
Abstract
Macrophage-mediated inflammation plays an important role in hypertensive cardiac remodeling, whereas effective pharmacological treatments targeting cardiac inflammation remain unclear. Lipoprotein-associated phospholipase A2 (Lp-PLA2) contributes to vascular inflammation-related diseases by mediating macrophage migration and activation. Darapladib, the most advanced Lp-PLA2 inhibitor, has been evaluated in phase III trials in atherosclerosis patients. However, the role of darapladib in inhibiting hypertensive cardiac fibrosis remains unknown. Using a murine angiotensin II (Ang II) infusion-induced hypertension model, we found that Pla2g7 (the gene of Lp-PLA2) was the only upregulated PLA2 gene detected in hypertensive cardiac tissue, and it was primarily localized in heart-infiltrating macrophages. As expected, darapladib significantly prevented Ang II-induced cardiac fibrosis, ventricular hypertrophy, and cardiac dysfunction, with potent abatement of macrophage infiltration and inflammatory response. RNA sequencing revealed that darapladib strongly downregulated the expression of genes and signaling pathways related to inflammation, extracellular matrix, and proliferation. Moreover, darapladib substantially reduced the Ang II infusion-induced expression of nucleotide-binding oligomerization domain-like receptor with pyrin domain 3 (NLRP3) and interleukin (IL)-1β and markedly attenuated caspase-1 activation in cardiac tissues. Furthermore, darapladib ameliorated Ang II-stimulated macrophage migration and IL-1β secretion in macrophages by blocking NLRP3 inflammasome activation. Darapladib also effectively blocked macrophage-mediated transformation of fibroblasts into myofibroblasts by inhibiting the activation of the NLRP3 inflammasome in macrophages. Overall, our study identifies a novel anti-inflammatory and anti-cardiac fibrosis role of darapladib in Lp-PLA2 inhibition, elucidating the protective effects of suppressing NLRP3 inflammasome activation. Lp-PLA2 inhibition by darapladib represents a novel therapeutic strategy for hypertensive cardiac damage treatment.
Similar content being viewed by others

Introduction
Hypertension is a significant public health challenge. Persistent hypertension leads to adverse cardiac remodeling, which is characterized by left ventricular hypertrophy and cardiac fibrosis and is a determinant of the clinical course of heart failure (HF) as well as one of the leading causes of death worldwide [1]. Despite its prevalence, efficacious therapies for inhibiting or reversing cardiac fibrosis are still lacking [2]. Therefore, identifying novel therapeutic strategies for pathological cardiac remodeling and dysfunction is of great importance.
Inflammation plays an important role in the pathogenesis of hypertensive cardiac remodeling [3]. Angiotensin (Ang) II, a key component of the renin–angiotensin system (RAS), mediates early infiltration of pro-inflammatory cells, especially monocytes/macrophages, in hypertensive cardiac remodeling [4]. Monocytes/macrophages release pro-fibrotic cytokines to stimulate the trans-differentiation of resident cardiac fibroblasts into myofibroblasts, leading to excess production of extracellular matrix (ECM) [5]. Previous studies have demonstrated that the balance between macrophage infiltration, polarization, and tissue clearance determines inflammation resolution and cardiac remodeling [6, 7]. Thus, targeting macrophages-associated inflammatory response may alleviate the progression of cardiac remodeling and improve cardiac function.
The inflammasome is an important mediator of innate immunity. The nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome is the best-characterized inflammasome, expressed mainly in monocytes and macrophages [8]. Once activated, NLRP3 initiates the formation of inflammasomes by interacting with the apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain (ASC), which recruits and activates pro-caspase-1 to generate the active caspase-1, which in turn activates the pro-inflammatory and pro-fibrotic cytokines, IL-1β and IL-18 [9]. Notably, NLRP3 inflammasomes play a key role in the progression of hypertension and are strongly linked to the development of hypertensive end-organ damage [10]. Thus, finding therapeutic interventions targeting NLRP3 inflammasome activation in macrophages may prove beneficial in preventing hypertensive cardiac remodeling.
Phospholipases A2 (PLA2s) are a superfamily of enzymes characterized by their ability to hydrolyze the ester bond at the sn-2 position of membrane glycerophospholipids and metabolize them into various inflammatory mediators [11]. Alterations in PLA2 expression or activity result in the progression of numerous inflammatory diseases, including arthritis, cardiovascular diseases, and autoimmune diseases [12, 13]. Thus, PLA2s are increasingly receiving attention as potential drug targets to treat inflammatory diseases.
Lp-PLA2 belongs to group VII of the PLA2 superfamily and is principally secreted by macrophages [14]. Lp-PLA2 hydrolyzes glycerophospholipids to generate bioactive lipids, many of which have pro-inflammatory and pro-oxidative activities. Lysophosphatidylcholine (LysoPC) is responsible for the majority of Lp‐PLA2‐derived proinflammatory effects. Elevated plasma Lp‐PLA2 levels are associated with a number of vascular diseases [15, 16]. Elevated Lp-PLA2 levels were also observed in hypertension patients [17]. However, the role of Lp-PLA2 in hypertensive cardiac inflammation and cardiac remodeling remains unknown.
Darapladib is a potent oral inhibitor of Lp-PLA2, which has been evaluated in phase III trials in atherosclerosis patients, demonstrating that darapladib is safe for use [18,19,20]. Several recent clinical and preclinical studies showed the promising therapeutic effects of Lp‐PLA2 inhibition in diabetic macular edema and Alzheimer’s diseases, indicating the potential pharmacological significance of targeting Lp-PLA2 [21, 22]. However, the effect of darapladib on hypertensive cardiac remodeling is still uncharacterized.
In the present study, we investigated the roles of the Lp-PLA2 inhibitor, darapladib, in hypertension-induced cardiac inflammation and fibrosis in a mouse model of hypertension induced by Ang II infusion, and we explored the underlying molecular mechanisms of darapladib’s protective effect. Our findings suggest that darapladib treatment may be a potential therapeutic approach to limit the progression of hypertension-induced cardiac remodeling and dysfunction through the downregulation of macrophage NLRP3 inflammasome activation. Thus, Lp-PLA2 inhibition may represent a novel therapeutic strategy for preventing cardiac fibrosis and the development of HF.
Materials and methods
Antibodies and reagents
Primary antibodies for immunohistochemistry (IH), immunofluorescence (IF), Western blotting (WB), and imaging flow cytometry analysis included rabbit anti-Lp-PLA2 (A9796) and rabbit anti-ASC (A11433) from ABclonal (Wuhan, China), mouse anti-Collagen I (bsm-33401m) and mouse anti-Collagen III (bsm-33129m) from Bioss (Beijing, China), rabbit anti-Mac-2 (ab76245; IH and IF), rabbit anti-IL-1β (ab9722; IF), rabbit anti-α-smooth muscle actin (α-SMA) (ab5694), and rabbit anti-transforming growth factor-β (TGF-β) (ab92486) from Abcam (Cambridge, MA, USA), mouse anti-Mac-2 (sc-32790; IF and WB), goat anti-NLRP3 (ab4207; IF), and mouse anti-caspase-1 (sc-56036) from Santa Cruz Biotechnology (CA, USA), rabbit anti-GAPDH (5174s), mouse anti-IL-1β (12242s; WB), and rabbit anti-NLRP3 (15101s; WB) from Cell Signaling Technology (Danvers, MA, USA), and rat anti-CD45 PE/Dazzle-594 (103146; Biolegend, San Diego, CA, USA) and rat anti-CD11b PE (557397; BD Bioscience, Franklin Lakes, NJ, USA).
Human angiotensin II (Ang II) and lipopolysaccharides (LPS) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Lp-PLA2 inhibitor darapladib was obtained from Biorbyt Ltd. (Cambridge, United Kingdom). NLRP3 inhibitor MCC950 was obtained from MedChem Express (Monmouth Junction, NJ, USA).
Animal maintenance and experimental treatments
We used the established hypertensive mouse model, as previously reported [6, 23]. Twelve-week-old male C57BL/6J mice (weight, 24–26 g) were provided by Beijing Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China). Mice were maintained under constant temperature and humidity with a 12-h light-dark cycle in a temperature-controlled environment, and they had free access to standard mouse chow and water [24]. Darapladib Lp-PLA2 inhibitor was dissolved in DMSO at a concentration of 100 mg/mL and then diluted in 0.5% CMC-Na to a final concentration of 5 mg/mL for oral administration. DMSO with similar dilutions as the darapladib solution was used as solvent control. Mice received a final volume of 1% of their body weight. Darapladib was administered by gavage (50 mg·kg−1·d−1) once per day for 3 days prior to Ang II infusion [20, 25]. Then, mice were infused for 7 days with saline or Ang II (1500 ng·kg−1·min−1) by osmotic mini-pumps (Alzet MODEL 1007D; DURECT, Cupertino, CA, USA) which were implanted subcutaneously [24]. During this process, mice were continuously given darapladib or solvent control once daily [20, 25]. Following experimental treatment, heart function and blood pressure were measured on day 7. Finally, mice were anesthetized with isoflurane (2%) and euthanized. The left ventricle was punctured and flushed with 20 mL of saline to remove blood from systemic circulation. Hearts were removed and prepared for further histological and molecular analyses. All of the animal protocols complied with the Animal Management Rule of the Ministry of Health, People’s Republic of China (Documentation no. 55, 2001) and the guidelines from EU Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. The protocols were also approved by the Institutional Animal Care and Use Committee of the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College.
Real-time polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from cardiac tissue or cells using an RNAeasyTM animal RNA isolation kit with spin column (Beyotime, Shanghai, China) in accordance with the manufacturer’s instructions. First-strand cDNA was synthesized using a PrimeScript RT reagent kit with gDNA Eraser (Takara Bio Inc., Shiga, Japan). Quantitative PCR was performed using TB Green Premix (Takara), and specific primers (Supplementary Table S1) using an Applied Biosystems 7900 (ABI, USA).
RNA sequencing analysis
Total RNA was extracted from mouse ventricular chambers. RNA quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA). Library construction and RNA sequencing were performed via the BGISEQ-500 platform at Beijing Genomics Institute (BGI, Shenzhen, China). The raw sequencing reads were filtered by removing low-quality reads, adapter-polluted reads, and reads with more than 10% of unknown bases. After filtering, clean reads were mapped to the mouse genome (GRCm38.p5) by HISAT2.
Gene expression was calculated by fragments per kilobase of transcript per million mapped reads (FPKM). Genes with the absolute value of Log2 fold change ≥ 0 and false discovery rate (FDR) ≤ 0.05 were considered to be significantly differentially expressed, and these genes are shown in Fig. 5. Heatmap analysis was conducted using the R package pheatmap. Molecular interaction network analysis was performed by protein–protein interaction (PPI) network analysis using STRING (https://www.string-db.org/) and ingenuity pathway analysis (IPA hereafter, Qiagen Redwood City Inc., CA, USA). Gene function and pathway enrichment were analyzed via the web-accessible program Database for Annotation, Visualization, and Integrated Discovery (DAVID) (https://david.abcc.ncifcrf.gov/), Cytoscape plug-in ClueGO, and CluePedia. The significance levels of terms and pathways were identified by FDR ≤ 0.05.
Histology, IH, and IF staining
Cardiac tissue samples were fixed in 4% paraformaldehyde, dehydrated, embedded in paraffin, and cut into 5-μm slides. Masson trichrome staining and hematoxylin & eosin (HE) staining were performed using standard procedures. The interstitial fibrotic areas were calculated as the ratio of the total area of interstitial fibrosis to the total area of the section using ImageJ.
For the IH staining, the heart sections were dewaxed, underwent antigen retrieval by high pressure in repairing citrate buffer, treated with 3% H2O2, were blocked with non-specific antigen with 10% goat serum or 10% bovine serum albumin, were incubated with primary antibodies for Lp-PLA2, α-SMA, TGF-β, IL-1β, and Mac-2 at 4 °C overnight, and finally were detected using the HRP-DAB detection method. For negative controls, the primary antibody was replaced with corresponding non-immune IgG (in all cases, negative controls exhibited non-significant staining).
For IF staining, cardiac sections were incubated with primary antibodies for Mac-2, Lp-PLA2, caspase-1, NLRP3, ASC, and α-SMA, followed by appropriate second antibodies, then counterstained with 4',6-diamidino-2-phenylindole (DAPI) and mounted with glycerol before imaging via confocal laser scanning microscopy (Leica, Wetzlar, Germany). For negative controls, the primary antibody was replaced with corresponding IgG. FITC-conjugated wheat germ agglutinin (WGA, Invitrogen) was used to evaluate the cardiomyocyte cross-sectional area. Cell area determinations were based on measurements of at least 200 cells per slide.
Imaging flow cytometry (Amnis)
Cardiac cell suspensions were prepared, as previously described [26]. The mouse hearts were perfused, removed, and washed in cold perfusion buffer. After removing the atria, the left ventricle chamber was minced into fine pieces and digested in 4 mL of 0.2% collagenase II and 1 U/mL dispase I (Sigma) by shaking (160 rpm) at 37 °C for 30 min. The obtained cell suspension was gently pipetted up and down and filtered through a 70-µm cell strainer. Cells were collected and re-suspended in DPBS. The cell suspensions were blocked with CD16/32 antibody (BD Biosciences) for 1 h at room temperature, then stained with rat anti-CD45 PE/Dazzle and rat anti-CD11b PE, as well as their homologous isotype-matched negative controls (BD Biosciences), at 4 °C for 30 min in the dark. Finally, the cells were washed, re-suspended, measured by ImageStream MarkII imaging flow cytometry, and analyzed by IDEAS statistical image analysis software (Amnis, EMD-Millipore, Seattle, WA, USA). Putative single cells were selected by gating only the area consistent with single cells based on a scatter plot of the bright field area versus the aspect ratio and cell imaging of each dot. The images and the percentage of CD45+CD11b+ cells were determined based on the frequency of the counts over isotype controls.
Magnetic resonance imaging
Magnetic resonance imaging (MRI) was performed after 7 days of Ang II infusion. Mice were anesthetized in a chamber (2–4% isoflurane mixed with 0.2-L/min 100% O2) and maintained with a face mask (1–2% isoflurane with 0.2-L/min 100% O2). We obtained cine images of the left ventricle short axis with a 7.0 T scanner (Bruker BioSpec, Bruker Medical, Ettlingen, Germany) using the following parameters: repetition time 8.0 ms, echo time 2.4 ms, field-of-view 25 mm × 25 mm, image size 192 mm × 192 mm, 0.8-mm thickness with no gap, bandwidth: 89285.7 Hz, 8 averages, and flip angle of 15°. We obtained 15 phases during the cardiac cycle. Continuous slices were applied from base to apex covering the entire heart. Images were analyzed using the software Segment (http://segment.heiberg.se). The end-diastolic volume (EDV), end-systolic volume (ESV), and ejection fraction (EF) were measured.
Cell culture
Bone marrow-derived macrophages (BMDMs) were obtained from the tibias and femurs of C57BL/6J mice. BM was flushed from the femur and tibia using a 1 mL syringe with DEME medium and purified through Ficoll–Paque gradient (Amersham Biosciences, Freiburg, Germany). Cells were incubated in DMEM medium with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin and stimulated with 10 ng/mL macrophage colony-stimulating factor (PeproTech, Rocky Hill, NJ, USA) for 3 days to differentiate to macrophages. Next, the medium was changed, and attached macrophages were obtained after 5 days. BMDMs were primed with LPS (100 ng/mL, Sigma) for 3 h before treatment with 100-nmol/L darapladib or 1-μmol/L MCC950 for 1 h, followed by Ang II (100 nmol/L) treatment for the indicated time [24]. Control cells received DMSO vehicle.
Cardiac fibroblasts were isolated from neonatal rats born within 48 h, as described previously [27]. All of the cardiac fibroblasts were used at the second to six passages. In brief, mouse hearts were isolated, washed, minced, and digested in a solution of 100 U/mL collagenase type I and 0.1% trypsin for sequential 10-min periods of digestion with constant stirring at 37 °C. The supernatant from each digestion was suspended in DMEM medium with 15% FBS and 1% penicillin and streptomycin, and the above digestion process was repeated until all of the pieces disappeared. Single-cell suspensions were filtered through nylon 40-µm size sterile filters, centrifuged at 1000 rpm for 5 min, re-suspended, and incubated in plates. After a 90-min incubation period, the cardiac fibroblasts were attached to culture plates, and the medium was changed to DMEM with 10% FBS. Cardiac fibroblasts were starved for 24 h in the DMEM medium with 5% FBS before treatment, then cultured in the BMDM culture supernatant or co-cultured with macrophages in transwell plates for 48 h.
Enzyme-linked immunosorbent assay (ELISA)
IL-1β levels were analyzed from BMDM culture supernatant using the ELISA MAX™ Deluxe Set Mouse IL-1β (Biolegend) in accordance with the manufacturer’s instructions.
Transwell migration assay
BMDMs (5 × 104 cells per chamber) were plated on the top chambers of a 6.5-mm transwell with an 8-µm pore polycarbonate membrane insert (Corning, NY, USA) and co-cultured with starved cardiac fibroblasts in 24-well plates. Cells were incubated with darapladib (100 nmol/L) or/and MCC950 (1 μmol/L) for 1 h before stimulation with Ang II (1 μmol/L). After a 48-h incubation period, the migrated cells at the bottom of the transwell membrane were detected by crystal violet staining (Shanghai Yuanye Bio-Technology Co. Ltd., Shanghai, China). The numbers of migrated cells were visualized and counted using a microscope (Nikon, Konan, Tokyo, Japan).
WB analysis
Total protein of cardiac tissue or macrophages was collected using lysis buffer (Cwbio, Beijing, China) plus protease inhibitor cocktail (Roche, Basel, Switzerland).
Protein was quantified by bicinchoninic acid protein assay (Pierce Biotechnology, Rockford, IL, USA), then separated by 10% or 12% sodium dodecyl sulphate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA). Membranes were incubated with special primary antibodies against NLRP3, IL-1β, Mac-2, GAPDH (1:1000), caspase-1, collagen I, and collagen III (1:500) at 4 °C overnight, followed by incubation with appropriate second antibodies. Images were obtained using an ImageQuant™ LAS 4000 luminescent image analyzer (GE, Boston, USA).
Measurement of serum Lp-PLA2 activity
Serum Lp-PLA2 activity was measured using a PAF Acetylhydrolase Assay Kit from Cayman Chemical (Ann Arbor, MI, USA). The assay uses 2-thio-PAF as the substrate. In brief, 10 μL of plasma was added to 5 μL of 1 mmol/L EGTA and 10 μL of 2 mmol/L 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB) in 0.1 mol/L Tris-HCl (pH 7.2) and then incubated for 30 min at room temperature to allow any free thiols in the sample to react with DTNB. Next, the reactions were initiated by adding a 200-µL substrate solution containing 200 µmol/L 2-thio-PAF. The absorbance was read once every minute at 414 nm. The Lp-PLA2 activity was calculated by the change in absorbance per minute.
Plasma lipid
At study endpoint, mice were anesthetized, and blood was collected immediately via cardiac puncture using a 25 G needle and a 1 mL syringe [28]. Total plasma cholesterol (TC), triglycerides (TG), and low-density lipoprotein (LDL) and high-density lipoprotein (HDL) levels were detected using kits from Zhong Sheng Bio-technology (Beijing, China) with an ACCUTE TBA-40 FR automated biochemical analyzer (Toshiba, Konan, Tokyo, Japan). Mice were not subjected to fasting before plasma lipid analysis [28, 29].
Blood pressure recordings
The blood pressure of mice infused with Ang II for 7 days was measured by the tail-cuff system (Softron BP-98A; Softron, Tokyo, Japan) [30].
Statistical analysis
All of the data were presented as the mean ± standard deviation for at least three independent experiments. Then, statistical analysis was performed using GraphPad software (GraphPad Prism version 7.0 for Windows, GraphPad Software, San Diego, CA, USA). The values were tested by the Shapiro–Wilk normality test to determine whether the data were normally distributed. If data were normally distributed, Student’s t test was used to determine the significant difference between the two groups, and one-way ANOVA followed by Tukey’s post hoc test was used for multiple comparisons. If the data were not normally distributed, the Mann–Whitney test was used for two-group comparisons, and Kruskal–Wallis followed by Dunn’s test was used for multiple comparisons. P < 0.05 was considered to be statistically significant.
Results
Ang II infusion increases Lp-PLA2 expression in mouse cardiac tissue
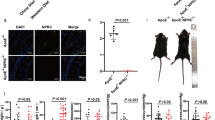
To investigate the role of Lp-PLA2 in hypertensive cardiac remodeling, we first performed RNA-seq analysis to examine PLA2 gene expression in Ang II-infused hearts (Ang II infusion 1500 ng·kg−1·min−1 for 1 week). Among the 23 PLA2 genes, Pla2g7 (also known as Lp-PLA2) was the only gene to be significantly upregulated in Ang II-infused hearts compared with control hearts (fold change ≥ 2 and probability ≥ 0.8 were considered statistically significant) (Fig. 1a; Supplementary Table S2). Notably, the fold changes of four housekeeping genes were within the range of 0.846–1.569 and showed no significant difference between the two groups, suggesting the high quality of our RNA-seq analysis and the specificity of Pla2g7 upregulation (Fig. 1a; Supplementary Table S2). The increased expression of Lp-PLA2 was validated by RT-PCR analysis and immunohistochemical staining (Fig. 1b, c). Moreover, plasma Lp-PLA2 activity was also significantly increased in Ang II-infused mice (Fig. 1d). Bioinformatics analysis showed that Lp-PLA2 was expressed almost exclusively by myeloid cells, especially by macrophages and monocytes (Supplementary Figs. S1 and S2), and Lp-PLA2 expression dramatically increases during the maturation of monocytes into macrophages [31]. RNA-seq analysis showed that the macrophage-specific gene, Lgals3 (galectin-3, also known as Mac-2), was upregulated in Ang II-infused hearts, indicating increased macrophage infiltration (Supplementary Table S3). IF staining showed the co-localization of Mac-2 and Lp-PLA2 in Ang II-infused hearts, indicating that macrophages are the major source of Lp-PLA2 in cardiac tissue (Fig. 1e). Ang II stimulation also increased macrophage Pla2g7 expression In vitro (Supplementary Fig. S3). Collectively, these results suggest that Lp-PLA2 was upregulated in the Ang II-infused mouse heart, and may play a critical role in hypertensive cardiac remodeling.

C57BL/6J mice were infused with Ang II (1500 ng·kg−1·min−1) or saline for 7 days before euthanasia. The hearts and plasma of these mice were collected for analysis. a RNA-seq analysis of mouse hearts. Volcano plot showing fold change and statistical probability value of PLA2 genes and four housekeeping genes (Gapdh, Eif5, Actb, and Nono) (n = 3; fold change ≥ 2 and probability ≥ 0.8 were considered statistically significant; black triangle, significantly increased PLA2 genes; white triangle, significantly decreased PLA2 genes; gray square, unchanged PLA2 genes; empty circle, control housekeeping genes). b RT-PCR analysis of the mRNA expression of Pla2g7 in mouse heart (n = 5). c Immunohistochemical staining of Lp-PLA2 in heart tissues and quantification (n = 5; scale bar = 200 μm). d Analysis of plasma Lp-PLA2 activity (n = 7). e Double immunofluorescence analysis of macrophage (Mac-2, red) and Lp-PLA2 (green) expression in hearts. Hoechst nuclear staining is shown in blue. Three independent experiments were performed. Scale bar = 25 μm. Data are presented as mean ± standard deviation, and n represents the number of animals. ***P < 0.001.
Inhibition of Lp-PLA2 by darapladib did not affect blood pressure or major plasma lipid levels
To evaluate whether pharmacological inhibition of Lp-PLA2 can prevent Ang II-induced cardiac remodeling, we used the highly selective small-molecule Lp-PLA2 inhibitor darapladib to block Lp-PLA2 activity in vivo [20]. The results showed that intragastric administration of darapladib (50 mg·kg−1·d−1) decreased Ang II-increased plasma Lp-PLA2 activity (Supplementary Fig. S4a). Ang II-induced cardiac Lp-PLA2 protein expression was also efficiently decreased by darapladib (Supplementary Fig. S4b). Besides, the in vitro experiment showed that darapladib decreased the Ang II-increased macrophage Pla2g7 expression (Supplementary Fig. S4c). These data indicated the successful inhibition of systemic and local Lp-PLA2 activation. We next sought to determine whether inhibition of Lp-PLA2 activity by darapladib could affect Ang II-induced hypertension. Systolic blood pressure was markedly elevated in mice undergoing Ang II infusion for 7 days compared to non-infused controls. Darapladib administration, however, did not affect systolic blood pressure in Ang II-infused mice (Supplementary Fig. S4d). As Lp-PLA2 mediates lipid metabolism, we also detected plasma lipid levels in Ang II-infused mice that underwent darapladib treatment. The results showed that Ang II significantly increased plasma TC, HDL, and LDL, but darapladib had no effects on these phenomena (Supplementary Fig. S4e). TG showed no significance across all groups (Supplementary Fig. S4e). Thus, darapladib significantly inhibited Lp-PLA2 activity without affecting blood pressure or major plasma lipid levels.
Inhibition of Lp-PLA2 by darapladib ameliorated Ang II infusion-induced collagen deposition and cardiac fibrosis
We examined whether darapladib treatment affects the progression of cardiac injury in response to Ang II. We found that Ang II infusion caused markedly increased fibrosis, indicated by Masson staining, whereas darapladib administration was associated with significantly lower fibrosis in Ang II-infused animals (Fig. 2a). Ang II-induced α-SMA-positive myofibroblasts and pro-fibrosis cytokine TGF-β were attenuated in darapladib-treated mice when compared with saline-infused mice (Fig. 2b). Furthermore, cardiac collagen deposition increased by Ang-II, which was detected by WB of collagen I and collagen III, was also reduced by darapladib treatment (Fig. 2c). Similarly, fibrotic gene expression of Ctgf, Col1a1, and Col3a1 in the heart was elevated by Ang II infusion and diminished upon darapladib treatment (Fig. 2d). Lp-PLA2 inhibition by darapladib thus suppressed cardiac myofibroblast trans-differentiation, decreased matrix overproduction, and reduced cardiac fibrosis associated with Ang II.

Inhibition of Lp-PLA2 by darapladib ameliorated Ang II infusion-induced collagen deposition and cardiac fibrosis. C57BL/6J mice received darapladib (50 mg·kg−1·d−1) or vehicle by gavage and were infused with saline or Ang II (1500 ng·kg−1·min−1) for 7 days. a Masson’s trichrome staining of myocardial fibrosis and quantification of fibrotic area (n = 5; scale bar = 100 μm). b Immunohistochemical staining of myofibroblasts with α-SMA and pro-fibrotic cytokine TGF-β. The quantifications are shown (n = 5; scale bar = 200 μm). c Western blot analysis of collagen I and collagen III protein levels in hearts, and the quantification of protein bands (n = 6). d RT-PCR analysis of Ctgf, Col1a1, and Col3a1 mRNA expression levels in heart tissues (n = 5). Ctgf connective tissue growth factor, Col1a1 collagen type I alpha 1, Col3a1 collagen type III alpha 1. Data are presented as the mean ± standard deviation, and n represents the number of animals. **P < 0.01, ***P < 0.001.
Inhibition of Lp-PLA2 attenuated Ang II infusion-induced cardiac hypertrophy and dysfunction in mice
We further assessed the effect of darapladib on myocardial histology. Ang II infusion-induced cardiac hypertrophy, as reflected by an increase in the heart size, and the heart weight to tibia length (HW/TL) ratio was markedly attenuated in darapladib-treated mice (Fig. 3a). The cardiomyocyte cross-sectional area was examined by FITC-labeled WGA staining. No significant effect was observed when darapladib was administered to saline-infused mice, but darapladib treatment significantly decreased cardiomyocyte transverse cross-sectional area after Ang II infusion (Fig. 3b). Atrial natriuretic factor (Anf) and brain natriuretic peptide (Bnp), the hypertrophic markers, were significantly increased in Ang II-infused mouse hearts and reduced upon darapladib treatment (Fig. 3c). To test the role of Lp-PLA2 in heart function, we performed magnetic resonance imaging in vivo in Ang II-infused mice treated with or without darapladib. Ang II infusion elevated the EF and reduced the EDV and the ESV, which were successfully rescued by darapladib treatment (Fig. 3d). Therefore, darapladib ameliorated Ang II-induced cardiac hypertrophy and dysfunction.

C57BL/6J mice received darapladib (50 mg·kg−1·d−1) or vehicle by gavage and were infused with saline or Ang II (1500 ng·kg−1·min−1) for 7 days. a HE staining of heart sections and the ratio of heart weight to tibia length (HW/TL) (n = 6; scale bar = 2 mm). b FITC-WGA staining of heart sections and the quantification of myocyte cross-sectional area (n = 5; scale bar = 50 μm). c RT-PCR analysis of the mRNA levels of Anf and Bnp in the hearts (n = 6). d MRI analysis of mouse heart. Representative images of end-systole and end-diastole are shown. EF ejection fraction, EDV end-diastolic volume, ESV end-systolic volume (n = 6). Data are presented as the mean ± standard deviation, and n represents the number of animals. *P < 0.05, **P < 0.01, ***P < 0.001.
Inhibition of Lp-PLA2 by darapladib attenuated cardiac inflammation in response to Ang II
To examine darapladib’s effects on Ang II-induced cardiac inflammation, we analyzed proinflammatory cell infiltration in cardiac tissues. ImageStream analysis revealed that the Ang II infusion-induced increase in accumulation of CD45+CD11b+ monocytes was markedly reduced by darapladib treatment (Fig. 4a). HE staining confirmed increased infiltration of pro-inflammatory cells in Ang II-infused heart tissue, which was significantly reduced by darapladib treatment (Fig. 4b). Moreover, immunohistochemical staining showed that the number of Mac-2 positive macrophages was markedly increased in Ang II-infused mice and reduced by darapladib administration (Fig. 4b). The decreased Mac-2 protein levels in cardiac tissue were also confirmed by WB analysis (Fig. 4c). In addition, the Ang II-stimulated expression of proinflammatory genes Mcp1, Tnfα, and Il12p40 were markedly lower in darapladib-treated mice (Fig. 4d). These data revealed that darapladib suppressed Ang II-induced cardiac inflammation.

C57BL/6J mice received darapladib (50 mg·kg−1·d−1) or vehicle by gavage and were infused with saline or Ang II (1500 ng·kg−1·min−1) for 7 days. a ImageStream analysis of CD45+CD11b+ monocytes in the hearts (n = 5; scale bar = 7 μm). b HE staining and immunohistochemical staining of macrophages (Mac-2) in the hearts (n = 5; scale bar = 200 μm). c Western blot analysis of Mac-2 protein level in the hearts and the quantification of protein bands (n = 6). d RT-PCR analysis of Mcp1, Tnfα, and Il12p40 mRNA expression levels in the heart tissues (n = 5). Mcp1 monocyte chemotactic protein 1, Tnfα tumor necrosis factor-alpha, Il12p40 interleukin 12b. Data are presented as the mean ± standard deviation, and n represents the number of animals. *P < 0.05, **P < 0.01, ***P < 0.001.
RNA sequencing identified transcriptomes of mouse cardiac tissue when Lp-PLA2 was inhibited by darapladib
To elucidate the transcriptional changes and identify the molecular events that give rise to the protective effects of Lp-PLA2 inhibition in a hypertensive mouse model, we performed RNA-sequencing experiments using heart samples from Ang II-infused mice with or without darapladib administration. RNA sequencing revealed that 243 genes were upregulated and 26 genes were downregulated in the Ang II group compared with the saline group, effects which were reversed in the Ang II+ darapladib group, suggesting that the Ang II-induced alteration of genes in pathological remodeling can be rescued by Lp-PLA2 inhibition with darapladib (Fig. 5a). We then compared the Ang II + darapladib group with the Ang II group, establishing differences in gene expression versus significance (P < 0.05) to visualize changes in gene expression (Fig. 5b). There were 363 differently expressed genes between the two groups, in which 75.2% (273 genes) were downregulated and 24.8% (90 genes) were upregulated in Ang II+ darapladib heart samples compared with Ang II heart samples. This indicates darapladib’s transcriptional suppression ability on Ang II-treated mouse hearts (Fig. 5b).

C57BL/6J mice received darapladib (50 mg·kg−1·d−1) or vehicle by gavage and were infused with saline or Ang II (1500 ng·kg−1·min−1) for 7 days. Hearts were collected for RNA-seq analysis. a Heatmap of differentially expressed genes that were both significantly changed in Ang II (compared to saline) and darapladib+Ang II (compared to Ang II) groups. Each column represents an individual replicate, with three replicates per group. Each row represents an individual gene. The top of the heatmap is the cluster of genes that are upregulated in Ang II-infused hearts compared with saline-infused hearts and those that are downregulated in darpladib + Ang II hearts, whereas the bottom is the cluster of genes that are downregulated in Ang II-infused hearts compared with saline-infused hearts and those that are upregulated in darapladib + Ang II hearts. The color bar represents the relative gene expression of log-transformed and normalized fragments per kilobase of transcript per million mapped reads (FPKM). Upregulated genes are shown in red, and downregulated genes are shown in blue. b Volcano plot shows the fold change and the significance of genes that were altered in darapladib+Ang II hearts vs. Ang II hearts. Upregulated genes are plotted in red, downregulated genes are plotted in blue, and genes not significant are plotted in gray. GO analysis of biological processes for the genes upregulated in the Ang II group compared with the saline group (c), and the genes downregulated in the darapladib+Ang II group compared with the Ang II group (d). Significantly changed ontology terms are mainly involved in inflammation, proliferation, fibrosis, and several fibrotic signaling pathways (c, d). KEGG pathway analysis of upregulated genes in the Ang II group compared with the saline group (e) and downregulated genes in the darapladib + Ang II group compared with the Ang II group (f). Significantly changed signaling pathways are mainly involved in inflammation, proliferation, and fibrosis (e, f). NS indicates not significant. P adj < 0.05 is considered to be different.
We compared RNA-seq of Ang II and saline groups and performed gene ontology (GO) analysis to determine functional changes that occurred during hypertensive cardiac remodeling. Upregulated genes included genes involved in proliferation, fibrosis, inflammation, and several signaling pathways (Fig. 5c). The activation of ECM organization and collagen fibril organization was consistent with findings of disruption and excess deposition of ECM in cardiac fibrosis (Fig. 5c). The significant upregulation of cell migration, the cytokine-mediated signaling pathway, and cellular responses to interleukin-1 (IL-1) confirmed the accumulation of immune cells and suggested that inflammatory response mediated by cytokine, especially IL-1, may play a critical role in cardiac pathological remodeling (Fig. 5c). Upregulated genes were also enriched in the TGF-β and ERK signaling pathways, which have been identified as critical pathways in cardiac fibrosis (Fig. 5c). Downregulated genes were involved in mitochondrial function, lipid metabolism, and amino acid metabolism (Supplementary Fig. S5a). We further compared RNA-seq between the Ang II + darapladib and Ang II groups to investigate the effects of darapladib on Ang II-induced alteration of genes in pathological remodeling. As expected, we found that downregulated genes were also enriched in proliferation, fibrosis, and inflammation, indicating the reversal effect of darapladib on Ang II-induced abnormal gene expression (Fig. 5d). Genes involved in Wnt, TGF-β, and MAPK signaling pathways were also decreased in the Ang II + darapladib group compared with the Ang II group (Fig. 5d). Upregulated genes were involved in the apoptotic process, mitochondrial function, and circadian rhythm (Supplementary Fig. S5b). We also performed the Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis, which further confirmed that Ang II upregulated the genes of proliferation, fibrosis, and inflammation, and darapladib showed suppressed effects on these processes (Fig. 5e, f). Supplementary Figure S6 shows the top 20 differentially expressed genes involved in inflammation, proliferation, and fibrosis, which were both significantly upregulated by Ang II and significantly downregulated in the presence of darapladib. We found that several genes were involved in more than one process, indicating the close connection between these processes. Taken together, the RNA-seq results support the role of darapladib in regulating a transcriptional profile that is consistent with its role in pathological cardiac remodeling and dysfunction.
Inhibition of Lp-PLA2 by darapladib blocked NLRP3-mediated inflammasome activation in cardiac tissue associated with Ang II infusion
C–C motif chemokine ligand 2 (CCL2, also known as MCP-1) regulates monocyte/macrophage migration. Cardiac Ccl2 gene expression was significantly upregulated by Ang II and markedly downregulated by darapladib (Figs. 4d, 6a; Supplementary Fig. S6), and the infiltration of cardiac monocytes/macrophages showed the same tendency (Fig. 4a, b). This indicates that darapladib may regulate monocyte/macrophage infiltration and the associated inflammatory response to influence Ang II-induced pathological damage. Excessive secretion of IL-1β resulting from inflammasome activation was found to be a key pathogenic mechanism for myofibroblast trans-differentiation and might account for the macrophage infiltration and subsequent remodeling seen in Ang II-infused hearts [24]. Therefore, we investigated darapladib’s effects on the expression of inflammasome components after Ang II infusion. RNA-seq analysis revealed that many inflammasome genes upregulated in Ang II-infused hearts showed downregulated tendencies in the presence of darapladib (Fig. 6a). Gene expression of Nlrp3, Il-1β, and Il-18 was also validated by RT-PCR (Fig. 6b). WB also confirmed that darapladib treatment attenuated Ang II-upregulated NLRP3, cleaved-caspase-1, pro-IL-1β, and mature IL-1β in cardiac tissues (Fig. 6c). Dual-IF staining revealed that the majority of cells in cardiac tissues expressing caspase-1 were macrophages and that darapladib treatment restored the upregulation (Fig. 6d). Immunostaining indicated that IL-1β was increased and localized to the inflammatory and fibrotic areas in Ang II mouse hearts, an effect that was attenuated by darapladib (Fig. 6e). These data indicate that darapladib attenuated Ang II-induced cardiac macrophage NLRP3 inflammasome activation as well as the production of IL-1β, which may contribute to the amelioration of cardiac inflammation.

C57BL/6J mice received darapladib (50 mg·kg−1d−1) or vehicle by gavage and were infused with saline or Ang II (1500 ng·kg−1·min−1) for 7 days. a Hearts were detected by RNA-seq analysis, and NLRP3 inflammasome-associated genes are shown in the heatmap (n = 3). The color bar represents the log-transformed and normalized relative fold change (FC) of genes. b RT-PCR analysis of Nlrp3, Il-1β, and Il-18 mRNA expression levels in the heart tissues (n = 5). c Western blot analysis of NLRP3 inflammasome-associated proteins (NLRP3, pro-caspase-1, cleaved-caspase-1, pro-IL-1β, and mature IL-1β) in hearts and the quantification of protein bands (n = 6). d Double immunofluorescence analysis of macrophage (Mac-2, green) and caspase-1 (red) expression in hearts. Hoechst nuclear staining is shown in blue. Three independent experiments were performed. Scale bar = 25 μm. e Immunohistochemical staining of IL-1β in the hearts (n = 5; scale bar = 200 μm). Data are presented as the mean ± standard deviation, and n represents the number of animals. P adj < 0.05 is considered to be different, *P < 0.05, **P < 0.01, ***P < 0.001.
Darapladib-treated macrophages inhibited myofibroblast trans-differentiation in vitro via inhibition of the NLRP3/Caspase-1/IL-1β secretion axis
To confirm darapladib’s effects on the NLRP3 inflammasome/caspase-1/IL-1β axis, we used BMDMs for further investigation. IF staining showed that Ang II increased the expression of NLRP3 and ASC in BMDMs, which were restored by darapladib treatment (Fig. 7a). Ang II-induced IL-1β intracellular expression and extracellular secretion were both inhibited by darapladib treatment (Fig. 7b, c). MCC950 is a selective NLRP3 inflammasome inhibitor. We found that Ang II-stimulated IL-1β secretion in BMDMs was downregulated to a similar extent by MCC950 or darapladib treatment, but did not further decrease when the two treatments were used in conjunction (Fig. 7c). Additionally, Ang II-promoted macrophage migration was also decreased to a similar extent by MCC950 and darapladib, and was not further decreased by the two when used in conjunction (Fig. 7d). To determine whether darapladib’s inhibition of Ang II-stimulated IL-1β production in BMDMs could affect myofibroblast transformation, we cultured fibroblasts with BMDM culture supernatant. Major markers of myofibroblasts, Sm22 and α-Sma, were strongly expressed by fibroblasts cultured in Ang II-stimulated BMDM supernatant (Fig. 7e, f). Darapladib or MCC950 inhibited myofibroblast trans-differentiation induced by Ang II-stimulated BMDM supernatant, but the inhibition was not further enhanced by treatment with darapladib plus MCC950 (Fig. 7e, f). Besides, we also detected the direct effects of darapladib and MCC950 on Ang II-stimulated fibroblasts. Ang II increased the expression of Sm22 and Tgfb, but neither MCC950 nor darapladib reversed these effects (Supplementary Fig. S7a, b). Cell proliferation was detected by CCK8 assay. We found that darapladib failed to decrease Ang II-induced fibroblast proliferation, but MCC950 showed inhibitory effects, and the inhibition was enhanced when MCC950 was used in combination with darapladib (Supplementary Fig. S7c). Thus, compared to fibroblasts, macrophages are more sensitive and more efficiently regulated by darapladib upon Ang II stimulation. These data indicate that darapladib attenuates myofibroblast trans-differentiation by suppressing macrophage NLRP3 inflammasome activation and IL-1β production.

BMDMs were primed with LPS (100 ng/mL) for 3 h, then treated with darapladib (10 or 100 nmol/L) for 1 h before stimulation with 100-nmol/L Ang II for 3 h to detect NLRP3 and ASC (a). Otherwise, they were stimulated with Ang II for 48 h to detect IL-1β (b) by immunofluorescence staining. Three independent experiments were performed. Scale bar = 10 μm in (a) and 40 μm in (b). c BMDMs were primed with LPS (100 ng/mL) for 3 h, then either treated with darapladib (100 nmol/L), MCC950 (1 μmol/L) or both for 1 h before stimulation with 100-nmol/L Ang II for 48 h to detect IL-1β in supernatants by ELISA (n = 6). d Macrophages were co-cultured with fibroblasts via transwell inserts. Cells were either treated with darapladib (100 nmol/L), MCC950 (1 μmol/L), or both for 1 h before stimulation with 1-μmol/L Ang II for 48 h to detect the migration of macrophages with crystal violet staining (n = 5; scale bar = 50 μm). Macrophages were treated as in (c), and the supernatants were collected. Starved fibroblasts were cultured in different macrophage supernatants for 24 h. The protein expression of α-SMA was detected by immunofluorescence staining (e) (scale bar = 40 μm). The mRNA levels of Sm22 and α-Sma were detected by RT-PCR (f). Sm22 smooth muscle protein 22-alpha. Data are presented as the mean ± standard deviation, and n represents the number of animals. NS indicates not significant. **P < 0.01, ***P < 0.001.
Darapladib regulated macrophage inflammation and myofibroblast trans-differentiation by targeting Lp-PLA2
To confirm whether darapladib targets Lp-PLA2 in the context of Ang II-induced cardiac inflammation and fibrosis, we used Lp-PLA2-knockout mice for further investigation. The expression of Pla2g7 was significantly reduced in the hearts of Lp-PLA2 knockout mice compared with that of WT mice (Supplementary Fig. S8a). We found that Ang II-stimulated Il-1β and Nlrp3 expression in WT BMDMs were downregulated in Lp-PLA2-knockout BMDMs but did not further decrease when darapladib was added (Supplementary Fig. S8b). Then, we cultured fibroblasts with different BMDM culture supernatants. The gene expression of Sm22 and α-Sma were upregulated in fibroblasts cultured in Ang II-stimulated WT-BMDM supernatants and reduced in Lp-PLA2-knockout-BMDM supernatants, but the reduction was not further enhanced by darapladib (Supplementary Fig. S8c). These data indicate that darapladib targets Lp-PLA2 to attenuate Ang II-induced macrophage inflammation and myofibroblast trans-differentiation in vitro.
Bioinformatics analysis revealed darapladib may regulate NLRP3 inflammasome activation by regulating lysoPC metabolism and the LDL receptor (LDLR) signaling pathway
In order to reveal the detailed molecular mechanisms of darapladib in NLRP3 inflammasome activation in Ang II-infused mouse heart, we conducted a detailed bioinformatics analysis using PPI analysis and IPA. Key genes regulated by darapladib in the heart and the associated genes found from related literature were put into STRING for network construction. PPI network showed NLRP3/IL-1β and PLA2G7 (Lp-PLA2) were connected via LDLR, which was downregulated by darapladib in response to Ang II (Supplementary Fig. S9). The LDLR pathway plays an important role in the regulation of cholesterol homeostasis. It can be activated by inflammation, RAS, and hyperglycemia, and it is associated with a series of diseases, such as atherosclerosis, nonalcoholic fatty liver disease, and kidney fibrosis [32,33,34]. IPA network further confirmed the interactions among NLRP3, IL-1β, PLA2G7, and LDLR and revealed that the interactions were associated with phospholipid/lysoPC metabolism (Fig. 8). Interestingly, in the network, PLA2G7 was found to affect NLRP3/IL-1β via lysoPC and LDLR, while IL-1β was found to affect PLA2G7, forming a regulatory loop (Fig. 8). These data indicate that darapladib may inhibit NLRP3 inflammasome activation by regulating phospholipid/lysoPC metabolism and the LDLR signaling pathway.

The interaction networks of DEGs regulated by darapladib and the associated molecules found from related literature were generated through the use of IPA. PLA2G7, NLRP3, IL-1β, CCL2, and LDLR are highlighted in green. Phospholipids and lysophosphatidylcholines are highlighted in blue. Key interactions associated with PLA2G7 and NLRP3 are highlighted in red.
Biological pathway networks mediated by darapladib in hypertensive cardiac remodeling
To understand the networks of significant biological pathways involved in darapladib’s protective effects on hypertensive cardiac remodeling, the key genes regulated by darapladib and validated in our study were submitted to the ClueGO tool for further analysis. As expected, these genes were clustered in three major aspects, including inflammation (regulation of leukocyte chemotaxis, regulation of mononuclear cell migration, cytokine production involved in immune response, regulation of cytokine production involved in immune response), proliferation (positive regulation of cell cycle process, regulation of fibroblast proliferation, positive regulation of fibroblast proliferation), and fibrosis and hypertrophy (ECM organization and regulation of cardiac muscle cell differentiation). They were closely linked by the cellular response to the interleukin-1 pathway. Inhibition of Lp-PLA2 by darapladib diminished Pla2g7, Nlrp3, IL-1β, and Ccl2-mediated leukocyte migration and cytokine production, especially the migration of mononuclear cells and the production of IL-1. Thus, darapladib suppressed the pathway networks that were involved in inflammation, proliferation, fibrosis, and hypertrophy, and centered on the cellular response to the interleukin-1 pathway as the core (Fig. 9).

ClueGO analysis of genes regulated by darapladib in Ang II-infused hearts. These networks are functionally grouped by enriched categories of target genes. Different functional groups of GO terms (biological process) are represented as nodes in different colors (pink nodes represent GO terms associated with inflammation; green nodes, proliferation; blue nodes, fibrosis, and hypertrophy). Shared genes are represented by yellow nodes, and Pla2g7 gene is represented by the red node. The edge between nodes reflects shared genes in different GO terms, or the connections between GO terms. Anxa2 annexin A2, Ccna2 cyclin A2, Ccnb1 cyclin B1, Cdc6 cell division cycle 6, Col14a1 collagen type XIV alpha 1, Dusp1 dual specificity phosphatase 1, Edn1 endothelin 1, E2f1 E2F transcription factor 1, Fn1 fibronectin 1, Igf1 insulin-like growth factor 1, Sfrp1 secreted frizzled-related protein 1.
Discussion
The present study identified the critical role of Lp-PLA2 in hypertensive cardiac fibrosis for the first time and demonstrated that pharmacological inhibition of Lp-PLA2 by darapladib prevented Ang II-induced cardiac remodeling and inflammation, which benefited from the suppression of NLRP3 inflammasome. Thus, darapladib could potentially be used as a therapeutic approach to address hypertensive cardiac damage.
Despite the prevalence of cardiac fibrosis, efficacious therapies are lacking. Current treatment of hypertensive cardiac fibrosis focusing on blood pressure control and blood volume reduction is intractable with adverse drug reactions, such as postural hypotension and hyperkalemia [35]. In recent years, inflammation has emerged as a critical biological process contributing to hypertensive cardiac remodeling [3]. Thus, finding new therapeutic strategies targeting cardiac inflammation is of great significance. Lp‐PLA2 is a calcium-independent serine lipase that belongs to group VII of the PLA2 superfamily, mediates vascular inflammation through the regulation of lipid metabolism, and is implicated in several vascular inflammation‐related diseases [15, 16]. Chimeric mice with bone marrow-derived leukocytes deficient in Lp-PLA2 (bmLp-PLA2−/− mice) developed smaller and fewer inflamed infarcts with reduced numbers of infiltrating neutrophils and inflammatory Ly-6Chigh monocytes after myocardial infarction [36]. Moreover, elevated Lp-PLA2 levels were observed in both the plasma of hypertension patients and the pathological tissues of fibrotic disease patients [17, 37, 38]. These studies indicate that Lp-PLA2 may be associated with hypertension and fibrosis, but the detailed effect of Lp-PLA2 on hypertensive cardiac damage is still unknown. In this study, we demonstrated that Ang II infusion highly increased Lp-PLA2 expression in hypertensive cardiac tissue, and the most advanced Lp‐PLA2 inhibitor, darapladib, inhibited Ang II-induced cardiac fibrosis, hypertrophy, and dysfunction, independently of its effects on blood pressure. These findings indicate that pharmacological inhibition of Lp-PLA2 by darapladib is a potential therapeutic approach in retarding the progression of hypertensive cardiac remodeling.
The protective effects of darapladib in atherosclerosis have been demonstrated previously both in preclinical and clinical studies [25, 39]. Unfortunately, darapladib subsequently failed to meet the primary endpoints of two large phases III trials in atherosclerosis patients cotreated with standard medical care in 2014 [18, 19]. However, there were several limitations in these clinical trials: nearly 95% of the patients received statins as the basic treatment, which could not only lower plasma lipid levels but also inhibit plasma Lp‐PLA2 levels [40]; all of the patients were screened based on their Lp-PLA2 activity levels, but the beneficial effects of darapladib on patients with higher baseline levels of Lp-PLA2 activity were not evaluated separately [41]. Although the discovery of an anti‐inflammatory drug for atherosclerosis is dim, the research on Lp‐PLA2 and darapladib has not been terminated [41]. Over the past few years, Lp‐PLA2 inhibition showed promising therapeutic effects in diabetic macular edema and Alzheimer’s disease [21, 22]. Darapladib lowered IL-1β and IL-6 levels in the kidney at type 2 diabetes mellitus [42]. The potential anti-glioma effects of darapladib were also investigated [43]. Moreover, mice deficient in bone marrow Lp-PLA2 had fewer ly-6Chigh monocytes in the peripheral blood and myocardium and showed ameliorated adverse left ventricle remodeling and improved heart function recovery after myocardial infarction [36]. These indicate the role of Lp-PLA2 in regulating myocardial inflammatory responses and heart function, which is distinct from the effects on atherosclerotic plaques [36]. In the present study, we revealed the protective effects of Lp-PLA2 inhibition by darapladib on hypertension-induced cardiac remodeling and dysfunction, illuminating a new potential application for darapladib. Considering the complex etiology mechanism of hypertensive cardiac fibrosis, it can be concluded that Lp-PLA2 may be one of the prime inflammatory factors in this process.
Lp-PLA2 is principally secreted by monocyte-derived macrophages and dramatically upregulated during macrophage differentiation [14]. Darapladib decreased macrophage Pla2g7 gene expression and macrophage accumulation in the heart. Thus, darapladib may reduce Lp-PLA2 levels in heart in two ways, i.e., by directly reducing Ang II-induced Lp-PLA2 expression and by decreasing macrophage accumulation. Meanwhile, inhibition of Lp-PLA2 by darapladib markedly reduced Ang II-induced cardiac pro-inflammatory cytokine secretion both in vivo and In vitro. These results indicate that the inhibition of Lp-PLA2 by darapladib may suppress hypertensive cardiac inflammation by decreasing macrophage migration and cytokine production in response to Ang II.
RNA-sequencing analysis showed that Ang II activated the biological process of cellular response to IL-1 in the mouse heart. IL-1β is a critical member of the IL-1 cytokine family, which has both pro-inflammatory and pro-fibrotic abilities. Il-1β activation and secretion is dependent on the inflammasome/caspase-1 axis, and previous studies have shown that Ang II induces NLRP3 inflammasome activation and IL-1β release in macrophages and mouse cardiac tissue [24, 44]. A recent study revealed that homocysteine elevated adipocyte lyso-PC generation, which was dependent on phospholipase A2 group 16, to activate the NLRP3 inflammasome in adipocyte tissue macrophages and also promoted insulin resistance, suggesting that PLA2 enzyme plays a role in regulating NLRP3 inflammasome activation [45]. Thus, we hypothesized that the protection against Ang II-induced cardiac inflammation and fibrosis by darapladib through Lp-PLA2 inhibition may depend on the NLRP3 inflammasome/IL-1β axis. RNA-sequencing showed that darapladib suppressed NLRP3 inflammasome-associated genes. WB analysis confirmed that darapladib inhibited Ang II infusion-increased protein levels of NLRP3, cleaved-caspase-1, and IL-1β, without affecting the upregulation of pro-caspase-1, suggesting that darapladib may affect inflammasome function via regulating gene expression and suppressing the assembly of inflammasome components. Furthermore, the suppression of darapladib on macrophage NLRP3 inflammasome assembly and IL-1β production was also confirmed in Ang II-stimulated BMDMs In vitro. We found that either NLRP3 inflammasome-specific inhibitor MCC950 or Lp-PLA2 inhibitor darapladib prevented the production of IL-1β in macrophages stimulated with Ang II, but the combination of the two showed no further suppression. These data indicate that darapladib-mediated Lp-PLA2 inhibition exerts anti-inflammatory properties in Ang II-induced cardiac remodeling via dampening NLRP3 inflammasome activation.
PPI and IPA revealed the possible molecular mechanisms of darapladib in NLRP3 inflammasome activation. NLRP3/IL-1β and LP-PLA2 were connected by phospholipid/lysoPC metabolism and the LDLR pathway. Lp‐PLA2 hydrolyzes oxidized LDL into two bioactive products, lysoPC and oxidized nonesterified fatty acids [41]. Accumulating evidence demonstrates that lyso-PC is a critical proinflammatory mediator in modulating NLRP3 inflammasome activation and IL-1β secretion [46, 47], and darapladib has been found to decrease IL-1β in the kidney at type 2 diabetes mellitus [42]. Darapladib decreased atherosclerotic plaque and the necrotic core area, with reduced lysoPC content and decreased expression of multiple genes associated with macrophage functioning [48]. Besides, the lysoPC content affects the affinity of LDL to LDLR, and the LDLR pathway is positively related to NLRP3 inflammasome activation in white adipose tissues and macrophages [49,50,51,52]. Thus, darapladib may inhibit NLRP3 inflammasome activation by regulating lysoPC metabolism and the LDLR pathway.
IL-1β exerts both pro-inflammatory and pro-fibrotic effects on Ang II-infused cardiac tissues. IL-1β initiates and propagates sterile inflammation, including macrophage recruitment, activation of the pro-inflammatory cytokine IL-6, and modulating chemokine expression [53,54,55]. Moreover, IL-1β upregulates the expression of TGF-β1, as well as other pro-fibrotic genes, and induces myofibroblast trans-differentiation and collagen production [56, 57]. As expected, we found that both Lp-PLA2 inhibition by darapladib and NLRP3 inflammasome inhibition by MCC950 decreased the migration of Ang II-promoted macrophages to fibroblasts. Moreover, supernatants of Ang II-stimulated macrophage promoted fibroblast transformation into myofibroblasts, which could be attenuated by either darapladib or MCC950 treatment, but the combination of the two showed no further effect. These results indicate that the protective effect of darapladib-mediated Lp-PLA2 inhibition on Ang II-stimulated macrophage-promoted myofibroblast trans-differentiation and fibrotic remodeling is dependent on NLRP3-regulated IL-1β secretion.
RNA sequencing and bioinformatics analysis further improved understanding of the molecular mechanisms underlying the morphology [58]. Most of the differentially expressed genes in darapladib + Ang II hearts vs. Ang II hearts were downregulated, indicating the suppressive effects of darapladib on Ang II-stimulated transcription. Enrichment analysis uncovered a number of darapladib-regulated signaling pathways upon Ang II stimulation, including Wnt, ERK, MAPK, and TGF-β receptor signaling pathways, which have been demonstrated to function in cardiac fibrosis [27, 59]. Moreover, the differentially expressed genes were mainly enriched in cellular processes involved in cardiac hypertrophy, fibrosis, proliferation, and inflammation. Biological processes involving cellular response to interleukin-1 seemed to function as a bridge linking the four aspects of inflammation, proliferation, fibrosis, and hypertrophy. Inhibition of Lp-PLA2 by darapladib regulated both leukocyte migration and pro-inflammatory cytokine production to affect Ang II-induced cardiac inflammation, which is consistent with the reduction of monocytes/macrophage and IL-1β observed by histological analysis. The dampened inflammatory response may lead to decreased cell proliferation, especially fibroblast proliferation, as well as reduction of ECM deposition and cardiac muscle cell differentiation, which were also demonstrated by histological analysis. Therefore, our findings at the physiological, histological, cellular, and molecular levels consistently and strongly indicate that the inhibition of Lp-PLA2 by darapladib has obvious protective effects on hypertensive cardiac inflammation, remodeling, and dysfunction. Lp-PLA2 inhibition by darapladib may represent a novel therapeutic strategy in combating cardiac diseases associated with adverse remodeling.
In addition, we also observed that darapladib decreased plasma Lp-PLA2 activity as well as Lp-PLA2 expression in cardiac tissues. Elevated circulating levels of Lp‐PLA2 are associated with a number of vascular diseases observed in a series of epidemiological studies, supporting the proinflammatory action of Lp-PLA2 [15, 16, 60, 61]. Lp-PLA2 affects endothelial cell functions and the homing of immune cells by generating proinflammatory mediators [62]. In the myocardial-infarction mouse model, bone marrow Lp-PLA2-deficiency reduced plasma Lp-PLA2 activity accompanied by reduced plasma inflammatory cytokines, fewer neutrophils, and Ly-6Chigh cells in both blood and ischemic myocardium [36]. In the atherosclerosis mouse model, silencing Lp-PLA2 by lentiviral vector delivery reduced plasma Lp-PLA2 levels and decreased MCP-1 and MMP-8 in both blood and plaques of ApoE−/− mice [63]. Thus, Lp-PLA2 inhibition may reduce both systemic and local inflammatory responses, which are closely linked to cardiovascular diseases. In the present study, the inhibition of Lp-PLA2 by darapladib decreased the migration capacity of Ang II-stimulated monocytes/macrophages and resulted in reduced monocytes/macrophages numbers and lower pro-inflammatory cytokine levels in cardiac tissues. Thus, the plasma Lp-PLA2 may contribute to Ang II-induced cardiac dysfunction by regulating peripheral monocytes/macrophages migration to cardiac tissues to amplify the local inflammatory response, and darapladib plays a protective role in this process. Future studies by our group will further investigate the underlying mechanisms.
In summary, we demonstrated that Lp-PLA2 is highly upregulated in the mouse heart and modulates cardiac inflammation and fibrosis in response to Ang II. Importantly, we shed light on the protective mechanism of darapladib, a selective Lp-PLA2 inhibitor, on Ang II-induced cardiac inflammation and fibrosis by inhibiting the NLRP3 inflammasome/IL-1β axis. In conjunction with the findings of phase III atherosclerosis trials that darapladib is a safe drug, our results may inform the development of novel strategies to treat hypertensive cardiac injury.
References
Gradman AH, Alfayoumi F. From left ventricular hypertrophy to congestive heart failure: management of hypertensive heart disease. Prog Cardiovasc Dis. 2006;48:326–41.
Park S, Nguyen NB, Pezhouman A, Ardehali R. Cardiac fibrosis: potential therapeutic targets. Transl Res. 2019;209:121–37.
McMaster WG, Kirabo A, Madhur MS, Harrison DG. Inflammation, immunity, and hypertensive end-organ damage. Circ Res. 2015;116:1022–33.
Zhu YC, Zhu YZ, Lu N, Wang MJ, Wang YX, Yao T. Role of angiotensin AT1 and AT2 receptors in cardiac hypertrophy and cardiac remodelling. Clin Exp Pharmacol Physiol. 2003;30:911–8.
O’Rourke SA, Dunne A, Monaghan MG. The role of macrophages in the infarcted myocardium: orchestrators of ECM remodeling. Front Cardiovasc Med. 2019;6:101.
Yang M, Zheng J, Miao Y, Wang Y, Cui W, Guo J, et al. Serum-glucocorticoid regulated kinase 1 regulates alternatively activated macrophage polarization contributing to angiotensin II-induced inflammation and cardiac fibrosis. Arterioscler Thromb Vasc Biol. 2012;32:1675–86.
Jia L, Li Y, Xiao C, Du J. Angiotensin II induces inflammation leading to cardiac remodeling. Front Biosci (Landmark Ed). 2012;17:221–31.
Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013;39:432–41.
Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411.
Liu D, Zeng X, Li X, Mehta JL, Wang X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res Cardiol. 2018;113:5.
Dennis EA, Cao J, Hsu YH, Magrioti V, Kokotos G. Phospholipase A2 enzymes: physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem Rev. 2011;111:6130–85.
Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A(2) research: from cells to animals to humans. Prog Lipid Res. 2011;50:152–92.
Nikolaou A, Kokotou MG, Vasilakaki S, Kokotos G. Small-molecule inhibitors as potential therapeutics and as tools to understand the role of phospholipases A2. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:941–56.
Tsimikas S, Tsironis LD, Tselepis AD. New insights into the role of lipoprotein(a)-associated lipoprotein-associated phospholipase A2 in atherosclerosis and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2094–9.
Munzel T, Gori T. Lipoprotein-associated phospholipase A(2), a marker of vascular inflammation and systemic vulnerability. Eur Heart J. 2009;30:2829–31.
Siddiqui MK, Kennedy G, Carr F, Doney ASF, Pearson ER, Morris AD, et al. Lp-PLA2 activity is associated with increased risk of diabetic retinopathy: a longitudinal disease progression study. Diabetologia 2018;61:1344–53.
Li Z, Liu J, Shen Y, Zeng F, Zheng D. Increased Lipoprotein-associated phospholipase A2 activity portends an increased risk of resistant hypertension. Lipids Health Dis. 2016;15:15.
O’Donoghue ML, Braunwald E, White HD, Lukas MA, Tarka E, Steg PG, et al. Effect of darapladib on major coronary events after an acute coronary syndrome: the SOLID-TIMI 52 randomized clinical trial. J Am Med Assoc. 2014;312:1006–15.
Investigators S, White HD, Held C, Stewart R, Tarka E, Brown R, et al. Darapladib for preventing ischemic events in stable coronary heart disease. N Engl J Med. 2014;370:1702–11.
Wang K, Xu W, Zhang W, Mo M, Wang Y, Shen J. Triazole derivatives: a series of Darapladib analogues as orally active Lp-PLA2 inhibitors. Bioorg Med Chem Lett. 2013;23:2897–901.
Maher-Edwards G, De’Ath J, Barnett C, Lavrov A, Lockhart A. A 24-week study to evaluate the effect of rilapladib on cognition and cerebrospinal fluid biomarkers of Alzheimer’s disease. Alzheimers Dement (N. Y). 2015;1:131–40.
Staurenghi G, Ye L, Magee MH, Danis RP, Wurzelmann J, Adamson P, et al. Darapladib, a lipoprotein-associated phospholipase A2 inhibitor, in diabetic macular edema: a 3-month placebo-controlled study. Ophthalmology. 2015;122:990–6.
Gan W, Ren J, Li T, Lv S, Li C, Liu Z, et al. The SGK1 inhibitor EMD638683, prevents Angiotensin II-induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1–10.
Gan W, Ren J, Li T, Lv S, Li C, Liu Z, et al. The SGK1 inhibitor EMD638683, prevents Angiotensin II-induced cardiac inflammation and fibrosis by blocking NLRP3 inflammasome activation. Biochim Biophys Acta. 2018;1864:1–10.
Hu MM, Zhang J, Wang WY, Wu WY, Ma YL, Chen WH, et al. The inhibition of lipoprotein-associated phospholipase A2 exerts beneficial effects against atherosclerosis in LDLR-deficient mice. Acta Pharmacol Sin. 2011;32:1253–8.
Li Y, Wu Y, Zhang C, Li P, Cui W, Hao J, et al. gammadeltaT Cell-derived interleukin-17A via an interleukin-1beta-dependent mechanism mediates cardiac injury and fibrosis in hypertension. Hypertension. 2014;64:305–14.
Wang L, Zhang YL, Lin QY, Liu Y, Guan XM, Ma XL, et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur Heart J. 2018;39:1818–31.
Tham YK, Bernardo BC, Huynh K, Ooi JYY, Gao XM, Kiriazis H, et al. Lipidomic profiles of the heart and circulation in response to exercise versus cardiac pathology: a resource of potential biomarkers and drug targets. Cell Rep. 2018;24:2757–72.
Hayek T, Attias J, Coleman R, Brodsky S, Smith J, Breslow JL, et al. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovasc Res. 1999;44:579–87.
Wang L, Li YL, Zhang CC, Cui W, Wang X, Xia Y, et al. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc Res. 2014;101:383–92.
Ferguson JF, Hinkle CC, Mehta NN, Bagheri R, Derohannessian SL, Shah R, et al. Translational studies of lipoprotein-associated phospholipase A2 in inflammation and atherosclerosis. J Am Coll Cardiol. 2012;59:764–72.
Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. 2015;21:37–46.
Liu J, Ma KL, Gao M, Wang CX, Ni J, Zhang Y, et al. Inflammation disrupts the LDL receptor pathway and accelerates the progression of vascular calcification in ESRD patients. PLoS One. 2012;7:e47217.
Liu J, Ma KL, Zhang Y, Wu Y, Hu ZB, Lv LL, et al. Activation of mTORC1 disrupted LDL receptor pathway: a potential new mechanism for the progression of non-alcoholic fatty liver disease. Int J Biochem Cell Biol. 2015;61:8–19.
Musso CG, Alfie J. Resistant hypertension in the elderly-second line treatments: aldosterone antagonists, central alpha-agonist agents, alpha-adrenergic receptor blockers, direct vasodilators, and exogenous nitric oxide donors. Cardiovasc Hematol Agents Med Chem. 2015;12:170–3.
He S, Chousterman BG, Fenn A, Anzai A, Nairz M, Brandt M, et al. Lp-PLA2 antagonizes left ventricular healing after myocardial infarction by impairing the appearance of reparative macrophages. Circ Heart Fail. 2015;8:980–7.
Triggiani M, De Marino V, Sofia M, Faraone S, Ambrosio G, Carratu L, et al. Characterization of platelet-activating factor acetylhydrolase in human bronchoalveolar lavage. Am J Respir Crit Care Med. 1997;156:94–100.
Mahmut A, Boulanger MC, El Husseini D, Fournier D, Bouchareb R, Despres JP, et al. Elevated expression of lipoprotein-associated phospholipase A2 in calcific aortic valve disease: implications for valve mineralization. J Am Coll Cardiol. 2014;63:460–9.
Serruys PW, Garcia-Garcia HM, Buszman P, Erne P, Verheye S, Aschermann M, et al. Effects of the direct lipoprotein-associated phospholipase A2 inhibitor darapladib on human coronary atherosclerotic plaque. Circulation. 2008;118:1172–82.
Mullard A. GSK’s darapladib failures dim hopes for anti-inflammatory heart drugs. Nat Rev Drug Discov. 2014;13:481–2.
Huang F, Wang K, Shen J. Lipoprotein-associated phospholipase A2: The story continues. Med Res Rev. 2020;40:79–134.
Wihastuti TA, Aini FN, Tjahjono CT, Sulfia YH, Sholichah Z, Heriansyah T. Lp-PLA2 selective inhibitor (Darapladib) effect in lowering the expression level of IL-1B and IL-6 In the renal at type 2 diabetes mellitus. Vasc Health Risk Manag. 2019;15:503–8.
Wang YJ, Chang SB, Wang CY, Huang HT, Tzeng SF. The selective lipoprotein-associated phospholipase A2 inhibitor darapladib triggers irreversible actions on glioma cell apoptosis and mitochondrial dysfunction. Toxicol Appl Pharmacol. 2020;402:115133.
Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler Thromb Vasc Biol. 2015;35:127–36.
Zhang SY, Dong YQ, Wang P, Zhang X, Yan Y, Sun L, et al. Adipocyte-derived lysophosphatidylcholine activates adipocyte and adipose tissue macrophage nod-like receptor protein 3 inflammasomes mediating homocysteine-induced insulin resistance. EBioMedicine. 2018;31:202–16.
Correa R, Silva LFF, Ribeiro DJS, Almeida RDN, Santos IO, Correa LH, et al. Lysophosphatidylcholine induces NLRP3 inflammasome-mediated foam cell formation and pyroptosis in human monocytes and endothelial cells. Front Immunol. 2019;10:2927.
Freeman L, Guo H, David CN, Brickey WJ, Jha S, Ting JP. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J Exp Med. 2017;214:1351–70.
Wilensky RL, Shi Y, Mohler ER 3rd, Hamamdzic D, Burgert ME, Li J, et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat Med. 2008;14:1059–66.
Benitez S, Villegas V, Bancells C, Jorba O, Gonzalez-Sastre F, Ordonez-Llanos J, et al. Impaired binding affinity of electronegative low-density lipoprotein (LDL) to the LDL receptor is related to nonesterified fatty acids and lysophosphatidylcholine content. Biochemistry. 2004;43:15863–72.
Cyr Y, Lamantia V, Bissonnette S, Burnette M, Besse-Patin A, Demers A, et al. Lower plasma PCSK9 in normocholesterolemic subjects is associated with upregulated adipose tissue surface-expression of LDLR and CD36 and NLRP3 inflammasome. Physiol Rep. 2021;9:e14721.
Faraj M. LDL, LDL receptors, and PCSK9 as modulators of the risk for type 2 diabetes: a focus on white adipose tissue. J Biomed Res. 2020;34:251–9.
Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–61.
Rider P, Carmi Y, Guttman O, Braiman A, Cohen I, Voronov E, et al. IL-1alpha and IL-1beta recruit different myeloid cells and promote different stages of sterile inflammation. J Immunol. 2011;187:4835–43.
McGeough MD, Pena CA, Mueller JL, Pociask DA, Broderick L, Hoffman HM, et al. Cutting edge: IL-6 is a marker of inflammation with no direct role in inflammasome-mediated mouse models. J Immunol. 2012;189:2707–11.
Natoli R, Fernando N, Madigan M, Chu-Tan JA, Valter K, Provis J, et al. Microglia-derived IL-1beta promotes chemokine expression by Muller cells and RPE in focal retinal degeneration. Mol Neurodegener. 2017;12:31.
Artlett CM. The role of the NLRP3 inflammasome in fibrosis. Open Rheumatol J. 2012;6:80–6.
Luo DD, Fielding C, Phillips A, Fraser D. Interleukin-1 beta regulates proximal tubular cell transforming growth factor beta-1 signalling. Nephrol Dial Transpl. 2009;24:2655–65.
Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res. 2019;125:662–77.
Wang M, Qian L, Li J, Ming H, Fang L, Li Y, et al. GHSR deficiency exacerbates cardiac fibrosis: role in macrophage inflammasome activation and myofibroblast differentiation. Cardiovasc Res. 2020;116:2091–102.
Acosta S, Taimour S, Gottsater A, Persson M, Engstrom G, Melander O, et al. Lp-PLA2 activity and mass for prediction of incident abdominal aortic aneurysms: a prospective longitudinal cohort study. Atherosclerosis. 2017;262:14–8.
Lehtinen L, Vainio P, Wikman H, Huhtala H, Mueller V, Kallioniemi A, et al. PLA2G7 associates with hormone receptor negativity in clinical breast cancer samples and regulates epithelial-mesenchymal transition in cultured breast cancer cells. J Pathol Clin Res. 2017;3:123–38.
Shi Y, Zhang P, Zhang L, Osman H, Mohler ER 3rd, Macphee C, et al. Role of lipoprotein-associated phospholipase A2 in leukocyte activation and inflammatory responses. Atherosclerosis. 2007;191:54–62.
Zhang H, Zhou W, Cao C, Zhang W, Liu G, Zhang J. Amelioration of atherosclerosis in apolipoprotein E-deficient mice by combined RNA interference of lipoprotein-associated phospholipase A2 and YKL-40. PLoS One. 2018;13:e0202797.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (Nos. 81973325 and 81903612).
Author information
Authors and Affiliations
Contributions
The study concept and design were done by MY and SLL. Administrative support was provided by MY. The mice model and in vivo experiments were done by SLL, ZFZ, and WQG. In vitro experiments were done by SLL and ZFZ. Bioinformatics analysis was created by SLL, WQG, and WQW. Collection and interpretation of data were done by SLL, WQW, ZFZ, WQG, and MY. Statistical advice and technical support were provided by TGL, YFH, ZY, and RXZ. Funding support was provided by MY and SLL. Paper writing and final approval were done by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Lv, Sl., Zeng, Zf., Gan, Wq. et al. Lp-PLA2 inhibition prevents Ang II-induced cardiac inflammation and fibrosis by blocking macrophage NLRP3 inflammasome activation. Acta Pharmacol Sin 42, 2016–2032 (2021). https://doi.org/10.1038/s41401-021-00703-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00703-7
Keywords
This article is cited by
-
Serum levels of lipoprotein-associated phospholipase A2 are associated with coronary atherosclerotic plaque progression in diabetic and non-diabetic patients
BMC Cardiovascular Disorders (2024)
-
Flaxseed oil fraction reverses cardiac remodeling at a molecular level: improves cardiac function, decreases apoptosis, and suppresses miRNA-29b and miRNA 1 gene expression
BMC Complementary Medicine and Therapies (2024)
-
Targeting myocardial inflammation: investigating the therapeutic potential of atrial natriuretic peptide in atrial fibrosis
Molecular Biology Reports (2024)
-
INPP5D regulates inflammasome activation in human microglia
Nature Communications (2023)
-
Mitochondria and NLRP3 inflammasome in cardiac hypertrophy
Molecular and Cellular Biochemistry (2023)
