bethesda, MD, USA).
Masson’s trichrome staining
Heart samples were collected from different groups, fixed in 4% paraformaldehyde, embedded in paraffin, and cut into 4-μm-thick slices. To determine the degree of cardiac fibrosis, slides were stained with Masson’s trichrome. The collagen tissue area was calculated using ImageJ software and is represented as the percentage of the full ventricle area.
Immunohistochemical (IHC) staining
Heart slices (4 μm) were treated with 4% paraformaldehyde to block endogenous peroxidase and then incubated with 1% bovine serum albumin (BSA) and anti-Cx43 (1:500; #71–0700, Thermo Fisher Scientific, Waltham, MA, USA), anti-caspase-1 (1:2500; #ab108362, Abcam, Cambridge, MA, USA), and anti-IL-1β (1:1000; #A11369, ABclonal, China) antibodies for 2 h and then with horseradish peroxidase (HRP)-conjugated anti-goat IgG (1:10000; #SA00001-1, Proteintech, China) for 60 min. Diaminobenzidine (DAB; #8801-4965-72, Invitrogen, Carlsbad, CA, USA) was used as the chromogen.
The cells fixed with 4% paraformaldehyde were washed and permeabilized with 0.1% Triton X-100. After incubation with 5% BSA for 1 h at room temperature, the cells were incubated with primary anti-Cx43 antibody overnight at 4 °C. Then, secondary antibody (Alexa Fluor 488) (1:200; #GB25301, Servicebio, China) was applied to the cells for 2 h at room temperature. Nuclei were stained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) (#S2110, Solarbio, China). Cells were analyzed using an EVOS™ M7000 Imaging System (Thermo Fisher Scientific).
Triphenyltetrazolium chloride (TTC) staining
Fresh hearts were frozen immediately at −20 °C for 20 min. Then, the heart was sectioned from the apex to the base into five sections. After 30 min of incubation in a 2% TTC solution at 37 °C, the uninfarcted myocardium was stained red, while the infarcted myocardium was stained white. ImageJ was used to measure the infarct area and the total area of each piece of tissue, calculate the ratio of the two, and record the average of the ratio of each layer of tissue as the infarct area of the heart.
Serum lactate dehydrogenase (LDH) and IL-1β levels
According to the manufacturer’s instructions, LDH levels in serum were measured using standardized commercially available kits (#A020-1-2, Jian Cheng, Nanjing, China), and IL-1β levels were measured using rat enzyme-linked immunoassay (ELISA) kits (#70-EK301B/3-96, Multi Sciences, Hangzhou, China).
Isolation and culture of primary rat neonatal cardiomyocytes and fibroblasts
Rat neonatal cardiomyocytes (RCMs) and fibroblasts (RCFs) were harvested according to a previously described method [25]. Briefly, newborn heart ventricles were minced into small fragments and then digested with 0.1 mg/mL trypsin (#T1300, Solarbio) and collagenase II (#LS004176, Worthington, Lakewood, NJ, USA) at 37 °C. The cell suspensions were plated for 2 h at 37 °C to separate rat cardiac fibroblasts from RCMs, and the RCM culture medium contained 0.1 mM 5-bromouracil (#51-20-7, Aladdin, Shanghai, China) to inhibit the growth of RCFs.
As an in vitro model of cellular inflammation, RCFs were divided into the NC group (blank control group), NC/VX765 group, LPS/ATP group, and LPS/ATP/VX765 group. VX765 (25 μM) was added to RCFs for 30 min in advance, and then, the cells were treated with LPS (1 μg/mL, #L2630, Sigma–Aldrich, St. Louis, MO, USA) for 12 h and ATP (5 mM, #A-2383, Sigma–Aldrich) for 1 h to activate the NLRP3 inflammasome. Subsequently, the supernatants from the four groups of RCFs were used as conditioned medium and incubated with RCMs for 24 h.
To verify the direct effect of IL-1β (#HY-P7097, Med Chem Express) on Cx43, we used exogenous IL-1β to stimulate RCMs for verification and used it in combination with the IL-1β inhibitor TLR1 (#HY-W011400, Med Chem Express) or the p-38 MAPK inhibitor SB203580 (#HY-10256, Med Chem Express). The cells were divided into the NC group, IL-1β group, IL-1β/TLR1 group and IL-1β/SB203580 group. TLR1 (40 μM) and SB203580 (3 μM) were added to RCMs 1 h in advance; IL-1β (80 ng/mL) was added for treatment for 24 h; and then, the cells were collected.
Transient siRNA transfection
Rat caspase-1 siRNA and negative control siRNA were obtained from Hanbio (Wuhan, China). RCFs were seeded in 6-well plates and transiently transfected with 40 nM siRNA using 3.75 µL Lipofectamine RNAiMAX (Invitrogen) following the manufacturer’s instructions. The following sequences were used in Table 1.
Scrape-loading/dye transfer (SL/DT) assay
The SL/DT technique was used to measure intercellular gap-junctional communication in cardiomyocytes. After treatment, the cells were rinsed gently three times with PBS. The dye was loaded into the cells by gently placing the tip of a surgical steel blade. Then, the cells were incubated with 0.5 mL of 0.1% (w/v) Lucifer Yellow CH (#L0144, Sigma–Aldrich) and 0.05% dextran conjugate-rhodamine B, 10,000 MW in PBS (#R9379, Sigma–Aldrich) at 37 °C for 10 min in the dark. The cells were then washed with PBS and fixed with 4% paraformaldehyde. Nuclei were stained with DAPI. The cells were viewed immediately using an Eclipse E100 fluorescence microscope (Nikon, Japan). The number of communicating cells in treated samples was compared with that in the controls. For each well, three random images were taken, and the means were calculated. For each treatment and control group, three separate experiments were carried out in triplicate.
Western blot analysis
Total protein from cardiac tissue or cells was collected using lysis buffer (#R0030, Solarbio) plus protease inhibitor cocktail (#HY-K0010, Med Chem Express) as described previously [25]. Briefly, membranes were incubated with primary antibodies against NLRP3 (1:2500; #ab214185, Abcam), ASC (1:1000; #AG-25B-0006, Adipogen, Liestal, Switzerland), caspase-1 (1:2500; #ab108362, Abcam), IL-1β (1:1000; #A11369, ABclonal), p-p38 MAPK (1:1000; #4511 S, Cell Signaling), p-38 MAPK (1:1000; #14064-1-AP, Proteintech), Cx43 (1:1000; #71-0700, Thermo Fisher Scientific), and GAPDH (1:10000; #60004-1-Ig, Proteintech) at 4 °C overnight, followed by incubation with appropriate secondary antibodies. The blots were analyzed and quantified using ImageJ analysis software.
Statistical analysis
Data are presented as the means ± standard deviations. All data were obtained and analyzed using SPSS 21.0 (SPSS Inc., Chicago, IL, USA). Statistical comparisons were performed using unpaired or paired t tests (P = 0.05). All data were tested using either a two-tailed Student’s t test or ANOVA followed by a two-tailed Dunnett’s test. P < 0.05 were considered statistically significant.
Results
Caspase-1 inhibition attenuates cardiac dysfunction post MI
First, H&E staining revealed that the cardiac histology in the sham group was normal, but the MI group showed different degrees of cell damage, including vacuolation and inflammatory infiltration. However, pretreatment with VX-765, a caspase-1 inhibitor, dramatically decreased MI-induced myocardial damage (Fig. 1a). Masson staining showed that the MI group had abundant collagen fibers and increased fibrosis, while the VX765 + MI group demonstrated improvement in the occurrence of cell damage and reduced cardiac fibrosis (Fig. 1b, c). We then used echocardiographic studies to measure heart function following MI (Fig. 1d). At 7 days after surgery, the LVEF (Fig. 1e) and LVFS (Fig. 1f) in the MI group were lower but significantly improved with VX765 pretreatment. Conversely, LVED in the MI group was substantially higher than that in the sham-operated group, and VX-765 pretreatment decreased LVED (Fig. 1g). As shown in Fig. 1h, VX765 pretreatment reduced the infarct size compared to that in the MI group (Fig. 1i). The level of LDH, an indicator of heart damage, was increased in the MI group and was significantly decreased after VX765 pretreatment (Fig. 1j). In general, VX765 attenuated MI-induced cardiac injury in rats.

a-b Representative images of hematoxylin-eosin (H&E) (a) and Masson (b) (20×) staining of cardiac tissues at 7 days after MI. As shown in the images, the left ventricular parts were defined as the ischemic zone, the 1-mm area around the ischemic zone was the border zone, and the area further to this was the remote zone. Bars = 100 μm. c Bar graph depicting the fibrillation region in the ischemic area 7 days after MI (n = 3 per group). Myocardial function was assessed using a Vevo 2100 high-resolution microimaging system 7 days post-MI. d M-mode echocardiographic imaging was acquired 7 days after MI. We further analyzed the echocardiographic parameters of left ventricular ejection fraction (LVEF) (e), left ventricular fractional shortening (LVFS) (f), and left ventricular internal dimension at end-diastole (LVED; d) (g) (n = 5). Triphenyltetrazolium chloride staining was performed for (h) comparison and (i) assessment of the ischemic area between the groups. Ischemic infarct regions look pale white, whereas normal tissues appear red (n = 3). j Serum lactate dehydrogenase (LDH) levels were assessed (n = 5 per group). *P < 0.05, **P < 0.01, ***P < 0.001 vs Sham. #P < 0.05, ##P < 0.01, ###P < 0.001 vs MI.
Caspase-1 inhibition shortens the elongated QT interval in rats with MI
A standard ECG was obtained before and after the procedure (Fig. 2a). The findings revealed that MI causes ST elevation and prolonged QT and QTc intervals. In the VX765 pretreatment group, ST elevation was decreased (Fig. 2b), the QT interval was shortened (Fig. 2c), and the QTc interval (Fig. 2d) was shortened compared to those parameters in the MI group.

a ECG waveforms before surgery, after surgery, and before sacrifice of the rats, with marked ST segments. Bar graph showing the height of the ST segment (b), QT interval (c), and QTc interval (d) (n = 5). *P < 0.05, **P < 0.01 vs Sham. #P < 0.05, ##P < 0.01 vs MI.
Caspase-1 inhibition decreases IL-1β secretion and increases Cx43 expression post MI
To explore the effects of VX765 on the inflammatory response, caspase-1 (Fig. 3a, b) and IL-1β (Fig. 3c, d) expression levels were determined by performing immunohistochemistry assays. The Western blotting results, as shown in Fig. 3e, indicated that the protein components of the NLRP3 inflammasome (ASC, NLRP3, pro-caspase-1) (Fig. 3f–h) and IL-1β (Fig. 3i) were considerably elevated in the ischemic region in the post-MI rat heart tissues compared to the sham group. However, VX765 pretreatment significantly reduced these protein levels. ELISA results indicated that the level of IL-1β in the MI group increased, whereas it declined in the MI + VX765 group (Fig. 3j).

a–d Representative immunohistochemistry images of cardiac samples 7 days post-MI were stained to detect caspase-1 (a) and IL-1β (c) (20×). Bars = 100 μm. The protein positivity rate for caspase-1 (b) and IL-1β (d) in ischemic, border, and remote zones 7 days post-MI is demonstrated (n = 3). e The levels of IL-1β (17 kDa), caspase-1 (20 kDa), ASC, NLRP3 and GAPDH in the ischemic area in rat heart tissue were determined by Western blotting 7 days post-MI. Bar graphs show the fold changes in the protein level in ischemic heart sections for NLRP3 (f), ASC (g), cleaved-caspase-1 (20 kDa) (h), and cleaved-IL-1β (17 kDa) (i) 7 days post-MI (n = 5). j Statistical graph of changes in the serum IL-1β concentration (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001 vs Sham. ##P < 0.01 vs MI.
To observe the effects of caspase-1 on Cx43 in MI tissue, we first evaluated the expression and distribution of Cx43 using immunohistochemistry assays (Fig. 4a). In the ischemic and border areas in the sham group, the Cx43 distribution was clustered, mainly in the end-to-end junctions of adjacent cells, and a small amount was present in the cell side junctions; in the MI group, Cx43 was hardly visible around the infarct area, and occasionally Cx43 was distributed in the cell-side junction or scattered on the cell surface; however, this abnormal distribution was significantly improved after VX765 pretreatment. The MI group had fewer Cx43 protein-positive cells than the sham group; however, this number increased after VX765 pretreatment (Fig. 4b). As shown in Fig. 4c, the protein expression level of Cx43 in ischemic tissue was considerably lower than that in the sham group, while VX765 pretreatment markedly increased the levels in comparison to those in the MI group (Fig. 4d).

a Representative immunohistochemistry images of cardiac sections 7 days post-MI stained for Cx43 (20×). Bars = 100 μm. b The Cx43 positive rate in ischemic, border, and remote zones 7 days after MI is demonstrated (n = 3 per group). c Cx43 expression was determined by Western blotting 7 days after MI in heart sections, and (d) bar graphs show the fold changes in Cx43 expression in ischemic sections (n = 5 per group). **P < 0.01 vs Sham. #P < 0.05, ##P < 0.01 vs MI.
Caspase-1 inhibition upregulates Cx43 expression and increases cell-to-cell communication in RCMs stimulated by supernatant from RCFs treated with LPS/ATP
Previous studies, including ours, have shown that activation of the NLRP3 inflammasome/caspase-1/IL-1β pathway occurs mostly in CFs, while NLRP3 inflammasome levels in CMs are minimal [24, 25]. In normal heart tissue, CMs and CFs are closely related in space and influence each other’s activities through paracrine activity [28]. We further investigated the effects of caspase-1 inhibition on IL-1β in RCFs and on Cx43 expression in RCMs via the paracrine pathway.
First, we observed that LPS/ATP mediated activation of the NLRP3 inflammasome (Fig. 5a) and formation of ASC specks (Supplementary Fig. S1) in RCFs, whereas VX765 downregulated the above changes and IL-1β secretion, as assessed by Western blotting (Fig. 5b–e) and ELISA (Fig. 5f).

a Determination of ASC, NLRP3, cleaved-IL-1β (17 kDa), and cleaved-caspase-1 (20 kDa) protein expression levels. RCFs and rat cardiac myocytes (RCMs) were isolated from 1-day-old rat neonates. RCFs were treated with VX765 (25 μM, added 30 min before other factors), lipopolysaccharide (LPS; 1 μg/mL, 12 h), and ATP (5 mM, 1 h). b–e Bar graphs showing fold-changes in NLRP3 (b), ASC (c), cleaved-caspase-1 (20 kDa) (d), and cleaved-IL-1β (17 kDa) (e) protein levels in RCFs after exposure to LPS/ATP (n = 3). f Levels of IL-1β in the supernatant were assessed in RCFs treated with or without VX765 (n = 3). *P < 0.05, **P < 0.01 vs NC/VX765. #P < 0.05, ##P < 0.01 vs LPS/ATP.
Next, we found that Cx43 expression in RCMs was decreased in the presence of LPS/ATP-stimulated RCFs supernatant (Fig. 6a), and VX765 pretreatment upregulated Cx43 expression (Fig. 6b). To preclude the possibility that the reported upregulation of Cx43 by VX765 in RCMs was attributable to unintended effects, caspase-1 expression was knocked down by transfection with siRNA (si-caspase-1-1, −2, and −3) in RCFs. Western blotting showed that all three siRNAs significantly inhibited caspase-1 expression (Fig. 6c). We chose si-caspase-1-3 for the follow-up experiments (Fig. 6d). Knockdown of caspase-1 in RCFs upregulated Cx43 expression in RCMs stimulated by the supernatant of RCFs treated with LPS/ATP. These findings demonstrate that pharmacological suppression of caspase-1 and knockdown of caspase-1 both increased Cx43 expression in RCMs.

a Cx43 expression was determined using Western blotting. The supernatant of lipopolysaccharide (LPS)/ATP-stimulated fibroblasts was transferred and used to stimulate cardiomyocytes for 24 h. b Bar graphs representing the fold changes in Cx43 protein levels in RCMs (n = 3). c Si-caspase-1 interference efficiency was examined by Western blotting. Rat cardiac fibroblasts (RCFs) were transiently transfected with si-NC or vehicle control and si-caspase-1 for 6 h before being cultured for 24 h. LPS/ATP was then used to expose the cells, and the supernatant was transferred to stimulate cardiomyocytes. d Cx43 expression was determined using Western blotting. e Images obtained from the scrape loading (SL)/dye transfer (DT) assay after application of lucifer yellow CH dilithium salt (LY, MW 457), which transfers via the functional gap junction channels, and rhodamine-dextran (RhD, MW 10,000), which was reserved in the scraped cells. Bars = 100 μm. f After eliminating RhD-stained areas, the net transfer of LY (the area through which LY diffuses) was used to assess gap junction intercellular communication (GJIC) function. To determine the fraction of the control (GJIC-FOC), the net LY areas in individual images were normalized to the averaged net area from negative or solvent control dishes. Each figure shows three independent experiments, all performed in triplicate (n = 3). (g) Immunostaining for total Cx43 and counterstaining of nuclei with DAPI (40 ×), Bars = 25 μm. *P < 0.05, **P < 0.01 vs NC. #P < 0.05, ##P < 0.01 vs LPS/ATP.
To test whether VX765 may influence Cx43-mediated cell-to-cell communication, we performed a fluorescent dye transfer assay on RCMs treated with LPS/ATP-stimulated RCFs supernatant (Fig. 6e). RCMs clearly showed lower lucifer yellow transfer, and VX765 pretreatment increased lucifer yellow transfer (Fig. 6f).
To determine the effect of VX765 on the internalization of Cx43, RCMs were cultured and exposed to supernatant from RCFs that had been treated with LPS/ATP, with or without VX765, followed by immunofluorescence staining (Fig. 6g). Cx43 expression was increased in the cytoplasm but reduced in the membranes of RCMs treated with LPS/ATP-stimulated RCFs supernatant. Additionally, VX765 pretreatment alleviated the Cx43 internalization changes in cells.
Caspase-1/IL-1β activates p38 MAPK, downregulates Cx43, and reduces cell-to-cell communication
In astrocytes, IL-1β has been shown to activate p38 MAPK and reduce dye coupling of Cx43-mediated gap junctions [29]. We further investigated p38 MAPK involvement in Cx43 regulation in MI. First, we observed that VX765 pretreatment decreased the ratio of p-p38 MAPK/p38 MAPK in rats with MI (Fig. 7a, b). Moreover, we observed that VX765 pretreatment also downregulated the ratio of p-p38 MAPK/p38 MAPK in RCMs treated with LPS/ATP-stimulated RCFs supernatant (Fig. 7c, d). In another experiment, RCMs were cultured with the supernatant of LPS/ATP-conditioned RCFs containing either TLR1 or SB203580. As shown in Fig. 7e, the supernatant of LPS/ATP-conditioned RCFs decreased the expression of Cx43 protein (Fig. 7f) and increased the ratio of p-p38 MAPK/p38 MAPK (Fig. 7g), while the effects were inhibited by SB203580 and TLR1.

Caspase-1 inhibition reduces the activation of p38 MAPK in myocardial infarction (MI) tissues and rat cardiac myocytes (RCMs). a Western blotting was used to assess the expression levels of p38 and p-p38 in ischemic heart sections obtained from rats with MI. b Bar graphs showing the fold-changes in the expression levels (n = 5). **P < 0.01 vs Sham. ##P < 0.01 vs MI. c The protein expression level of p38 and p-p38 in RCMs was stimulated by supernatant from rat cardiac fibroblasts (RCFs). d Fold-changes are shown by bar graphs (n = 3). Supernatant from RCFs was used to stimulate RCMs for 24 h and used in combination with the IL-1β inhibitor TLR1 (40 μM) or p-38 MAPK inhibitor SB203580 (3 μM). **P < 0.01 vs NC. ##P < 0.01 vs LPS/ATP. e Western blotting was used to identify changes in the protein expression levels of Cx43 and p38 MAPK in RCMs stimulated with the supernatant from RCFs. f, g Bar graphs showing fold-changes in Cx43 (f) and p-p38 MAPK (g) protein levels. **P < 0.01 vs NC. #P < 0.05, ##P < 0.01 LPS/ATP/TLR1 vs LPS/ATP. &P < 0.01, &&P < 0.001 LPS/ATP/SB vs LPS/ATP.
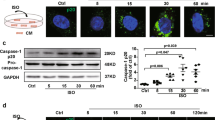
Next, we directly stimulated RCMs with exogenous IL-1β. According to our findings, IL-1β increased the levels of phosphorylated p-p38 MAPK (Fig. 8a). p38 MAPK activation induces a decrease in Cx43. Furthermore, SB203580 (Fig. 8b) and TLR1 (Fig. 8c) significantly reduced the effect of IL-1β on Cx43. In addition, treatment of RCMs with IL-1β reduced lucifer yellow transfer (Fig. 8d). Pretreatment of RCMs with SB203580 or TLR1 increased lucifer yellow transfer (Fig. 8e). SB203580 and TLR1 both alleviated the internalization of Cx43 induced by IL-1β (Fig. 8f).

a Western blotting was used to detect alterations in the protein expression of p38 MAPK and Cx43 in RCMs stimulated with supernatant from rat cardiac fibroblasts (RCFs). Exogenous IL-1β (80 ng/mL) was used to stimulate RCMs for 24 h and was used in combination with the IL-1β inhibitor TLR1 (40 μM) or p-38 MAPK inhibitor SB203580 (3 μM). b, c Bar graphs showing fold changes in Cx43 (b) and p38 MAPK (c) protein levels. d Images obtained from the scrape loading (SL)/dye transfer (DT) assay in which lucifer yellow (LY) and rhodamine-dextran (RhD) were applied. Bars = 100 μm. **P < 0.01 vs NC. #P < 0.05, ##P < 0.01 IL-1β/SB vs IL-1β. &P < 0.05 IL-1β/TLR1 vs IL-1β. e To obtain the fraction of the control (GJIC-FOC), the net LY areas in individual images were normalized to the averaged net area from negative or solvent control dishes. Each figure shows three independent experiments, all performed in triplicate (n = 3). **P < 0.01 vs NC. ##P < 0.01 IL-1β/TLR1 vs IL-1β. &&P < 0.01 IL-1β/SB vs IL-1β. (f) Immunostaining for total Cx43 and counterstaining with DAPI to visualize nuclei (40 ×). Bars = 25 μm.
Discussion
Inflammation is a key pathogenic hallmark of MI damage. The main proinflammatory cytokine implicated in inflammation is IL-1β. Caspase-1 converts pro-IL-1β into mature and active IL-1β, which is subsequently released into the extracellular environment to trigger an inflammatory response [30].
Recently, based on extensive knowledge of the mechanisms of inflammasome activation, the development of therapies that target these pathways or mechanisms has been accelerated [31]. For example, currently, three IL-1 inhibitors, anakinra, canakinumab, and rilonacept, are being used in clinical practice [32]. In addition, small-molecule NLRP3 inflammasome inhibitors have been identified and are under clinical trial [33,34,35]. Treatments aimed at inhibiting IL-1 activity in MI, heart failure, pericarditis, and other chronic inflammatory heart diseases have been developed [32].
VX765, quickly converted to an active metabolite in vivo by nonspecific esterases VRT-043198, is a selective caspase-1 inhibitor [36]. VX765 is currently being investigated in humans in a phase 2b clinical study for drug-resistant partial epilepsy and has been reported to be effective in humans [37]. Moreover, VX765 has been reported to have a protective effect against acute ischemia–reperfusion (I/R) injury in an in vivo rat model [26]. This particular subclass of caspase-1 inhibitors might be a viable option for future cardioprotective therapies [38]. However, the role of VX765 in myocardial electrical remodeling and its underlying molecular mechanisms have not been fully elucidated. We have provided evidence that selectively inhibiting caspase-1 with VX765 can rescue cardiac histological morphology and cardiac dysfunction and promote Cx43 expression in a rat MI model (Fig. 1). Moreover, this cardioprotective effect was mediated by preventing IL-1β secretion from RCFs and inhibiting the IL-1β/p38 MAPK/Cx43 signaling pathway in RCMs.
In this study, we found that VX765 alleviated abnormal changes in cardiac morphology and reduced cardiac dysfunction caused by MI. These findings are in accordance with those of a previous study showing that inhibition of caspase-1 protects rat hearts from MI [39]. One of the primary variables impacting the propagation of electrical impulses through the heart is the degree of cell-to-cell communication via gap junction (GJ) channels. The QT interval has been demonstrated to be lengthened by faulty electrical impulse propagation [40]. If left untreated, a long QT can cause torsades de pointes and eventually ventricular fibrillation, which can lead to abrupt cardiac death [41]. The QT interval duration is determined by ventricular depolarization and repolarization. Prolongation of the QT interval reflects ventricular conduction delay, a substrate for arrhythmogenicity [42]. We found that the QTc interval was significantly longer in the MI rat model than in the sham group (Fig. 2). These findings support a previous report indicating that MI can lead to prolongation of the QTc interval [43]. It has been demonstrated that modifications in electrical coupling through GJ channels can lead to aberrant conduction and arrhythmogenesis in the heart [9]. Cx43, the most prevalent cardiac GJ protein [44], maintains a steady beating rhythm by mediating ventricular conduction of cardiac action potentials. Our data revealed that inhibition of caspase-1 could shorten the QT interval prolongation caused by MI. These results indicate that inhibition of caspase-1 may shorten the conduction process by improving GJs between cardiomyocytes. Therefore, we investigated the effects of VX765 on Cx43 following MI in rats (Fig. 4). First, we observed reduced NLRP3 inflammasome activation in rats with MI after VX765 pretreatment. The NLRP3 inflammasome is an essential line of defense for the innate immune system against infection, but it is also linked to a variety of chronic inflammatory and metabolic disorders, including gout, atherosclerosis, human inflammatory bowel disease, diabetes, neuroinflammation, and cancer [45,46,47,48,49].
We observed that inhibiting caspase-1 significantly decreased the protein levels of NLRP3, ASC, caspase-1, and IL-1β. Why does treatment with VX765 cause downregulation of NLRP3 and ASC protein expression levels in rats with MI (Fig. 3)? It has been reported that the suppression of caspase-1 by VX765 is due to a negative feedback mechanism involving the NLRP3 inflammasome activation step or activation of signaling pathways implicated in other priming processes, such as interacting with NLRP3 inflammasomes [50, 51]. In addition, Cx43 expression decreased in the MI group, and VX765 pretreatment upregulated Cx43 expression. Our findings are in accordance with previous research showing that the total amount of Cx43 is decreased after cardiac ischemia [52], MI [53], and heart failure [54].
CFs are well known for their function in deposition of the extracellular matrix that forms a proteinaceous network around cells, reinforcing the cardiac architecture [55]. More evidence has shown that CFs also secrete signaling molecules to the surrounding immune cells and cardiomyocytes, which affect the inflammatory response, contraction, and hypertrophy [56, 57].
Recent findings, including those from our study, have shown that the NLRP3 inflammasome is significantly upregulated and subsequently induces IL-1β production in CFs during the intense inflammatory response to MI [25, 58]. Therefore, we investigated whether and how NLRP3 inflammasome activation and IL-1β secretion in CFs after MI affects Cx43 expression in CMs through the paracrine pathway. We used a cell model that activated the NLRP3/caspase-1 pathway via LPS/ATP stimulation in RCFs. We incubated RCMs with the supernatant from RCFs treated with LPS/ATP to simulate this paracrine effect, as previously described [59]. Our results showed that after LPS/ATP stimulation, the NLRP3 inflammasome was activated, and IL-1β secretion in the supernatant was enhanced; however, VX765 reduced NLRP3 inflammasome activation and IL-1β secretion in RCFs (Fig. 5). Subsequently, we found that VX765 pretreatment upregulated Cx43 expression in RCMs (Fig. 6). To exclude the off-target effects of the inhibitor VX765, we collected the supernatant of LPS/ATP-stimulated RCFs and knocked down caspase-1 via siRNA to observe Cx43 expression in RCMs. The results revealed that inhibiting caspase-1 pharmacologically and physiologically upregulates Cx43 expression.
Activation of the MAPK signaling pathway appears to significantly contribute to the proinflammatory response in the myocardium [60]. It has also been reported that LPS stimulates the activation of p38 MAPK and JNK, which promotes the release of inflammatory mediators followed by cardiac dysfunction [60]. We found that VX765 pretreatment also decreased p38 MAPK phosphorylation (Fig. 7). This suggests that VX765 pretreatment may upregulate Cx43 expression in RCMs via suppressing the IL-1β/p38 MAPK pathway. To confirm this hypothesis, we cultured RCMs with the supernatant of LPS/ATP-conditioned RCFs containing either TLR1 (an IL-1β inhibitor) or SB203580 (a p38 MAPK inhibitor). The results showed that TLR1 and SB203580 reversed the effects on Cx43 protein expression as well as on the ratio of p-p38/p38 MAPK in RCMs in the presence of the supernatant of LPS/ATP-conditioned RCFs.
Furthermore, Cx43 GJ intercellular communication has been found to be decreased as a result of changes in the Cx43 expression level, phosphorylation status, and membrane localization [61]. To determine whether caspase-1 affects cell-to-cell communication, junctional transfer of the tracer lucifer yellow (anionic) was assessed in a scrape-loading experiment utilizing RCM cells treated with the supernatant from RCFs treated with LPS/ATP. RCMs clearly showed lower lucifer yellow transfer, and VX765 pretreatment increased lucifer yellow transfer in the LPS/ATP-stimulated RCFs supernatant (Fig. 6). Moreover, immunofluorescence analysis of RCMs treated with supernatant from RCFs treated with LPS/ATP, with and without caspase-1, indicated that the positive effects seen in the presence of VX765 were due to an increase in Cx43 levels in GJ plaques.
To further validate the above experiment, we conducted another experiment. We used exogenous IL-β combined with TLR1 or SB203580 to directly stimulate RCMs (Fig. 8). The composition of the LPS/ATP-stimulated RCFs supernatant is complex; therefore, the above results alone did not directly verify that Cx43 expression in RCMs was caused by IL-1β from NLRP3 inflammasome activation. The purpose of this experiment was to further verify that caspase-1 inhibitors indeed upregulate Cx43 expression by inhibiting the IL-1β/p38 MAPK pathway. Consistent with these results, it has been stated that IL-1β activates p38 MAPK, downregulates Cx43 levels, and suppresses cell–cell communication in the early stages of experimental autoimmune myocarditis in rats [62]. Furthermore, the results showed that targeting NLRP3/caspase-1/IL-1β activation in RCFs can increase Cx43 expression in RCMs via inhibiting the p38 MAPK pathway.
This study has several limitations. In many tissues, coupling between cells of the same cell type (homocellular coupling) and cells of distinct cell type (heterocellular coupling) is ubiquitous [63]. In the current study, we studied cell–cell communication between cardiomyocytes. In addition to NLRP3 inflammasome activation, caspase-1 is implicated in the activation of additional inflammasomes, including the AIM2, NLRP1, and NLRC4 inflammasomes. Studies on the NLRP3 inflammasome are relatively extensive; thus, in this work, we solely investigated the impact of VX765 on Cx43 during NLRP3 inflammasome activation.
Conclusions
In this study, we found that caspase-1 inhibition reduced activation of the NLRP3 inflammasome and IL-1β secretion in RCFs, which subsequently led to upregulation of Cx43 expression and improved cell–cell communication in RCMs through inhibition of the IL-1β/p38 MAPK pathway, thus alleviating cardiac dysfunction (Fig. 9). Therefore, caspase-1 inhibition might be a promising therapeutic approach for management of MI.

Targeting caspase-1 upregulates Cx43 expression and improves communication between cells after myocardial infarction (MI) in rats.
References
Yellon DM, Hausenloy DJ. Myocardial reperfusion injury - reply. N Engl J Med. 2007;357:2409–10.
Henkel DM, Witt BJ, Gersh BJ, Jacobsen SJ, Weston SA, Meverden RA, et al. Ventricular arrhythmias after acute myocardial infarction: a 20-year community study. Am Heart J. 2006;151:806–12.
Chen JG, Li M, Yu YW, Wu XL, Jiang R, Jin YY, et al. Prevention of ventricular arrhythmia complicating acute myocardial infarction by local cardiac denervation. Int J Cardiol. 2015;184:667–73.
Heijman J, Voigt N, Dobrev D. New directions in antiarrhythmic drug therapy for atrial fibrillation. Future Cardiol. 2013;9:71–88.
Frommeyer G, Eckardt L. Drug-induced proarrhythmia: risk factors and electrophysiological mechanisms. Nat Rev Cardiol. 2016;13:36–47.
Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM, et al. Cardiac cell–cell junctions in health and disease: electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23–31.
Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–400.
Basheer W, Shaw R. The “tail” of Connexin43: an unexpected journey from alternative translation to trafficking. Biochim Biophys Acta. 2016;1863:1848–56.
van Rijen HVM, Eckardt D, Degen J, Theis M, Ott T, Willecke K, et al. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. 2004;109:1048–55.
Desplantez T. Cardiac Cx43, Cx40 and Cx45 co-assembling: involvement of connexins epitopes in formation of hemichannels and gap junction channels. BMC Cell Biol. 2017;18:3.
Hood AR, Ai X, Pogwizd SM. Regulation of cardiac gap junctions by protein phosphatases. J Mol Cell Cardiol. 2017;107:52–7.
Michela P, Velia V, Aldo P, Ada P. Role of connexin 43 in cardiovascular diseases. Eur J Pharmacol. 2015;768:71–6.
Rusiecka OM, Montgomery J, Morel S, Batista-Almeida D, Van Campenhout R, Vinken M, et al. Canonical and non-canonical roles of connexin43 in cardioprotection. Biomolecules. 2020;10:1225.
Nahrendorf M, Pittet MJ, Swirski FK. Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation. 2010;121:2437–45.
Takahashi M. Role of NLRP3 inflammasome in cardiac inflammation and remodeling after myocardial infarction. Biol Pharm Bull. 2019;42:518–23.
Hartman MHT, Groot HE, Leach IM, Karper JC, van der Harst P. Translational overview of cytokine inhibition in acute myocardial infarction and chronic heart failure. Trends Cardiovasc Med. 2018;28:369–79.
Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. 2018;15:203–14.
Toldo S, Marchetti C, Mauro AG, Chojnacki J, Mezzaroma E, Carbone S, et al. Inhibition of the NLRP3 inflammasome limits the inflammatory injury following myocardial ischemia-reperfusion in the mouse. Int J Cardiol. 2016;209:215–20.
Sun Q, Scott MJ. Caspase-1 as a multifunctional inflammatory mediator: noncytokine maturation roles. J Leukoc Biol. 2016;100:961–7.
Cavalli G, Pappalardo F, Mangieri A, Dinarello CA, Dagna L, Tresoldi M. Treating life-threatening myocarditis by blocking interleukin-1. Crit Care Med. 2016;44:e751–4.
De Jesus NM, Wang L, Lai J, Rigor RR, Stuart SDF, Bers DM, et al. Antiarrhythmic effects of interleukin 1 inhibition after myocardial infarction. Heart Rhythm. 2017;14:727–36.
Van Tassell BW, Raleigh JM, Abbate A. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr Heart Fail Rep. 2015;12:33–41.
Emmi G, Urban ML, Imazio M, Gattorno M, Maestroni S, Lopalco G, et al. Use of interleukin-1 blockers in pericardial and cardiovascular diseases. Curr Cardiol Rep. 2018;20:61
Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta. 2013;1833:945–53.
Wang S, Su X, Xu L, Chang C, Yao Y, Komal S, et al. Glycogen synthase kinase-3beta inhibition alleviates activation of the NLRP3 inflammasome in myocardial infarction. J Mol Cell Cardiol. 2020;149:82–94.
Yang XM, Downey JM, Cohen MV, Housley NA, Alvarez DF, Audia JP. The highly selective caspase-1 inhibitor VX-765 provides additive protection against myocardial infarction in rat hearts when combined with a platelet inhibitor. J Cardiovasc Pharmacol Ther. 2017;22:574–8.
Chang C, Wang SH, Xu LN, Su XL, Zeng YF, Wang P, et al. Glycogen synthase kinase 3 beta inhibitor SB216763 improves Kir2.1 expression after myocardia infraction in rats. J Interv Card Electrophysiol. 2021. https://doi.org/10.1007/s10840-021-00963-7.
Pellman J, Zhang J, Sheikh F. Myocyte-fibroblast communication in cardiac fibrosis and arrhythmias: mechanisms and model systems. J Mol Cell Cardiol. 2016;94:22–31.
Esen N, Shuffield D, Syed MM, Kielian T. Modulation of connexin expression and gap junction communication in astrocytes by the gram-positive bacterium S. aureus. Glia. 2007;55:104–17.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Savic S, Caseley EA, McDermott MF. Moving towards a systems-based classification of innate immune-mediated diseases. Nat Rev Rheumatol. 2020;16:222–37.
Abbate A, Toldo S, Marchetti C, Kron J, Van Tassell BW, Dinarello CA. Interleukin-1 and the inflammasome as therapeutic targets in cardiovascular sisease. Circ Res. 2020;126:1260–80.
Coll RC, Robertson AAB, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
He HB, Jiang H, Chen Y, Ye J, Wang AL, Wang C, et al. Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun. 2018,9:2550.
Jiang H, He HB, Chen Y, Huang W, Cheng JB, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214:3219–38.
Wannamaker W, Davies R, Namchuk M, Pollard J, Ford P, Ku G, et al. (S)-1-((S)-2-{[1-(4-amino-3-chloro-phenyl)-methanoyl]-amino}-3,3-dimethyl-butanoy l)-pyrrolidine-2-carboxylic acid ((2R,3S)-2-ethoxy-5-oxo-tetrahydro-furan-3-yl)-amide (VX-765), an orally available selective interleukin (IL)-converting enzyme/caspase-1 inhibitor, exhibits potent anti-inflammatory activities by inhibiting the release of IL-1beta and IL-18. J Pharmacol Exp Ther. 2007;321:509–16.
Li J, Hao JH, Yao D, Li R, Li XF, Yu ZY, et al. Caspase-1 inhibition prevents neuronal death by targeting the canonical inflammasome pathway of pyroptosis in a murine model of cerebral ischemia. CNS Neurosci Ther. 2020;26:925–39.
Kloner RA, Brown DA, Csete M, Dai WD, Downey JM, Gottlieb RA, et al. New and revisited approaches to preserving the reperfused myocardium. Nat Rev Cardiol. 2017;14:679–93.
Do Carmo H, Arjun S, Petrucci O, Yellon DM, Davidson SM. The caspase 1 inhibitor VX-765 protects the isolated rat heart via the RISK pathway. Cardiovasc Drug Ther. 2018;32:165–8.
Sato S, Suzuki J, Hirose M, Yamada M, Zenimaru Y, Nakaya T, et al. Cardiac overexpression of perilipin 2 induces atrial steatosis, connexin 43 remodeling, and atrial fibrillation in aged mice. Am J Physiol Endocrinol Metab. 2019;317:E1193–E204.
Gilbert S, Singh D, Jesuraj ML, Long QT. syndrome and torsades de pointes complicating mitral valve replacement. Indian Heart J. 2016;68:S210–S1.
Wildburger NC, Laezza F. Control of neuronal ion channel function by glycogen synthase kinase-3: new prospective for an old kinase. Front Mol Neurosci. 2012;5:80.
Ahnve S, Helmers C, Lundman T, Rehnqvist N, Sjogren A. QTc intervals in acute myocardial infarction: first-year prognostic implications. Clin Cardiol. 1980;3:303–8.
Severs NJ, Bruce AF, Dupont E, Rothery S. Remodelling of gap junctions and connexin expression in diseased myocardium. Cardiovasc Res. 2008;80:9–19.
Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481:278–86.
Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism. 2017;74:1–9.
Zhao Z, Wang Y, Zhou R, Li Y, Gao Y, Tu D, et al. A novel role of NLRP3-generated IL-1beta in the acute-chronic transition of peripheral lipopolysaccharide-elicited neuroinflammation: implications for sepsis-associated neurodegeneration. J Neuroinflammation. 2020;17:64.
Loukovaara S, Piippo N, Kinnunen K, Hytti M, Kaarniranta K, Kauppinen A. NLRP3 inflammasome activation is associated with proliferative diabetic retinopathy. Acta Ophthalmol. 2017;95:803–8.
Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer. 2018;17:158.
Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285:9792–802.
Sun Z, Nyanzu M, Yang S, Zhu X, Wang K, Ru J, et al. VX765 attenuates pyroptosis and HMGB1/TLR4/NF-kappaB pathways to improve functional outcomes in TBI mice. Oxid Med Cell Longev. 2020;2020:7879629.
Ando M, Katare RG, Kakinuma Y, Zhang DM, Yamasaki F, Muramoto K, et al. Efferent vagal nerve stimulation protects heart against ischemia-induced arrhythmias by preserving connexin43 protein. Circulation. 2005;112:164–70.
Peters NS, Green CR, Poole-Wilson PA, Severs NJ. Reduced content of connexin43 gap junctions in ventricular myocardium from hypertrophied and ischemic human hearts. Circulation. 1993;88:864–75.
Poelzing S, Rosenbaum DS. Altered connexin43 expression produces arrhythmia substrate in heart failure. Am J Physiol Heart Circ Physiol. 2004;287:H1762–70.
Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, et al. Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation. 2018;137:707–24.
Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;120:254–65.
Nagaraju CK, Dries E, Gilbert G, Abdesselem M, Wang N, Amoni M, et al. Myofibroblast modulation of cardiac myocyte structure and function. Sci Rep. 2019;9:8879.
Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119:91–112.
Zhang W, Xu X, Kao R, Mele T, Kvietys P, Martin CM, et al. Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: the role of NLRP3 inflammasome activation. PLoS One. 2014;9:e107639.
Li C, Wan WG, Ye TX, Sun YZ, Chen XL, Liu X, et al. Pinocembrin alleviates lipopolysaccharide-induced myocardial injury and cardiac dysfunction in rats by inhibiting p38/JNK MAPK pathway. Life Sci. 2021;277:119418.
Kieken F, Mutsaers N, Dolmatova E, Virgil K, Wit AL, Kellezi A, et al. Structural and molecular mechanisms of gap junction remodeling in epicardial border zone myocytes following myocardial infarction. Circ Res. 2009;104:1103–12.
Zhong CL, Chang H, Wu Y, Zhou L, Wang Y, Wang MY, et al. Up-regulated Cx43 phosphorylation at Ser368 prolongs QRS duration in myocarditis. J Cell Mol Med. 2018;22:3537–47.
Truskey GA. Endothelial cell vascular smooth muscle cell co-culture assay for high throughput screening assays for discovery of anti-angiogenesis agents and other therapeutic molecules. Int J High Throughput Screen. 2010;2010:171–81.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81670311) and the Henan Province Science and Technology Research Project (No. 182300410010).
Author information
Authors and Affiliations
Contributions
SNH, LRZ, and XLS conceived and designed the experiments. XLS, SHW, LGC, SK, and RCN performed the experiments. XLS and SHW analyzed the data. XLS, SK, and SNH contributed to the writing of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Su, Xl., Wang, Sh., Komal, S. et al. The caspase-1 inhibitor VX765 upregulates connexin 43 expression and improves cell–cell communication after myocardial infarction via suppressing the IL-1β/p38 MAPK pathway. Acta Pharmacol Sin 43, 2289–2301 (2022). https://doi.org/10.1038/s41401-021-00845-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00845-8
Keywords
This article is cited by
-
Post translational modifications of connexin 43 in ventricular arrhythmias after myocardial infarction
Molecular Biology Reports (2024)
-
IL-1 signaling pathway, an important target for inflammation surrounding in myocardial infarction
Inflammopharmacology (2024)
-
Histological Features of Uterine Myometrial Dysfunction: Possible Involvement of Localized Inflammation
Current Medical Science (2024)
