
Abstract
Many G-protein-coupled receptor (GPCR) agonists have been studied for transactivating epidermal growth factor receptor (EGFR) signaling through extracellular or intracellular pathways. Accumulated evidence has confirmed that GPCR transactivation participates in various diseases. However, the clinical application of GPCR transactivation has not been explored, and more translational studies are needed to develop therapies to target GPCR-mediated EGFR transactivation. In cancer patients treated with EGFR inhibitors (EGFRi), especially afatinib, a unique acneiform rash is frequently developed. In this study, we first established the connection between GPCR transactivation and EGFRi-induced skin disease. We examined the ability of three different GPCR agonists to reverse signaling inhibition and ameliorate rash induced by EGFRi. The activation of different agonists follows unique time and kinase patterns. Rats treated with EGFRi show a similar skin phenotype, with rash occurring in the clinic; correspondingly, treatment with GPCR agonists reduced keratinocyte apoptosis, growth retardation and infiltration of inflammatory cytokines by transactivation. This phenomenon demonstrates that EGFR inhibition in keratinocytes regulates key factors associated with rash. Our findings indicate that maintaining EGFR signaling by GPCR agonists might provide a possible therapy for EGFR inhibitor-induced skin toxicities. Our study provides the first example of the translational application of GPCR transactivation in treating diseases.
Similar content being viewed by others


Introduction
The capacity of G-protein-coupled receptor (GPCR) agonists to stimulate the signaling activity of epidermal growth factor receptor (EGFR) and its downstream effector kinase extracellular signal–regulated kinase (ERK) has been well described. EGFR transactivation has been verified in various cell types [1,2,3,4,5]. The major route of EGFR transactivation is through the MMP-mediated shedding of heparin binding-EGF (HB-EGF) from its membrane-spanning precursor, at which point cleaved EGF-like ligands are released to the extracellular space and stimulate EGFR [1, 5, 6]. Activation of EGFR by its ligands HB-EGF has been shown to regulate normal keratinocyte proliferation, differentiation, migration and survival [6]. In some situations, EGFR transactivation has been identified independent of HB-EGF [1, 7], suggesting an intracellular pathway through the activation of tyrosine kinases. The ligand-independent mechanisms focus on Src kinase, which has been proposed to directly phosphorylate and activate EGFR through the formation of a multimeric protein complex [8, 9].
Most of the research in the field of EGFR transactivation has reported the identification of transactivation progress by a GPCR, such as newly recognized GPCR ligands [10,11,12], additional molecular mechanisms underlying the transactivation [13,14,15], or demonstration of the role in various diseases [16,17,18,19,20]. However, there is a need to explore the clinical relevance and to develop therapies based on targeting GPCR-mediated EGFR transactivation. Since GPCR agonists have the potential to stimulate EGFR signaling, we speculate their therapeutic potential to rescue EGFR-retardant diseases.
Upregulation of EGFR is common in many solid tumors, causing cancer cells to proliferate in an uncontrolled manner, therefore inspiring a number of EGFR inhibitors to be developed for anticancer therapies. A specific adverse effect common to these agents is papulopustular rash. Little is known about the etiology of rash induced by EGFRi, and the current clinical management is not very effective. Broad-spectrum antibiotics and corticosteroids are most widely used as symptomatic treatments, which can only alleviate some of the symptoms and do not serve as a prophylactic treatment based on the cause of this disease. Among clinical trials aimed at treating rash, several agents showing clinical benefit were verified to reverse EGFR inhibition, such as cyclosporine [21] and vitamin K3 [22]. In addition, genetic ablation of epidermal EGFR in mice demonstrated abnormal keratinocytes, follicular differentiation and local inflammation, similar to the clinical symptoms [23,24,25,26]. Taken together, these data indicate a central pathogenic role of keratinocyte-leading impairment in skin homeostasis caused by EGFR inhibition; thus, restoring the phosphorylated expression of EGFR in the epidermis may be essential for maintaining skin homeostasis and may be prevention for rash. Therefore, we propose a hypothesis that transactivation of EGFR and its downregulation pathway might complement the damage to cell functions, promote further tissue regeneration and ultimately ameliorate EGFRi-induced rash.
We selected three GPCR agonists to test our hypothesis based on several principles: (1) the stimulation of well-described GPCR receptors that have been reported to transactivate EGFR or ERK, such as angiotensin receptor, adrenergic receptor and lysophospholipid receptor; (2) they have relatively small molecule weights to aid skin penetration when applied as topical ointment; and (3) they are in the advanced stage of clinical development for general safety profiles. One of the first GPCR agonists identified to crosstalk with EGFR is lysophosphatidic acid; [4] its receptor belongs to the lysophospholipid receptor family, which also contains the sphingosine-1-phosphate (S1P) receptor. Currently, many selective S1P receptor modulators are under development, and the representative drug ozanimod (OZ) has shown clinical remission in several autoimmune diseases [27, 28]. The angiotensin II (ANG) and ANG receptor axes interact primarily with Gq/11 proteins, leading to increases in Ca2 + [29], ROS [30] or EGFR ligand [1, 31] and involving c-Src [8], c-Abl [32] or MMP [1, 31], depending on the cell type. The effect of β-adrenergic receptor-mediated EGFR transactivation has also been shown in cardiomyocytes and noncardiac cells. Isoproterenol (ISO), an agonist of the β-adrenergic receptor, was shown to be independent of EGFR activation [7].
Here, we used keratinocytes and a rat model treated with EGFRi to unravel the mechanism by which ISO/ANG/OZ activates EGFR signaling and to examine the effect on the relief of EGFRi-induced skin toxicities. We found that the activations induced by ANG and OZ both require c-Src, whereas ANG is also sensitive to metalloproteinase (MMP). Interestingly, ISO activates in a more elusive way that shows no response to either MMP or Src inhibitor. Our study demonstrates that compensating EGFR signaling is essential and effective for maintaining the cellular functions of keratinocytes, and treatment with ISO/ANG/OZ reduced keratinocyte apoptosis, growth retardation and infiltration of inflammatory cytokines by transactivation. Consistent with the in vitro results, restored EGFR signal transduction by topical application of GPCR agonists ameliorated abnormal skin phenotypes caused by EGFRi.
Materials and methods
Agents
Afatinib, erlotinib and ozanimod were purchased from Shanghai Goyic Pharmaceutical & Chemical Co., Ltd. Isoprenaline hydrochloride was from Sigma. Angiotensin II was purchased from Hangzhou Taijia Biotech Co. Ltd. PEG400, PEG3350, lanolin were obtained from Xi’an Tianzheng Medicinal Materials Co. Ltd. Anti-phospho-EGFR (Y1068), anti-phospho-AKT (Ser473), anti-phospho-ERK1/2 (Thr202/Tyr204), anti-EGFR, anti-AKT, anti-ERK1/2 antibodies were from Cell Signaling Technology Inc. Anti-phospho-c-Src antibody was from Abcam. Anti-GAPDH antibody was from Proteintech. Secondary antibodies conjugated to horseradish peroxidase were from Beyotime Biotechnology. Pertussis toxin was from List Biologicals.
Cell culture
HaCaT, A431 and PC9 cells were grown in DMEM/high glucose, HUVEC cells was grown in EBM-2, all supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin in an incubator under a humidified atmosphere of 5% CO2 at 37 °C. HaCaT cells were pretreated with 100 ng/mL PTX in serum-free DMEM for 16 h prior to stimulations for immunoblotting.
Cell counting Kit-8 assay
Cells were cultured in 96-well plate and treated with different agents. Twenty-four hours after treatments, cells were counted using Cell Counting Kit-8 (Beyotime).
EdU proliferation assay
EdU staining was performed using a EdU Cell Proliferation Assay (Beyotime) according to the manafacturer’s protocol. Briefly, cells were stimulated with Afa or GPCR agonists and then incubated with 10 mM EdU for 2 h before fixation, permeabilization, and EdU staining. The nuclei were stained with DAPI Fluoromount-G (Yeasen). The proportion of nucleated cells incorporating EdU was determined by fluorescence microscopy.
Cell migration assay
HaCaT and HUVEC migration assay was performed using transwell chamber with 8 μm pore size (Falcon) for 24-well plates. A total of 500 μL of cultured medium containing 10% FBS was loaded in the lower chamber. Cell suspension in a volume of 20 μL in medium containing 1% FBS was added to the upper chamber. Then, test compounds were added to the upper chamber and incubated at 37 °C for 24 h or 48 h in a CO2 incubator. The membranes were fixed with methanol for 10 min at room temperature and then stained with crystal violet for another 10 min. The number of migratory cells was counted in five random fields of microscope.
Measurement of MMP activity
MMP activity was measured by incubation of cells with fluorescent MMP substrate following the manufacturer’s instructions (AATbio, USA). Briefly, HaCaT cells were grown in 96-well plates and stimulated with ISO/ANG/OZ with or without GM6001, then 50 μL of MMP GreenTM substrate working solution was added to each well of the assay plate. The fluorescence intensities were monitored with a fluorescence plate reader at Ex/Em = 490/525 nm. In separate experiments, the amount of HB-EGF shed after ISO/ANG/OZ stimulation was measured by a ELISA kit following the manufacturer’s instructions (Mlbio, China).
Immunoblotting
Cells lysates containing equivalent amounts of proteins were electrophoresed on SDS-PAGE (8%–12%) gradient gel and transferred to polyvinylidine difluoride membranes. The membranes were blocked by 5% nonfat dry milk in TBST followed by overnight incubation at 4 ℃ with primary antibodies and washed three times with TBST before probing with secondary antibody for 1 h at room temperature. Following three 10-min washes, membranes were then visualized with ECL Western blotting substrate (ThermoFisher). In some cases, blots were stripped and reprobed with other antibodies.
Generation of c-Src knockdown HaCaT cells
For lenti-virus production, HEK293FT cells were transfected with 10 µg lentiviral vectors along with delta 8.91, VSVG vectors. After 24 h, the viral supernatants were collected and filtered. Following infection of the cells with high-titration lentivirus, stable cell lines derived from HaCaT cells were cultured in selection medium containing 2 μg/mL puromycin. Successful knockdown of the target was verified by immunoblotting.
Colony formation assay
A431 and PC9 cells were seeded into 6-well plate with seeding density of 1000 cells/well. After culture for 7 days, the cells were treated with Afa or GPCR agonists for 2 days and then stained with crystal purple.
Animal study
Female 7–8 weeks old SD rats were obtained from SLAC, Shanghai. The animals were housed and maintained within the animal center at Shanghai Jiao Tong University, in accordance with the Guidelines for Animal Experimentation of Shanghai Jiao Tong University, China. The hair on the back was removed gentle with a shaver before the experiment. Rats were randomly divided in to different groups: (1) Control animals received no EGFRi and ointment; (2) Vehicle group received vehicle ointment; (3) GPCR agonists group recived 0.1% ISO, 0.1% ANG or 1% OZ. Each group (except control group) received 40 mg/kg afatinib per os daily for 7 days to develop rash model, after gavage, rats were applied with different ointment on their back and fixed in a cylinder, after 4 h, rats were freed and wiped residual ointment with a soft tissue paper. Body weight was recorded every day and photographs were taken under inhalational anesthesia with isoflurane.
Rash grade
Grade 0: normal.
Grade1: papules and/or pustules covering <10% surface area of shaved-back skin.
Grade2: papules and/or pustules covering 10%–30% surface area of shaved-back skin.
Grade3: papules and/or pustules covering a 30% surface area of shaved-back skin.
Scratching behavior
A scratching movement by the hind paw that was directed toward the rash-appearing site was defined as a scratching bout. The total number of scratching bouts was counted during 30 min.
Immunohistochemical staining
Back skin tissues of rash were fixed in 4% neutral buffered formalin and embedded in paraffin. Five-micrometer sections were deparaffinized and rehydrated, and then antigen unmasking was performed by heating the slides for 3 min in citrate antigen retrieval solution. Sections were treated with 3% hydrogen peroxide for 25 min and blocked with 3% BSA solution for 30 min. Primary anti-phospho-EGFR, anti-phospho-ERK and Ki67 (Servicebio) antibodies were stained at 4 °C overnight and were detected using a biotin-conjugated secondary antibody. Slides were then incubated for 15 min at room temperature with a DAB kit (DAKO). Nuclei was counterstained using a solution of hematoxylin. Finally the sections were mounted and image analysis was performed using an Aperio ScanScope (Leica Biosystems).
TUNEL staining
TUNEL staining was performed using an Apoptosis Kit (Beyotime) according to the manafacturer’s protocol. Briefly, pre-stimulated HaCaT cells were fixed, permeabilized, and stained with TUNEL working solution, sections prepared as described above were treated with proteinase K (Beyotime) and then stained with TUNEL working solution. Cell nucleus were stained with DAPI Fluoromount-G (Yeasen). The proportion of TUNEL-positive cells was determined by fluorescence microscopy.
Cytokine ELISA of rat samples
Rat IL-4, IL-10, IL-6, IL-1β and TNFα cytokine assays (Boster, China) were performed according to the manufacturer’s instructions with supernatants collected from total skin lysates.
Statistics analysis
Values are expressed as means ± SEM. Two-tailed Student’s t test was used to evaluate the differences between two groups. One-way ANOVA analysis was applied when there are more than two groups in the independent variable. Kaplan–Meier analysis was performed for rash incidence analysis, and significance was determined by a log rank test. P < 0.05 was considered to be statistically significant.
Results
Different GPCR agonists transactivate EGFR signaling pathway in diverse time patterns
Based on the literature, GPCR agonists induce ERK phosphorylation in a rapid, transient period within 3–10 min in different cell lines [1, 7]. To investigate whether GPCR agonists share the same response in keratinocytes, HaCaT cells were treated with ISO, ANG and OZ for 3 min. ISO and ANG treatment of HaCaT cells induces phosphorylation of ERK in a rapid manner over a broad range of doses (Supplementary Fig. 1a). As the induced phosphotyrosine signal was relatively weaker under basal conditions, EGFR and AKT activation could not be distinguished, and the maximal induction concentration was not determined. OZ-mediated signal transduction did not show any transactivation effect in this setting.
Some studies have suggested that ERK activation by isoproterenol occurs independently of EGFR phosphorylation in HEK293 cells [7], whereas the activation effect of other GPCR agonists was significantly suppressed by AG1478, a specific EGFR inhibitor [1, 4]. To dissect the requirement of EGFR in HaCaT cells, we treated HaCaT cells with the second-generation EGFR kinase inhibitor afatinib (Afa). Pretreatment of cells with Afa did not block ANG-induced activation of AKT and ERK or even EGFR (Fig. 1a). Interestingly, although ISO cannot activate obvious EGFR phosphorylation in the presence of Afa, it still activated AKT and ERK (Fig. 1a).

a HaCaT cells were pretreated with 100 nM Afa for 20 min and then stimulated with increasing concentrations of ISO/ANG/OZ for 3 min. The concentrations of ISO were 1, 10, and 100 µM, those of ANG were 100 nM, 1 and 10 µM, and those of OZ were 100 nM and 1 µM. Western blotting was conducted on cell lysates. b Time courses from 3 min to 12 h following 10 μM ISO, 1 μM ANG and 100 nM OZ stimulation in the presence of 100 nM Afa. c Relative protein levels compared with GAPDH are shown as line graphs. The results shown are obtained from three independent experiments and are presented as the mean ± SEM (n = 3).
Considering the long circulation time of Afa after oral administration and an inhibitory activity analysis of Afa in A431 cellular assays showing an ability to inhibit EGFR phosphorylation for >8 h [33], EGFR inhibition on keratinocytes should last for hours. Therefore, we wondered whether the transactivation response could be sustained in a long-term pattern. We extended the treatment time of Afa to 12 h and tested several time points from 3 min to 12 h. Surprisingly, OZ showed immense activation of all three proteins after 6 h (Fig. 1b, c). For ISO, activation of AKT and ERK was not synchronous, and the time course experiments revealed rapid and simultaneous tyrosine phosphorylation by ANG, though it was maintained for only ~1 h (Fig. 1b, c). The transactivating signals of ISO and ANG were inhibited by pertussis toxin (PTX), an inhibitor of α subunits of Gi/o protein, indicating that the ISO/ANG-mediated EGFR transactivation in HaCaT is dependent upon PTX-sensitive G proteins (Supplementary Fig. 1b). Whereas OZ-mediated activation utilized a PTX-insensitive pathway to transactivate AKT (Supplementary Fig. 1b). Taken together, ISO/ANG/OZ elicited the activation of EGFR and its downstream pathway during EGFR inhibition caused by EGFRi. The different time courses of ISO/ANG/OZ functions might point to different pathophysiological mechanisms, and we speculate that competitive binding and activation to the receptor might contribute to the EGFR-independent phenomenon.
Complicated intracellular and extracellular pathways are involved in ISO/ANG/OZ transactivation
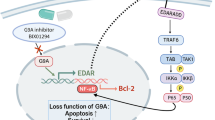
The canonical cascade of EGFR transactivation suggested a major role for the MMP in mediating extracellular HB-EGF-induced EGFR transactivation and Src family kinases in modulating direct intracellular activation (Fig. 2a). The released EGF-like ligands stimulate EGFR directly, while Src kinase mediates the phosphorylation of EGFR. To further elucidate the mechanism by which intracellular or extracellular effects were involved in ISO/ANG/OZ-induced EGFR transactivation, we employed the specific MMP antagonist GM6001 and the Src inhibitor PP1. In the present study, we could not interfere with the transactivation of ISO by either GM6001 or PP1 (Fig. 2b), suggesting its weak stimulation of EGFR and the ability of GPCRs to transactivate other receptor tyrosine kinases (RTKs) [5]. It can be hypothesized that this process occurs via a feedback loop that involves other RTKs, such as platelet-derived growth factor receptor (PDGFR) [34] or fibroblast growth factor receptor (FGFR) [35]. As expected, Afa inhibited the phosphorylation of c-Src; correspondingly, we detected a significant activation of c-Src following cellular stimulation with ANG and OZ (Fig. 2c, d). In the presence of PP1, inhibition of ANG/OZ-induced Src phosphorylation and EGFR pathway signaling was observed (Fig. 2c, d). Furthermore, ANG but not OZ also showed a response to GM6001 (Fig. 2c, Supplementary Fig. 1c). To examine whether transactivated effect depends on Src kinase activity, a lentriviral vector to knock down Src expression was created. HaCaT cells were transduced with scramble or shRNA-SRC by lentiviral infection (Supplementary Fig. 1d). Consistent with the previous results of pharmacological Src inhibitor PP1, knockdown of Src kinase blocked the activation by ANG and OZ, while did not interfere with the effect of ISO (Fig. 2e, Supplementary Fig. 1e). Together, these data suggest that different GPCR agonists transactivate EGFR signaling pathway via different work patterns and that, in addition to classical modes of action, alternative pathways may also participate in this process.

a Schematic diagram of the canonical cascade of EGFR transactivation. b, c, d Transactivated HaCaT cells were treated with GM6001 for 20 min or PP1 for 3 min. Concentrations in (b) for GM6001 and PP1 were 0.1, 1, and 10 µM. The final concentration for GM6001 and PP1 was 1 µM. e Effects of ANG or OZ on scramble or shSRC infected HaCaT cells. f Effects of ISO/ANG/OZ and GM6001 on MMP activity in HaCaT cells as measured by cleavage of fluorescent MMP substrate (n = 3). g HB-EGF shedding by stimulation with ISO/ANG/OZ and GM6001. HaCaT cells were stimulated for 12 h, 20 min and 6 h for ISO, ANG and OZ, respectively. Then, MMP activity was measured, and cell supernatants were collected for HB-EGF ELISA detection (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
To evaluate the roles of MMP and HB-EGF in ISO/ANG/OZ-induced transactivation more deeply, we measured the MMP activity and HB-EGF concentration in cell culture supernatants. ANG and OZ stimulated enzyme activity and their effects were reduced by GM6001 (Fig. 2f). Consistent with the Western blot analysis (Fig. 2b), ISO had no effect on MMP activity or HB-EGF release (Fig. 2f, g). In addition, ANG significantly released HB-EGF after Afa treatment, while OZ represented a different response with or without EGFR inhibition (Fig. 2g). These data indicate that among these three GPCR agonists, ANG might have a single MMP-HB-EGF activation axis with direct Src-mediated phosphorylation. Furthermore, although OZ enhanced MMP activity when added alone, EGFR inhibition probably disturbed the MMP activation induced by OZ when it was treated with Afa. Moreover, ISO is likely to act downstream of EGFR kinase or an alternative signaling pathway, in view of its relatively weak transactivation effect and separate stimulation of ERK and AKT (Fig. 1b) and the independent mechanisms of MMP and Src (Fig. 2f, g).
GPCR agonists restore cell functions
Activation of EGFR has been shown to regulate keratinocyte proliferation, differentiation, migration and survival. Previous study have reported that treatment of normal human keratinocytes with EGFRi induces apoptosis and growth arrest [36, 37], suggesting growth arrest and apoptosis might play a more crucial role in rash caused by EGFRi. Single GPCR agonist stimulation for proliferation was significant for ISO at different concentrations, while ANG and OZ did not stimulate proliferation (Fig. 3a). To investigate whether this enhanced proliferation will be blocked by EGFRi, we pretreated HaCaT cells with Afa and another EGFR inhibitor, erlotinib. The results showed that ISO significantly rescued cell viability in a dose-dependent manner (Fig. 3a, Supplementary Fig. 2a). By adding GM6001 or PP1, the enhanced proliferation was impaired, similar to the immunoblot signal (Fig. 3b). We next detected DNA synthesis of proliferating cells, based on the incorporation of 5-ethynyl-2′-deoxyuridine (EdU). Very few EdU-incorporated cells existed in the Afa group, all three GPCR agonists recovered DNA synthesis in different degrees (Fig. 3c, d), correspondingly, the effects of ANG and OZ were interfered by GM6001 and PP1 (Supplementary Fig. 3b, c). Treatment of HaCaT cells with Afa also induced apoptosis, which destroyed the skin structure and stimulated rash development, addition of ISO, ANG, and OZ decreased cell death (Fig. 3e, f). The remissions caused by ANG and OZ were blocked by PP1, unexpectedly, GM6001 increased apoptosis in all groups (Supplementary Fig. 3d, e). Taken together, these results indicate that inhibition of proliferation and induction of apoptosis caused by EGFR blockade could be rescued through GPCR agonist stimulation.

a, b Effects of Afa, ISO/ANG/OZ and GM6001/PP1 on HaCaT cell proliferation (n = 5). c Representative images of EdU staining. Scale bar: 130 μm. d Quantitative results of EdU-positive cells per filed (n = 5). e Representative images of TUNEL staining. f Quantitative results of TUNEL-positive cells per field (n = 5). Scale bar: 130 μm. g Representative images of the migration assay showing the stimulatory effects of ISO/ANG/OZ on HUVECs treated with 30 nM Afa for 24 h. Scale bar: 30 μm. h Quantitative results of migrating HUVECs. The results are shown as the mean ± SEM, n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 vs Con.
In addition to proliferation and apoptosis, stimulation by ANG/OZ dramatically enhanced the migration of HaCaT cells, and cells treated with ANG had roughly twice the capacity of migration compared to control cells (Supplementary Fig. 3a, b). However, ANG/OZ-stimulated HaCaT migration was attenuated by Afa treatment after incubation for both 24 and 48 h (Supplementary Fig. 3c, d). Some research has reported that stimulation by GPCR agonists could promote migration in smooth muscle cells and endothelial cells, which perform major migration functions in angiogenesis for tissue remodeling and healing [6]. To investigate whether transactivation could reverse EGFR inhibition on the migration of endothelial cells, we performed a migration assay on HUVECs. Stimulation by ISO/ANG enhanced the migration of HUVECs after Afa treatment (Fig. 3g, h). Thus, improved migration of HUVECs by EGFR transactivation may partly contribute to rash remission. In summary, these results show that EGFR transactivation by ISO/ANG/OZ can further improve keratinocyte and endothelial cell functions upon EGFR inhibition and might have the potential to maintain skin homeostasis in EGFR inhibitor-induced rash.
Considering the improved cell functions by EGFR transactivation might have an effect on tumor cells, we performed cell viability and colony assays to evaluate whether GPCR agonists interfere with the anti-tumor effect of Afa. EGFR-overexpressed skin squamous cell carcinoma cell line A431 and EGFR-mutated lung carcinoma cell line PC9 were chosen for investigation. The PC9 cell line was more sensitive to Afa, the IC50 for cell viability was 10 nM. Addition of OZ did not influence the effect of Afa in two cell lines, although ANG showed a slight proliferation tendency in A431 at a narrow dose range, the concentration we used having no interfering effect (Supplementary Fig. 4a, b). ISO seemed promoting cell viability in PC9 but without significant difference (Supplementary Fig. 4b), and the colony formation abilities decreased by Afa of both two cell lines were not disturbed by ISO (Supplementary Fig. 4c, d). In addition, neither ANG nor OZ affects the colony formation ability (Supplementary Fig. 4c, d). Together these data showed that GPCR agonists had no effect on the anti-tumor effect of Afa.
GPCR agonists are effective at reducing EGFRi-induced rash
Following the results seen in vitro, which showed that EGFRi suppressed proliferation, survival, and migration while GPCR agonists enhanced them, we used a rat model of rash to confirm the properties of GPCR agonists in vivo, as data from clinical trials showed that above 80% of Afa-treated patients developed rash [38, 39]. To develop a rat model to study rash, we repeated oral gavage of Afa once daily for seven days. Rats treated with Afa replicated similar skin features as clinical patients, including wavy and rough fur, hair loss, scaly skin, and swollen and encrusted muzzle (Fig. 4a). Then, we carried out a prophylactic experiment. In this setting, all three GPCR agonists decreased the rash occurrence and severity grade (Fig. 4a–c). ANG and OZ significantly delayed the occurrence of rash, with median time of 5 and 6 days compared with 3 days in the vehicle control, while ISO showed no efficiency in delaying occurrence (Fig. 4c). The incidence of grade ≥2 rash declined in all three groups, with barely any occurrence with treatment with ANG or OZ (Fig. 4c). In addition, histopathological observations revealed neutrophilic pustules, dilated blood vessel, and abnormal differentiated hair follicles occurred in vehicle group, while after ISO, ANG, or OZ ointment treatments, only small amounts of infiltration of inflammatory cells and normal differentiated hair follicles and basal keratinocytes were observed (Fig. 4d). As pruritus was also accompanied by rash, we observed the scratching behavior of rats. The mean scratching bonuses decreased in the ISO/ANG/OZ treatment groups compared to the vehicle group (Fig. 4e).

Afa (40 mg/kg) was orally administered daily to rats for 7 days to induce an experimental rash model. Vehicle, ISO, ANG, or OZ ointment was applied to the shaved backs of the rats for 4 h daily. a Representative images of back rash on day 7. b Rash grade assessment. c Kaplan–Meier analysis of time to first rash and grade ≥2 rash in rats. Log-rank P (ISO/ANG/OZ) = ns/ns/0.0446 (left) and 0.0124/0.0048/0.0193 (right) vs vehicle. d Representative micrographs and partially enlarged details of H&E-stained sections of skin tissues. Neutrophilic pustules (white asterisks) and dilated blood vessels (black asterisks) are highlighted. Scale bar: 300 and 200 μm (from top to bottom). e Pruritus analysis by counting scratching bonuses. Data are expressed as the mean ± SEM (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
In addition to the pathological improvement in skin rash, the number of TUNEL-positive cells decreased in the epidermis and hair follicle (Fig. 5a, b, Supplementary Fig. 5a) treated with ISO/ANG/OZ. In line with the enhanced proliferation effect in vitro (Fig. 3a), more Ki67 + cells appeared in the epidermal basal layer (Fig. 5c, d, Supplementary Fig. 5b) in GPCR agonists treament groups. As expected, the reduced expression levels of phospho-EGFR and phospho-ERK were reversed in skin treated with GPCR agonists compared to vehicle controls (Fig. 5e–g, Supplementary Fig. 5c, d), consistent with the transactivation ability verified by Western blot (Fig. 1a). Furthermore, the inflammatory cytokines IL-1β, IL-6 and TNFα, which were confirmed to be elevated in EGFRi-induced rash, were normalized or reduced in ISO/ANG/OZ-treated animals, with no changes in the anti-inflammatory cytokines IL-4 and IL-10 (Fig. 5h). Taken together, these results showed that GPCR agonists have the potential to ameliorate EGFRi-induced rash by transactivating the EGFR pathway and subsequently improving cell proliferation, migration, apoptosis and inflammation.

a Representative images of TUNEL-stained skin sections in each group. Scale bar: 130 μm. b Quantification of TUNEL-positive cells. Representative images of c Ki67-, e p-EGFR- and f p-ERK-immunostained skin sections. Scale bar: 100 μm. d Quantification of Ki67 intensity. g Quantification of pEGFR/pERK intensity. h Levels of IL-1β, IL-6, TNFα, IL-4 and IL-10 in skin homogenates. Scale bar, 100 μm. Data are expressed as the mean ± SEM (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Agents that target EGFR have shown promise for various malignancies, inhibiting pathways that are crucial for cancer cell survival and proliferation. Although treatment with EGFRi agents has fewer systemic toxicities than chemotherapy and radiotherapy, a specific dermatologic adverse effect rash occurs instead. This kind of dermatologic adverse event significantly impairs the process of treatment and quality of life, leading to dose interruption or even discontinuation.
Currently, no clinical management offers a targeted approach for EGFRi-induced rash due to the lack of understanding of the etiology. It is widely believed that the EGFR system plays a significant role in the epidermis by clinical histopathological analysis and genetic research in murine models. EGFR is primarily expressed in basal keratinocytes and hair follicles, and inhibition of EGFR signaling pathways induces growth arrest, decreasing migration, inflammation and apoptosis in keratinocytes [36, 37, 40]. Epidermal EGFR genetic mice recapitulate the skin phenotype of EGFRi-treated patients with keratin plugs, leukocyte infiltration, abnormal hair follicles and sebaceous glands [23, 41]. Cancer patients treated with EGFR inhibitors show early inflammatory infiltration dominated by dendritic cells, macrophages, granulocytes, mast cells, and T cells. EGFRi induced the expression of various cytokines and chemokines, such as IL-6, IL-1β, CCL2, and CCL5 in epidermal keratinocytes [23, 24]. As a result, EGFR downregulation in the epidermis was thought to be the major reason for EGFRi-induced rash, and EGFR signaling in keratinocytes has been thought of as regulating key factors involved in skin inflammation [24]. Therefore, finding a complementary way to upregulate EGFR signaling may be an effective intervention for rash.
GPCR agonists have been validated to activate tyrosine kinases in many cell lines. In addition, some of them are independent of EGFR. Recent clinical progress supports the role of GPCR-mediated EGFR transactivation in various diseases. For example, the prostanoid receptor regulates the induction of cyclooxygenase-2 and its downstream metabolite prostaglandin E2, and this induction appears to be an early event in colorectal cancer [18]. In additionally, lysophosphatidic acid is involved in stimulating migration in oral carcinoma cells through EGFR transactivation [20]. Moreover, some studies investigating EGFR transactivation have proven to be both harmful and beneficial in cardiovascular diseases. Therapeutic blockade of EGFR is protective in ANG-induced cardiac hypertrophy [42], whereas β-arrestin-mediated cardiac EGFR activation offers a cardioprotective effect [43]. As EGFR activation can be both protective and detrimental, it could be proposed that, at least in some pathological systems, such as ablation or blockade of EGFR, transactivation may be an option for treatment. Moreover, the clinical application of GPCR-mediated transactivation was not explored, and further translational studies are needed to develop therapies for GPCR-mediated EGFR transactivation.
EGFR expression is lost in EGFRi-induced cutaneous toxicities, and the importance of EGFR signaling in epidermal function and inflammation modulation is highlighted in genetic mice that have decreased EGFR activity [23, 24, 26]. Thus, we hypothesized that using GPCR agonists might ameliorate EGFRi-induced rash. In this study, we first investigated this hypothesis in vitro. We demonstrated that Afa decreased the phosphorylation of EGFR and ERK and that GPCR agonists were able to attenuate this decrease in a dose- and time-dependent manner. However, this cross-communication between GPCRs and EGFR is not a universal process. Although the reasons for these differential stimulation patterns are obscure, the present findings indicate that both induction of MMP-HB-EGF and activation of c-Src are necessary for ANG, while for OZ, c-Src plays a more dominant role. In contrast to classical GPCR transactivation signals that are transmitted via EGFR kinase, our data suggest that ISO-mediated activation of the AKT and ERK cascades is independent of EGFR. A further point that requires investigation is the possible activation of other latent RTKs, PDGFR [34] and FGFR [35]. In addition, more work is required to study the effect of ISO on EGFR downstream proteins and the interplay of EGFR transactivation and parallel signaling tracks. In addition to its effect on signal transduction, ISO, ANG and OZ also promote cell migration and proliferation, which are thought to be useful for healing and tissue regeneration [6]. Regarding migration, although transactivation has a stimulatory effect on HaCaT cells, it cannot attenuate the inhibitory effect caused by Afa but is available to circumvent EGFR inhibition in HUVECs. This phenomenon indicates that the beneficial effects of EGFR transactivation can contribute to other types of cells and cell-cell interactions.
Given this evidence, we then determined whether GPCR agonists were effective for Afa-induced rash in the rat model. Consistent with the in vitro observations, GPCR agonists showed a significant preventive effect on rash progression. Moreover, considering the synchronous time pattern of ANG/OZ with their more effective treatment in vivo, we speculated that transactivating AKT and ERK simultaneously may be a more efficient treatment system for rash. Further studies need to be tailored to explore the relationship between the transactivation time pattern and treatment efficacy. In addition, among these three GPCR agonists, OZ is the most powerful agent in activating EGFR and its downstream signaling pathway and also shows the best remission of EGFRi-induced skin toxicities. This evidence confirms the therapeutic concept based on compensating EGFR signaling.
One major limitation of application of GPCR agonists is that besides EGFR transactivation effect in common, these agents have other special pharmacologic actions. This additional pharmacological role raises the concern of whether transactivation indeed dominates in EGFRi-induced skin toxicity. The S1P is a sphingolipid whose major physiological functions focuses on controlling lymphocyte trafficking and vascular tone. Agonists of S1P receptors induce internalization and degradation of the recptors, rendering lymphocytes from secondary lymphoid organs [44]. The modulators of S1P have been widely used in immune-related diseases, such as ulcerative colitis, relapsing multiple sclerosis, and atherosclerosis [27, 28]. They also play an active role in vascular maturation, angiogenesis and endothelial cell migration [45]. It therefore seems that the additional pharmacological actions of OZ provide positive additive effects on skin inflammation. As for ANG, histopathological examination showed obvious vasodilation in either patients or rodents receiving EGFRi, it is reasonable that a vasoconstrictor might have some potential therapeutic effects. The adrenergic receptors are critical regulators of cardiac functions, topical application excludes the physiological outcome from cardiac tissues. Hence, we propose OZ as the most ideal therapeutic agent for prevention of skin toxicity caused by EGFR inhibiton, in view of its immense activation of EGFR signaling and anti-inflammatory potential.
In conclusion, our work discovered the advantages of EGFR transactivation in a specific disease without effective management and transferred this potential for pharmacological targeting. We confirmed that reversing EGFR signaling in keratinocytes by GPCR-mediated EGFR transactivation is a feasible prevention method for EGFRi-induced rash, thus providing further preclinical evidence for the potential prophylactic use of GPCR agonists in the treatment of EGFRi-induced skin toxicities. Our findings may also provide insights into extracutaneous adverse effects caused by EGFR inhibitors, such as gastrointestinal (diarrhea, vomiting, nausea) and oral-nasal responses (oral mucositis, cheilitis, epistaxis). These side effects also account for a large proportion of EGFRi-associated complications. Moreover, a syndrome characterized by neonatal inflammatory skin and bowel lesions has been linked to ADAM17 deletion, an EGFR ligand sheddase [46]. Thus, in addition to suggesting potential adjuvant drugs for improving the quality of life of EGFRi patients, our study may also provide targets for other EGFR-inhibited diseases.
References
Shah BH, Yesilkaya A, Olivares-Reyes JA, Chen H-D, Hunyady L, Catt KJ. Differential pathways of angiotensin II-induced extracellularly regulated kinase 1/2 phosphorylation in specific cell types: role of heparin-binding epidermal growth factor. Mol Endocrinol. 2004;18:2035–48.
Forrester SJ, Kawai T, Elliott KJ, Brien SO, Thomas W, Harris RC, et al. EGFR transactivation: mechanisms, pathophysiology and potential therapies in cardiovascular system. Annu Rev Pharmacol Toxicol. 2017;2:627–53.
Kranenburg O, Moolenaar WH. Ras-MAP kinase signaling by lysophosphatidic acid and other G protein-coupled receptor agonists. Oncogene. 2001;20:1540–6.
Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–60.
Wetzker R, Böhmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol. 2003;4:651–7.
Yang X, Zhu MJ, Sreejayan N, Ren J, Du M. Angiotensin II promotes smooth muscle cell proliferation and migration through release of heparin-binding epidermal growth factor and activation of EGF-receptor pathway. Mol Cell. 2005;20:263–70.
Schmitt JM, Stork PJS. β2-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein Rap1 and the serine/threonine kinase B-Raf. J Biol Chem. 2000;275:25342–50.
Bokemeyer D, Schmitz U, Kramer HJ. Angiotensin II-induced growth of vascular smooth muscle cells requires an Src-dependent activation of the epidermal growth factor receptor. Kidney Int. 2000;58:549–58.
Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, et al. The β2-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem. 2000;275:9572–80.
Cheng CY, Tseng HC, Yang CM. Bradykinin-mediated cell proliferation depends on transactivation of EGF receptor in corneal fibroblasts. J Cell Physiol. 2012;227:1367–81.
Feng PH, Hsiung TC, Kuo HP, Huang CD. Cross-talk between bradykinin and epidermal growth factor in regulating IL-6 production in human airway smooth muscle cells. Chang Gung Med J. 2010;33:92–9.
Cattaneo F, Iaccio A, Guerra G, Montagnani S, Ammendola R. NADPH-oxidase-dependent reactive oxygen species mediate EGFR transactivation by FPRL1 in WKYMVm-stimulated human lung cancer cells. Free Radic Biol Med. 2011;51:1126–36.
Akhtar S, Yousif MHM, Dhaunsi GS, Chandrasekhar B, Al-Farsi O, Benter IF. Angiotensin-(1-7) inhibits epidermal growth factor receptor transactivation via a Mas receptor-dependent pathway. Br J Pharmacol. 2012;165:1390–400.
Zhong S, Yin H, Liao Y, Yao F, Li Q, Zhang J, et al. Lung tumor suppressor GPRC5A binds EGFR and restrains its effector signaling. Cancer Res. 2015;75:1801–14.
Ratchford AM, Baker OJ, Camden JM, Rikka S, Petris MJ, Seye CI, et al. P2Y2 nucleotide receptors mediate metalloprotease-dependent phosphorylation of epidermal growth factor receptor and ErbB3 in human salivary gland cells. J Biol Chem. 2010;285:7545–55.
Lin X, Zhong S, Ye X, Liao Y, Yao F, Yang X, et al. EGFR phosphorylates and inhibits lung tumor suppressor GPRC5A in lung cancer. Mol Cancer. 2014;13:233.
Moody TW, Ramos-Alvarez I, Moreno P, Mantey SA, Ridnour L, Wink D, et al. Endothelin causes transactivation of the EGFR and HER2 in non-small cell lung cancer cells. Peptides.2017;90:90–9.
Yoshida K, Fujino H, Otake S, Seira N, Regan JW, Murayama T. Induction of cyclooxygenase-2 expression by prostaglandin E2 stimulation of the prostanoid EP4 receptor via coupling to Gαi and transactivation of the epidermal growth factor receptor in HCA-7 human colon cancer cells. Eur J Pharmacol. 2013;718:408–17.
Moody TW, Mantey SA, Moreno P, Nakamura T, Lacivita E, Leopoldo M, et al. ML-18 is a non-peptide bombesin receptor subtype-3 antagonist which inhibits lung cancer growth. Peptides.2015;64:55–61.
Brusevold IJ, Tveteraas IH, Aasrum M, Ødegård J, Sandnes DL, Christoffersen T. Role of LPAR3, PKC and EGFR in LPA-induced cell migration in oral squamous carcinoma cells. BMC Cancer. 2014;14:432.
Lopez-Ilasaca M, Schiene C, Küllertz G, Tradler T, Fischer G, Wetzker R. Effects of FK506-binding protein 12 and FK506 on autophosphorylation of epidermal growth factor receptor. J Biol Chem. 1998;273:9430–4.
Perez-Soler R, Zou Y, Li T, Tornos C, Ling Y. Topical vitamin K3 (Vit K3, Menadione) prevents erlotinib and cetuximab-induced EGFR inhibition in the skin. J Clin Oncol. 2006;24:18S.
Mascia F, Lam G, Keith C, Garber C, Steinberg SM, Kohn E, et al. Genetic ablation of epidermal EGFR reveals the dynamic origin of adverse effects of anti-EGFR therapy. Sci Transl Med. 2013;5:199ra110.
Lichtenberger BM, Gerber PA, Holcmann M, Buhren BA, Amberg N, Smolle V, et al. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci Transl Med. 2013;5:199ra111.
Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, et al. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–41.
Klufa J, Bauer T, Hanson B, Herbold C, Starkl P, Lichtenberger B, et al. Hair eruption initiates and commensal skin microbiota aggravate adverse events of anti-EGFR therapy. Sci Transl Med. 2019;11:1–18.
Sandborn WJ, Feagan BG, Wolf DC, D’Haens G, Vermeire S, Hanauer SB, et al. Ozanimod induction and maintenance treatment for ulcerative colitis. N Engl J Med. 2016;374:1754–62.
Comi G, Kappos L, Selmaj KW, Bar-Or A, Arnold DL, Steinman L, et al. Safety and efficacy of ozanimod versus interferon beta-1a in relapsing multiple sclerosis (SUNBEAM): a multicentre, randomised, minimum 12-month, phase 3 trial. Lancet Neurol. 2019;18:1009–20.
Che Q, Carmines PK. Angiotensin II triggers EGFR tyrosine kinase-dependent Ca2+ influx in afferent arterioles. Hypertension. 2002;40:700–6.
Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res. 2002;91:406–13.
Eguchi S, Dempsey PJ, Frank GD, Motley ED, Inagami T. Activation of MAPKs by angiotensin II in vascular smooth muscle cells. Metalloprotease-dependent EGF receptor activation is required for activation of ERK and p38 MAPK but not for JNK. J Biol Chem. 2001;276:7957–62.
Ushio-Fukai M, Zuo L, Ikeda S, Tojo T, Patrushev NA, Alexander RW. cAbl tyrosine kinase mediates reactive oxygen species- and caveolin-dependent AT1 receptor signaling in vascular smooth muscle: role in vascular hypertrophy. Circ Res. 2005;97:829–36.
Center for drug evaluation and research. Pharmacol Rev (201292Orig1s000). 2013.
Herrlich A, Daub H, Knebel A, Herrlich P, Ullrich A, Schultz G, et al. Ligand-independent activation of platelet-derived growth factor receptor is a necessary intermediate in lysophosphatidic, acid-stimulated mitogenic activity in L cells. Proc Natl Acad Sci USA. 1998;95:8985–90.
Peng H, Myers J, Fang X, Stachowiak Ewa K, Maher PA, Martins GG, et al. Integrative nuclear FGFR1 signaling (INFS) pathway mediates activation of the tyrosine hydroxylase gene by angiotensin II, depolarization and protein kinase C. J Neurochem. 2002;81:506–24.
Rodeck U, Jost M, Kari C, Shih DT, Lavker RM, Ewert DL, et al. EGF-R dependent regulation of keratinocyte survival. J Cell Sci. 1997;110:113–21.
Peus D, Hamacher L, Pittelkow MR. EGF-receptor tyrosine kinase inhibition induces keratinocyte growth arrest and terminal differentiation. J Invest Dermatol. 1997;109:751–6.
Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–22.
Sequist LV, Yang JCH, Yamamoto N, O’Byrne K, Hirsh V, Mok T, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34.
Mascia F, Mariani V, Girolomoni G, Pastore S. Blockade of the EGF receptor induces a deranged chemokine expression in keratinocytes leading to enhanced skin inflammation. Am J Pathol. 2003;163:303–12.
Hansen LA, Alexander N, Hogan ME, Sundberg JP, Dlugosz A, Threadgill DW, et al. Genetically null mice reveal a central role for epidermal growth factor receptor in the differentiation of the hair follicle and normal hair development. Am J Pathol. 1997;150:1959–75.
McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. 2012;18:1429–33.
Tilley DG, Rockman H. G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res. 2011;109:217–30.
Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–63.
Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, et al. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell. 1999;99:301–12.
Blaydon DC, Biancheri P, Di WL, Plagnol V, Cabral RM, Brooke MA, et al. Inflammatory skin and bowel disease linked to ADAM17 deletion. N Engl J Med. 2011;365:1502–8.
Acknowledgements
This work was, in part, supported by the Youth Thousand Talents Program of China, start-up grants from the Shanghai Jiao Tong University (WF220408211). This work was also supported by the grants from the State Key Laboratory of Onco-genes and Related Genes (90-17-02) and from the Interdisciplinary Program of Shanghai Jiao Tong University (YG2017MS18).
Author information
Authors and Affiliations
Contributions
LYC designed and performed most of the experiments. QY and SHL performed the shRNA silence experiment and colony formation assay. DZL performed part of the animal experiments. LYC analyzed data, LYC and SYZ wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Chen, Ly., You, Q., Lv, Dz. et al. GPCR-mediated EGFR transactivation ameliorates skin toxicities induced by afatinib. Acta Pharmacol Sin 43, 1534–1543 (2022). https://doi.org/10.1038/s41401-021-00774-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00774-6
