
Abstract
Individual differences in the development of uncontrollable fear in response to traumatic stressors have been observed in clinic, but the underlying mechanisms remain unknown. In the present study we first conducted a meta-analysis of published clinical data and found that malondialdehyde, an oxidative stress biomarker, was significantly elevated in the blood of patients with fear-related anxiety disorders. We then carried out experimental study in rats subjected to fear conditioning. We showed that reestablishing redox homeostasis in basolateral amygdale (BLA) after exposure to fear stressors determined the capacity of learned fear inhibition. Intra-BLA infusion of buthionine sulfoximine (BSO) to deplete the most important endogenous antioxidant glutathione (GSH) blocked fear extinction, whereas intra-BLA infusion of dithiothreitol or N-acetylcysteine (a precursor of GSH) facilitated extinction. In electrophysiological studies conducted on transverse slices, we showed that fear stressors induced redox-dependent inhibition of NMDAR-mediated synaptic function, which was rescued by extinction learning or reducing agents. Our results reveal a novel pharmacological strategy for reversing impaired fear inhibition and highlight the role of GSH in the treatment of psychiatric disorders.
Similar content being viewed by others

Introduction
Fear-related anxiety disorders, such as social phobia, posttraumatic stress disorder and panic disorder, are highly prevalent psychiatric conditions that are still inadequately treated. Exposure to traumatic events provokes an uncontrollable state of fear, which associates traumatic events with previous sensitizing experiences to inhibit memory extinction and is a characteristic of fear-related diseases [1,2,3]. While somewhere between 50% and 84% of the general population will experience a traumatic event, most individuals are resilient to these stressors, and ~28% of the population will go on to develop anxiety [4, 5]. Like humans, animals also exhibit individual differences in Pavlovian extinction paradigms [6, 7]. However, much less is known about the mechanism underlying individual differences in fear inhibition.
Fear extinction does not erase the original fear memories [8, 9]; rather, it allows the old memories to be updated based on new information [10], a process that relies on interactions between the hippocampus and emotional input provided by the basolateral amygdala (BLA) [11]. It is generally believed that after fear conditioning, memory-encoding synaptic circuits, such as the lateral amygdala (LA), are persistently but reversibly modified and become less plastic [12,13,14]. N-methyl-D-aspartate receptors (NMDARs) in the BLA have been identified to be crucial in learning during fear memory acquisition, consolidation and extinction [15,16,17]. Pharmacological antagonism of NMDARs profoundly impairs extinction [18, 19], whereas augmenting NMDARs through d-cycloserine and brain-derived neurotrophic factor (BDNF) facilitates extinction [20,21,22]. It should be noted that although NMDAR-dependent long-term potentiation (LTP) in the thalamo-LA pathway is an essential factor in auditory fear conditioning [23,24,25,26], fear-induced persistent potentiation in glutamatergic synaptic transmission [23,24,25] occludes further LTP [27, 28], which may hinder the remaining capacity for new information storage. To generate additional plastic synaptic circuits for learned fear inhibition, depotentiation of LTP modified by fear may be beneficial [29]. Our previous work identified potential extinction-promoting agents that induce chemical LTP or facilitate LTP depotentiation in the BLA, such as calcitonin gene-related peptide and leptin [30, 31].
Oxidative stress, which results from an imbalance between antioxidant defenses and reactive oxygen species (ROS), contributes to age-related memory deficits [32, 33]. Reports from our laboratory and other groups indicate that oxidative stress impairs NMDAR function in the hippocampus and replenishes cellular reducing ability through thiol agents such as dithiothreitol (DTT) and glutathione (GSH), reversing aging-associated synaptic dysfunctions [34,35,36]. Regarding fear-related behavior, some redox-related genes, such as GSH reductase 1 and 3-mercaptopyruvate sulfurtransferase, are involved in the genesis of anxiety [37, 38]. Interestingly, in recent years, increasing evidence from both animal models and clinical data has suggested that oxidative stress may underlie the pathophysiological mechanisms of psychiatric diseases, including depression, schizophrenia and drug addiction [39,40,41,42,43,44]. In human anxiety disorders such as panic disorder, obsessive-compulsive disorder, specific phobias and generalized anxiety disorder, there are many conflicting clinical findings about oxidative biomarkers [45,46,47]. In the present study, we found that redox homeostasis controls fear extinction, providing new insight into fear-related anxiety disorder therapy.
Materials and methods
Animals
Male Sprague-Dawley rats (3–4 months old, weighing 250–300 g) were used in this study. All of the animals were housed under a controlled 12 h light/dark cycle at a constant temperature (22 ± 2 °C) and humidity (50% ± 10%) and were given ad libitum access to food and water. The use of animals for all experimental procedures in accordance with the Guide for Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health and approved by the Animal Welfare Committee of Huazhong University of Science and Technology.
Agents
DTT, 5-5′-dithio-bis (2-nitrobenzoic acid) (DTNB) and N-acetylcysteine (NAC) were purchased from Sigma (St. Louis, MO, USA). Buthionine sulfoximine (BSO) and diamide were purchased from J&K (Beijing, China). Other general reagents were purchased from commercial suppliers. NAC and DTNB were prepared as a stock solution in DMSO. The final concentration of DMSO was <0.05%. DTNB and DTT were diluted directly into the bath solution to achieve the final concentration.
Fear conditioning
All training and testing were carried out in a sound-attenuating box (32 cm × 26 cm × 30 cm), and electric foot shock was delivered via the stainless steel grid floor as described previously [30]. During the experiment, a video camera was used to record each rat’s behavior. Before the experiment, rats were randomly separated into groups. Each group received a drug or the vehicle according to the schemes shown in the figures. The experiment was conducted over 3 days. On day 1 (habituation and fear conditioning training day), the rats were exposed to box A (context A) for 3 min. Then, rats were presented with four tones (conditional stimulus (CS), 65 dB, and 29 s), each terminating with a 0.75/1.2 mA foot shock lasting 1 s (unconditional stimulus (US)); the inter trial interval between each CS–US was 30 s. After training, rats were transferred from the box to their home cage immediately. Day 2 (extinction training day): rats were given 24 presentations of the CS tone (65 dB, 30 s; every set of 4 tones was considered a trial for data analysis) in the absence of the US (in context B), and the inter trial interval was 10 s. Day 3 (extinction recall day): rats were exposed to the CS (in context B) for 2.5 min along with 4 presentations of the CS tone (65 dB, 30 s).
The percentage of time spent freezing was measured during the tone presentation, which was recorded by Anilab version 3.1 software (Anilab Corporation, Ningbo, China). During the experiment, the activity of each rat was recorded continuously using the software described above [48]. Each chamber rested on a load cell that recorded chamber displacement in response to the motor activity of each rat, and the load cell activity was digitized at 3 Hz. In the experiment, freezing behavior was quantified before foot shock in the pre-US period and after foot shock on the conditioning day, as well as in the 10 min extinction training and test. Freezing behavior was scored only if the rat was immobile for at least 1 s.
Surgery and injection
Rats were anesthetized with sodium pentobarbital [60 mg/kg, intraperitoneally (i.p.)] and then mounted in a stereotaxic apparatus. A 22-gauge stainless steel guide cannula was bilaterally implanted dorsal to the LA region (−2.8 AP, ± 5.0 ML, −7.2 DV from bregma) and secured to the skull with steel jewelers’ screws and dental acrylic. Rats were allowed 5–7 days for recovery before experiments. When intracranial injection was performed, a 33-gauge injection cannula was used to replace the inner sealing wire and protruded 0.5 mm beyond the guide cannula. Drugs were infused into the LA at a rate of 0.25 μL/min with a total volume of 0.5 μL/side. The injection cannula was left for an additional 1.5 min before withdrawal to minimize back flow of the injected liquid along the injection track.
Measurement of pain threshold
Rats were placed individually into the conditioning chamber with an electric grid. After 3 min of retention, electric foot shocks (1 s) were applied starting with an intensity of 0.1 mA. The intensity was increased gradually by 0.1 mA (with pauses of 30 s between successive stimuli) until the animal showed the first signs of pain (flinching or jumping), and the corresponding intensity was considered the pain threshold [49].
Measurement of enzymatic and nonenzymatic antioxidants
After training or testing, the BLA tissue was dissected, homogenized, and analyzed. For the naïve group, BLA tissue was collected 0.5 h after home cage adaptation. For the Cond group, BLA tissue was collected 0.5 h after fear conditioning. For the Ext and Ext deficit groups, BLA tissue was collected 0.5 h after extinction recall. Tissues were stored at −80 °C, and the levels of oxidative biomarkers in all groups were assayed in lysates from BLA tissues simultaneously. Commercial kits purchased from Biovision, Inc. (Mountain View, CA, US) were used to examine the levels of GSH and glutathione disulfide (GSSG); kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) were used to assess the levels of malonaldehyde (MDA) and the activities of superoxide dismutase (SOD), GSH peroxidase (GPx) and catalase (CAT) as previously described [34, 50]. All procedures were conducted in accordance with the manufacturer’s instructions.
Total thiol and protein concentration assays
The total thiol assay was performed using the DTNB method described in our previous research [34]. In brief, protein samples were extracted from rat amygdala slices; then, the rat amygdala was homogenized in ice-cold extraction buffer consisting of 50 mM Tris-base (pH 7.4), 100 mM NaCl, 1% NP-40, 10 mM EDTA, 20 mM NaF, 1 mM PMSF, 3 mM Na3VO4 and protease inhibitors. The reaction was started by the addition of DTNB and then oxidized (disulfide), generating a yellow derivative (TNB) whose absorption was measured at 412 nm. The protein concentration of the supernatant was determined using a BCA protein assay kit (Pierce Biotechnology, Rockford, IL, USA) The results were reported as μmol TNB per mg of protein.
Electrophysiological recording
Transverse LA slices (350 μm) were obtained from adult rats as described in our previous studies with some modifications [49]. Briefly, brain slices were cut with a vibratome (VT 1000 S; Leica, Wetzlar, Germany) after recovery of the tissue in artificial cerebrospinal fluid (aCSF) for at least 1 h at room temperature (25 ± 1 °C). Then, an individual slice was transferred to a submerged recording chamber and superfused with aCSF at room temperature (30 ± 1 °C) at a rate of 3–4 mL/min. Field excitatory postsynaptic potentials (fEPSPs) were recorded in thalamic inputs in response to stimuli at a frequency of 0.033 Hz using a 3 M NaCl-filled glass electrode (3–5 MΩ) placed within the LA. To evoke LTP, high-frequency stimulation (HFS) was induced after recording fEPSPs for at least 15 min. HFS consisted of three trains of 100 pulses at 100 Hz separated by 30 s and delivered at test intensity. A test with a single pulse was then resumed for 90 min to determine the level of stable LTP. LTP data were acquired without parallel recordings from a nontetanized control pathway. NMDAR-dependent LTD was induced by low-frequency stimulation (LFS) (900 pulse/1 Hz) after recording at baseline for 20 min. Then, fEPSPs were continuously recorded from the brain slices for another 60 min. The input–output relationship (IOR) for synaptic transmission was constructed by varying the intensity of single-pulse stimulation. Paired stimuli (25-, 50-, 75-, and 100 ms intervals) were delivered to the lateral nucleus, and paired-pulse facilitation (PPF) was calculated as the ratio between the mean slope of the second fEPSP (fEPSP2) and that of the first fEPSP (fEPSP1).
Whole-cell patch-clamp recordings from BLA neurons were visualized with infrared optics using an upright microscope equipped with a 40× water-immersion lens and an infrared-sensitive CCD camera (BX51WI; Olympus) as previously described [51]. EPSCs were evoked by electrical stimulation of axons in the stratum radiatum (0.033 Hz) in the presence of picrotoxin (100 μM). The pipette (input resistance: 5–7 MΩ) solution consisted of 105 mM K-gluconate, 30 mM KCl, 10 mM HEPES, 10 mM phosphocreatine, 4 mM ATP-Mg, 0.3 mM GTP-Na, 0.3 mM EGTA, and 5 mM QX314 (pH 7.35). TTX (1 µM) was added to the bath solution when recording mEPSCs. NMDA-mediated EPSCs were recorded at −70 mV in 0.1 mM Mg2+ aCSF supplemented with the AMPAR antagonist 6-cyano-7-nitroquinoxaline- 2,3-dione (CNQX, 20 μM). Data were collected when series resistance fluctuated within 20% of the initial values (20–40 MΩ), were filtered at 1 kHz, and were sampled at 10 kHz.
Meta-analysis
Two independent researchers, G XL and W PF, searched for available studies published through March 2018 in the PubMed, Embase, Web of Science, CNKI and Wangfang databases. The search was performed by using the search terms: (Anxiety Disorder OR Disorder, Anxiety OR Disorders, Anxiety OR Neuroses, Anxiety OR Anxiety Neuroses OR Anxiety States, Neurotic OR Anxiety State, Neurotic OR Neurotic Anxiety State OR Neurotic Anxiety States OR State, Neurotic Anxiety OR States, Neurotic Anxiety) AND (Peroxidase, Glutathione OR Selenoglutathione Peroxidase OR Peroxidase, Selenoglutathione OR Glutathione Lipoperoxidase OR Lipoperoxidase, Glutathione) OR (Propanedial OR Malonyldialdehyde OR MDA OR Malonylaldehyde OR Sodium Malondialdehyde OR Malondialdehyde, Sodium) OR (Dismutase, Superoxide OR Erythrocuprein OR Hemocuprein OR Mn-SOD OR Mn SOD OR Manganese SOD OR Dismutase, Manganese Superoxide OR SOD, Manganese OR Mn-SOD OR Mn SOD) OR (Catalase T OR Mn Catalase OR Manganese Catalase OR Catalase A). The titles and abstracts of studies found by the search strategy were screened to determine whether the studies were potentially eligible for inclusion in this meta-analysis.
The studies fulfilling the initial screening were examined based on the inclusion criteria used in this analysis, including studies that (1) enrolled patients with anxiety disorder diagnoses such as social phobia, obsessive-compulsive disorder, PTSD, panic disorder and general anxiety disorder; (2) used blood samples to measure any of the following oxidative stress indices: GPx, MDA, SOD or CAT; (3) had a dataset that did not overlap with other studies; (4) enrolled patients diagnosed by a psychiatrist; and (5) assayed the blood biomarkers in HC as well.
The first purpose of the current meta-analysis was to compare levels of oxidative stress markers, including GPx, MDA, SOD and CAT, between patients with anxiety disorders and healthy control subjects. Second, we examined the biomarker levels of anxiety patients and relevant control subjects using subgroup analysis. For all analyses, P < 0.05 was considered statistically significant. All meta-analyses were completed with the statistical software R (http://www.r-project.org) with the meta package.
Statistical analysis
All analyses were performed using SPSS 18.0 software (SPSS Inc., Chicago, Illinois, USA), and data are presented as the mean ± SEM. All statistical results and tests used are included in the figure legends. Sample size was chosen based on prior experience. Data were evaluated by an independent samples t-test or one-way analysis of variance (ANOVA) as appropriate. To compare freezing rate at the time points relative to fear extinction or administration of drugs between groups, two-way repeated measures ANOVA was used. After ANOVA, least significant difference (LSD) post hoc analysis was used to compare the differences between groups. The correlation between GSSG/GSH and freezing rate was compared with Pearson correlation analysis. Other results from the fear conditioning test and the level of stable LTP(or LTD) were statistically evaluated using Student’s t test. A P value < 0.05 was considered significant.
Results
A meta-analysis of published clinical data supports a link between redox homeostasis and fear-related anxiety disorders
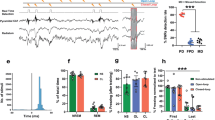
We first searched online databases for records about the association between redox homeostasis and fear-related anxiety disorders, including obsessive-compulsive disorder, posttraumatic stress disorder, general anxiety disorder, social phobia, and panic disorder. The search strategy yielded 275 results. There were 229 records left after duplicates were removed. Of these, 185 records were excluded because the study used nonhuman subjects (n = 161), the topic was irrelevant to our work (n = 9), or the article was not published in English (n = 15). Forty-four full-text studies were downloaded for eligibility screening. However, of these 44 studies, eight studies did not provide original data, five studies did not include healthy controls (HCs), eight studies were not published in peer-reviewed journals, and two studies used the same set of patients. Finally, 21 studies that assessed the association between oxidative stress biomarkers and fear-related anxiety disorders were included (the basic information is shown in Table S1). The process of the meta-analysis is presented in Fig. 1a.

a Flow diagram of the screened and included studies. b Forest plot of the studies on the association between MDA content in blood samples and fear-related anxiety by meta-analysis with the random-effects analysis. c Forest plot of the studies on the association between GPx activity in blood samples and fear-related anxiety by meta-analysis with the random-effects analysis. SMD = standardized mean differences.
MDA is an important biomarker of oxidative stress. Nineteen studies examined the association between MDA and fear-related anxiety disorders. In total, 643 patients and 638 healthy controls (HCs) were included. The difference between patients and HCs is shown as standardized mean differences (SMDs). The meta-analysis results indicated that patients with anxiety disorders had higher levels of MDA than HCs (SMD = 2.41, 95% CI, 1.58–3.24, P < 0.01, shown in Fig. 1b). The influential analysis showed that the result remained unchanged when any individual study was omitted (shown in Fig. S1), indicating that the result was stable. Next, we used a funnel plot to display the publication bias, as shown in Fig. S1. Egger’s linear regression test was used to evaluate the asymmetry of the funnel plot (t = 1.7179, df = 15, P value = 0.1064), which showed no significant publication bias. The I2 value showed large heterogeneity, so we used a random-effects model to examine the SMD of the analysis. We next used subgroup analysis to measure the MDA content in each subtype of anxiety disorder and found that all anxiety disorders, except obsessive-compulsive disorder (SMD = 1.16, 95% CI, −0.47–2.78), exhibited a strong and significant association with MDA content (shown in Table S2).
Ten studies were included in the analysis of the association between GPx activity and anxiety disorders. The results showed no significant difference in GPx activity between patients and HCs (SMD = 0.04, 95% CI, −0.91–0.98, P = 0.94, shown in Fig. 1c). As shown in Fig. S2a-b, no significant difference was found in the meta-analysis of either rCAT or SOD (SMD = −0.62, 95% CI, −1.54–0.30, P = 0.19; SMD = −0.22, 95% CI, −1.15–0.70, P = 0.67).
Sustained imbalance of redox homeostasis in the BLA of extinction-deficit rats
We next sought to determine the correlation between fear behavior and GSH redox homeostasis in the BLA. The scheme of the experiment for fear conditioning is shown in Fig. 2a. Rats were trained in context A on Day 1 (termed Cond) and in extinction training in context B on Day 2. The percentage of time spent freezing in context B was measured during tone presentation on day 3 (that of naïve or Cond was also measured). However, a proportion (12 out of 29) of rats that were trained on 1.2 mA exhibited a high freezing rate after extinction (freezing rate ≥50%, Fig. 2b). In our experiments, the mean freezing after extinction learning was 49% ± 4%.We defined animals with an average freezing duration greater than the median (50%) as having an extinction deficit (Ext deficit) according to previous studies [6] and use freezing >50% after extinction learning as a standard of extinction deficit. Previous reports also indicated that extinction training induced a fast decline in freezing in most animals, while a freezing level >50% persisted in high-anxiety rats or a genetic mouse model with a profound impairment in fear extinction [52, 53]. According to this standard, approximately one-third (12 out of 29) of rats were defined as having an extinction deficit, and this rate was close to that in a published study [7]. To eliminate the presence of the current intensity effect on the freezing response, we applied milder foot shock conditioning (0.75 mA) and examined extinction learning (Fig. S3a-b). The results were similar to those seen when stronger conditioning was employed: rats showed normal conditioning, and only a proportion of the rats (5 out of 20 rats) showed Ext deficit.

a Experimental protocols of the cue-induced fear conditioning paradigm. b The percentage of freezing during 4 CS-alone trials. The rats that showed a high freezing rate (≥50%) were defined as Ext Deficit. c, d GSH and GSSG contents in rat BLA tissues after fear conditioning were detected (n = 10). e, f GSH and GSSG contents were assayed in the hippocampus or mPFC of rats after fear conditioning (n = 8). g–k Oxidative biomarkers, including GPx, MDA, SOD, CAT and total thiol, were measured in rat BLA tissues after fear conditioning (n = 8–10). All data are expressed as the mean ± SEM. *P < 0.05, **P < 0.01 vs. the naïve group; #P < 0.05, ##P < 0.01 vs. the Cond group; $P < 0.05, $$P < 0.01 vs. the Ext group. Data were analyzed by one-way ANOVA with a post hoc LSD test.
We found that changes in the GSH level in fear-conditioned rats (Fig. S4a, ANOVA F(3,36) = 69.48, P < 0.01) were characterized by a significant and sustained decline at 0.5 (post hoc LSD, t = 13.13, P < 0.01 compared to the naïve group), 4 (post hoc LSD, t = 11.70, P < 0.01 compared to the naïve group) and 24 h after conditioning (post hoc LSD, t = 9.045, P < 0.01 vs. the naïve group). On the other hand, fear conditioning induced a significant increase in the GSSG level at 0.5 h after conditioning (ANOVA F(3,36) = 1.42, P > 0.05; post hoc LSD, t = 3.03, P < 0.01 compared to controls), but this level decreased (post hoc LSD, t = 2.33, P < 0.05 compared to that at 0.5 h) 24 h after conditioning (Fig. S4b), which may have resulted from a depletion of endogenous GSH levels.
Then, we collected the BLA tissue 0.5 h after home cage adaptation for the naïve group, 0.5 h after fear conditioning for the Cond group, and 0.5 h after extinction recall for the Ext and Ext deficit groups to observe the effect of extinction on oxidative biomarkers, including GSH, GSSG, GPx, MDA, total thiol, SOD and CAT. Tissues were stored at −80 °C, and the levels of oxidative biomarkers in all groups were assayed in lysates from BLA tissues. We found that after fear conditioning, most oxidative biomarkers examined in the BLA were altered, including a decrease in GSH levels (7.21 ± 0.52 nmol/mg protein in the naïve group vs. 3.60 ± 0.98 nmol/mg protein in the Cond group, n = 10, ANOVA F(3,36) = 49.10, P < 0.01, post hoc LSD, t = 11.48, P < 0.01, Fig. 2c) and an increase in GSSG levels (0.14 ± 0.04 nmol/mg protein in the naive group vs. 0.23 ± 0.03 nmol/mg protein in the Cond group, n = 10, ANOVA F(3,36) = 11.13, P < 0.01, post hoc LSD, t = 5.58, P < 0.01, Fig. 2d). Moreover, GSH and GSSG levels remained unchanged in the hippocampus and mPFC in response to fear training (Fig. 2e, t = 0.29 and 0.16, p > 0.05; Fig. 2f, t = 0.27 and 1.28, P > 0.05; n = 8). GPx activity (32.5 ± 4.2 pmol·min−1·mg−1 protein in the control group vs. 48.7 ± 3.2 pmol·min−1·mg−1 protein in the Cond group, n = 10, ANOVA F(3,36) = 32.55, P < 0.01, post hoc LSD, t = 9.08, P < 0.01, Fig. 2g) and MDA content (1.23 ± 0.17 nmol/mg protein in the control group vs. 1.72 ± 0.25 nmol/mg protein in the Cond group, n = 6, ANOVA F(3,20 = 4.06, P < 0.05; post hoc LSD, t = 2.83, P < 0.05, Fig. 2h) in the BLA also increased after fear training. The activity levels of two additional antioxidant enzymes, SOD and CAT (Fig.2i-j), were examined. Only the activity level of CAT exhibited a significant increase in response to fear conditioning (3.6 ± 0.7 U/mg protein in control group vs. 5.2 ± 0.7 U/mg protein in the Cond group, n = 8, ANOVA, F(3,28) = 4.50, P < 0.05; post hoc LSD, t = 2.59, P < 0.05). After extinction, the changes in the majority of altered oxidative biomarkers were reversed (Fig. 2c-k). However, in the Ext deficit groups, a decreased GSH level (6.11 ± 0.61 nmol/mg protein in the Ext group and 4.37 ± 0.53 in the Ext deficit group, n = 10, post hoc LSD, t = 3.94, P < 0.01, Fig. 2c), increased GPx activity (36.1 ± 3.2 pmol·min−1mg−1 protein in the Ext group and 42.3 ± 3.5 pmol·min−1·mg−1 protein in the Ext deficit group, n = 10, post hoc LSD, t = 2.59, P < 0.05, Fig. 2g) and reduced total thiol content (52.3 ± 3.7 μmol/mg protein in the Ext group and 44.2 ± 3.2 in the Ext deficit group, n = 8, ANOVA F(3,28) = 8.52, P < 0.01; post hoc LSD, t = 2.16, P < 0.05, Fig. 2k) were observed compared to those in the Ext group. These data indicate that altered oxidative biomarkers may underlie resistance to fear extinction.
Redox imbalance in BLA through disruption of GSH homeostasis impairs extinction
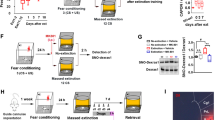
Previous reports indicated that GSH peroxidase and/or GSH reductase play important roles in the genesis of anxiety [37, 45,46,47]. The decrease in GSH levels after fear stressor exposure lasted for at least 24 h (Fig. S4). We normalized the ratio of GSSG/GSH levels in different groups and found that the GSSG/GSH ratio in the extinction deficit group was higher than that in the extinction group (Fig. 3a, 1.63 ± 0.22 in the Ext Deficit group vs. 1.26 ± 0.20 in the Ext group, n = 10, ANOVA, F(3,36) = 21.02, P < 0.01, post hoc LSD, t = 2.34, P < 0.05). Next, we performed a Pearson’s correlation test and found that the correlation between GSSG/GSH levels and freezing rate was significant (Fig. 3b, Pearson correlation coefficient = 0.516, P < 0.01), indicating that rats with a high GSSG/GSH ratio exhibited a high freezing rate. To determine whether altered GSH homeostasis in the BLA contributes to deficits in fear extinction, we used chemical tools to deplete GSH levels in the BLA. BSO is an inhibitor of γ-glutamylcysteine synthetase, the rate-limiting enzyme in the GSH synthesis pathway, that leads to GSH depletion [54, 55]. After fear training, aCSF or BSO was injected into the BLA (once per day, with the first time being 24 h after training and the last time being half an hour before extinction, as shown in Fig. 3c), and extinction was performed after 3 days. BSO injection significantly depleted GSH in the BLA (from 6.91 ± 0.52 nmol/mg protein in the aCSF group to 4.98 ± 0.31 nmol/mg protein in the BSO group, n = 12, t = 11.32, P < 0.01, Fig. 3d). Two-way repeated measures ANOVA revealed a main effect of trials (F(5,18 = 21.547, P < 0.01), with post hoc tests confirming a significant increase in freezing in the last 3 trial blocks (Fig. 3e, P < 0.05, P < 0.01). The freezing rates on extinction recall days increased from 45% ± 7% in the aCSF group to 72% ± 8% in the BSO group (n = 12, t = 6.19, P < 0.01, Fig. 3f). Diamide, a thiol-oxidizing agent that increases protein glutathiolation [56, 57], was used to evaluate the relationship between the GSH system and fear extinction. Injection of diamide significantly depleted GSH in the BLA (Fig. 3g, from 7.34 ± 0.62 nmol/mg protein in the aCSF group to 5.21 ± 0.57 in the diamide group, n = 12, t = 7.77, P < 0.01). Repeated measures ANOVA identified a main effect of trials (Fig. 3h, F(5,18) = 27.382, P < 0.01), with post hoc tests confirming a significant increase in freezing in the last 2 trial blocks (P < 0.01). The freezing rates on the extinction testing day increased from 47% ± 6% in the control group to 80% ± 5% in the diamide group (n = 12, t = 10.70, P < 0.01, Fig. 3i). These data suggest that GSH homeostasis may play a critical role in fear extinction.

a The normalized ratio of GSSG/GSH is illustrated by the column graph (n = 10, **P < 0.01 vs. the naïve group, #P < 0.05 vs. the Cond group). b The association between the freezing rate and the GSSG/GSH ratio was assayed by Pearson correlation test (r = 0.516, P < 0.01). c The experimental protocols of BSO (1 mM, 0.5 μL) or diamide (300 μM, 0.5 μL) intra-BLA infusion. Rats received an infusion once per day (the first time was 24 h after training, and the last time was half an hour before extinction). d The GSH assay was performed 30 min after extinction recall (n = 12, **P < 0.01 vs. the aCSF group). e, f Rats received fear extinction in context B at 24 h after the last intra-BLA infusion of BSO or aCSF (n = 12, F(5,18) = 21.547, P < 0.01 for time × group interaction, post hoc LSD, *P < 0.05, **P < 0.01). The freezing rate on extinction recall days was measured (n = 12, **P < 0.01 vs. aCSF group). g The GSH assay was performed at 30 min after extinction recall (n = 12, **P < 0.01 vs. the aCSF group). h Rats received fear extinction in context B at 24 h after the last intra-BLA infusion of diamide or aCSF (n = 12, F(5,18) = 27.382, P < 0.01 for time × group interaction, post hoc LSD, **P < 0.01). i Fear memory was assessed at 24 h after the extinction session by placing the animals in context B and measuring conditional freezing during tone presentation (n = 12, **P < 0.01 vs. aCSF group). All data are expressed as the mean ± SEM. Data were analyzed by one-way ANOVA with a post hoc LSD test.
Redox imbalance in the BLA triggers NMDAR hypofunction
In our previous study, we found that oxidative status is important for NMDAR-dependent synaptic plasticity [34]. Herein, we tested whether the disruption of GSH homeostasis underlies the impairment of LTP in the BLA. After incubation with aCSF, DTT or DTNB for 30 min, the GSH/GSSG content of BLA tissue was measured. The GSH content changed from 7.42 ± 0.52 nmol/mg protein to 10.23 ± 1.21 nmol/mg protein (DTT group) and 4.78 ± 1.76 nmol/mg protein (DTNB group) (Fig. 4a, n = 8–10 from at least three rats, F(2,24) = 77.41, P < 0.01; post hoc LSD t = 2.50, P < 0.05 vs. control; t = 4.96, P < 0.01 vs. control). GSSG levels changed significantly in the presence of DTT and DTNB, from 0.12 ± 0.03 nmol/mg protein in the control group to 0.07 ± 0.03 nmol/mg protein and 0.21 ± 0.05 nmol/mg protein, respectively (Fig. 4b, n = 8–10, ANOVA F(2,24) = 26.88, P < 0.01; post hoc LSD, t = 2.35, P < 0.05 vs. control and t = 4.99, P < 0.01 vs. control).

a–b The GSH/GSSG contents in rat BLA slices were measured after 30 min of perfusion in aCSF with DTT or DTNB (n = 8–10, *P < 0.05, **P < 0.01). c Slices were treated with DTT (50 μM) or DTNB (100 μM) 10 min after recording. Then, the time-course of long-term potentiation (LTP) was induced by HFS in the BLA (n = 8–10 slices). d Bar graph illustrating the mean increase in fEPSPs slope averaged from the last 15 min in each group (**P < 0.01 vs. control). e I-O curve of DTT-, DTNB- or vehicle-incubated slices is shown (n = 8 slices from 3 rats at least). f Paired-pulse facilitation of DTT-, DTNB- or vehicle-incubated slices is shown (n = 8 slices from 3 rats at least). Insets are representative fEPSPs evoked by paired pulses delivered 25 ms apart (n = 8 slices). g Example data showing that DTT (50 μM) or DTNB (100 μM) application significantly changed evoked NMDAR-mediated currents in the thalamo-LA pathway. h NMDAR-EPSCs recorded at the time points indicated on the graph (n = 8). i Bar diagram showing the averaged amplitudes of NMDAR-EPSCs in each group (n = 8 slices from 5 rats for each group; **P < 0.01 vs. control). All data are mean ± SEM. Data were analyzed by one-way ANOVA with a post hoc LSD test.
As shown in Fig. S5a-b, bath application of DTT (50 μM) or DTNB (100 μM) did not induce any alterations in basal fEPSPs in rat slices (n = 8 slices from at least 3 rats). Next, we confirmed this finding using HFS, consisting of five trains at 100 Hz for 1 s with 90 s intermission between trains to introduce LTP into the BLA. A significant change in NMDAR-dependent LTP in the BLA in response to DTT or DTNB treatment (control 133% ± 4%; DTT 162% ± 3%; DTNB 102% ± 4%,ANOVA F(2,21) = 290.40, P < 0.01; post hoc LSD, t = 12.37 or 11.73, P < 0.01 vs. control group, Fig. 4c-d) was observed. The IOR curves were compared among the control, DTT-treated and DTNB-treated groups, and no significant change was observed (Fig. 4e). PPF, an indicator of presynaptic neurotransmitter release, was measured, and no obvious change was observed (Fig. 4f), indicating that the impairment of LTP is unlikely to be ascribed to the alteration of presynaptic neurotransmitter release but rather to a change in postsynaptic responsiveness. We found that DTT significantly increased the amplitude of NMDAR currents to 168% ± 19% of baseline (n = 8 from 5 rats, P < 0.01). Moreover, NMDAR currents in the DTNB-treated group decreased to 58% ± 6% of baseline (n = 6 from 3 rats, ANOVA F(2, 16) = 14.27, P < 0.01; post hoc LSD, t = 3.02 or 2.87, P < 0.01, Fig. 4g-i).
Reversal of NMDAR hypofunction in the BLA by reestablishing redox homeostasis predicts extinction
NMDAR function is essential for fear extinction. Basolateral synapses on intercalated neurons can express NMDA-dependent LTP and LTD, which may serve as a critical locus of plasticity for the extinction of conditioned fear responses [25, 58]. Previous reports have indicated that fear conditioning inhibits NMDAR-dependent LTP in the BLA [28]. In our study, we also confirmed this finding using HFS-induced LTP in the BLA. Consistent with previous reports, a significant suppression of NMDAR-dependent LTP in the BLA was observed after fear conditioning (control 142% ± 3%; Cond group 117% ± 8%, ANOVA F(3, 32) = 38.12, P < 0.01; post hoc LSD, t = 7.51, P < 0.01 vs. control group, Fig. 5a, b). Furthermore, we found that extinction regenerated NMDAR-dependent LTP in the BLA, but in the Ext deficit rats, sustained inhibition of NMDAR-dependent LTP was observed (control 142% ± 3%; Ext deficit group 113% ± 5%, n = 10, post hoc LSD, t = 8.58, P < 0.01 vs. control group, Fig. 5a, b). Considering the possible role of redox status in extinction defects, we investigated the effect of DTT on LTP inhibition in extinction-deficit rats. As shown in Fig. 5c and d, DTT rescued LTP from 115% ± 8% in the Ext deficit group to 139% ± 9% in the Ext deficit + DTT group (n = 8, t = 3.78, P < 0.05). The IOR was not significantly altered compared to that of the naïve group (n = 8 slices from at least 3 rats; Fig. S6a). The PPF was measured, and no obvious change was observed (Fig. S6b), indicating that presynaptic neurotransmitter release was not significantly altered.

a Time course of LTP induced by HFS in naïve rats (n = 8 slices), rats that received fear conditioning (Cond, n = 8 slices) and rats that received extinction learning (Ext, n = 10 slices). b Bar graph illustrating the mean increase in fEPSP slope averaged from the last 15 min in each group (**P < 0.01 vs. naïve, #P < 0.05 vs. Cond). c Time course of long-term potentiation (LTP) induced by HFS in rats in the Ext group (n = 10 slices) or Ext deficit + DTT group (n = 10 slices). d Bar graph illustrating the mean increase in fEPSP slope averaged from the last 15 min in each group (**P < 0.01 vs. Ext deficit). e Time-course of long-term depression (LTD) induced by LFS in naïve rats (n = 8 slices) and in rats that received fear conditioning (Cond, n = 8 slices). f Bar graph illustrating the mean fEPSPs slope averaged from last 15 min in each group (**P < 0.01 vs. Naïve group). g Time-course of LTD induced by LFS in rats in the Ext group (n = 8 slices), Ext deficit group (n = 8 slices), or Ext deficit + DTT group (n = 8 slices). h Bar graph illustrating the mean fEPSPs slope averaged from the last 15 min in each group (**P < 0.01 vs. Ext group, ##P < 0.01 vs. Ext deficit group). i, j GSH and GSSG contents were detected 30 min after treatment with different drugsfor 30 min (n = 8 slices, **P < 0.01 vs. saline group, #P < 0.05, ##P<0.01 vs. BSO group). k Time course of LTP induced by HFS in rat brain slices treated with saline (n = 8 slices), BSO (n = 8 slices), diamide or BSO + DTT (n = 8 slices). l Bar graph illustrating the mean increase in fEPSP slope averaged from the last 15 min in each group (**P < 0.01 vs. saline, ##P < 0.01 vs. BSO). All slices used in the electrophysiological experiments were harvested from at least 4 rats. Student’s t test and one-way ANOVA with post hoc LSD test were used for analysis as appropriate. All data are expressed as the mean ± SEM.
Then, we observed the effect of fear conditioning and extinction on LTD. After fear conditioning, we failed to induce NMDAR-dependent LTD in the BLA by a LFS at 1 Hz for 15 min (naïve group 82% ± 5%, Cond group 100% ± 3%; n = 8, t = 10.80, P < 0.01 vs. control group, Fig. 5e, f). Likewise, extinction regenerated NMDAR-dependent LTD in the BLA, but in the Ext deficit rats, sustained inhibition of NMDAR-dependent LTD was observed (Ext group 79% ± 4%; Ext deficit group 94% ± 5%, n = 8, ANOVA F(2, 21) = 25.84, P < 0.01; post hoc LSD, t = 5.20, P < 0.01 vs. control group, Fig. 5g, h). Considering the possible role of redox status in extinction defects, we investigated the effect of DTT on LTD impairments in extinction-deficit rats. DTT rescued LTD from 94% ± 5% in the Ext deficit group to 76% ± 4% in the Ext deficit + DTT group (n = 8, post hoc LSD, t = 6.89, P < 0.05, Fig. 5g, h).
We further investigated whether redox imbalance mimics the effect of fear conditioning on LTP. BSO (1 mM) or diamide (300 μM) was added to the perfusion buffer of rat brain slices 30 min before recording. The GSH content changed from 7.34 ± 0.62 nmol/mg protein to 5.31 ± 0.71 nmol/mg protein (BSO group) and 5.21 ± 0.57 nmol/mg protein (diamide group) (Fig. 5i, n = 8 from 3 rats at least, ANOVA F(3, 28) = 18.11, P < 0.01; post hoc LSD, t = 5.40 or 5.36, P < 0.01). GSSG levels significantly increased in the presence of BSO or diamide, from 0.12 ± 0.03 nmol/mg protein in the control group to 0.21 ± 0.04 nmol/mg protein and 0.24 ± 0.06 nmol/mg protein, respectively (Fig. 5j, n = 8–10 from 3 rats, ANOVA F(3,31) = 13.31, P < 0.01; post hoc LSD, t = 39.58 or 55.34, P < 0.01). However, neither BSO nor diamide changed the basal fEPSP in rat slices (Fig. S5c-d, n = 8 slices from at least 3 rats).
After BSO/diamide incubation, a significant change in NMDAR-dependent LTP in the BLA was observed (control 133% ± 2%; BSO 107% ± 9%; diamide 102% ± 6%, n = 8 slices from at least 3 rats, ANOVA F(3, 28) = 38.69, P < 0.01; post hoc LSD, t = 6.55 or 7.47, P < 0.01 vs. control group, Fig. 5k, l). Next, we tested whether DTT could reverse GSH depletion by BSO. As shown in Fig. 5g and h, DTT increased the GSH content from 5.31 ± 0.71 nmol/mg protein to 7.78 ± 1.26 (n = 8, post hoc LSD, t = 5.06, P < 0.01, BSO + DTT group vs. BSO group) and decreased the GSSG content from 0.21 ± 0.04 nmol/mg protein to 0.14 ± 0.02 (n = 8, post hoc LSD, t = 2.41, P < 0.05, BSO + DTT group vs. BSO group). Next, bath application of the reductant DTT (50 μM) 10 min after recording reversed the BSO-induced impairment of plasticity in brain slices. The amplitude of LTP was reversed from 107% ± 9% to 137% ± 2% (n = 8 from 4 rats, F(3, 28) = 38.69, P < 0.01; post hoc LSD, t = 7.64, P < 0.01, BSO + DTT group vs. BSO group, Fig. 5k-l). Moreover, the IOR and PPR of the BSO, diamide and BSO + DTT groups remained unchanged (n = 8–10, Fig. S6c-d).
Pharmacologically reestablishing GSH homeostasis in the BLA facilitates extinction of cued fear memory
Subsequently, we investigated whether targeting the GSH imbalance in the BLA facilitates the extinction of cued fear memory. First, we observed the effect on fear extinction by supplementation with NAC in drinking water, which has been suggested to increase GSH in vivo [59, 60]. As shown in Fig. 6a, NAC (200 mg/L) or vehicle was added to drinking water, which was supplied ad libitum for 3 days after fear conditioning. After the rats drank water with NAC or vehicle for 3 days, extinction was performed. The GSSG content in the NAC group (0.14 ± 0.03 nmol/mg protein) was significantly lower than that in the vehicle group (0.18 ± 0.02 nmol/mg protein, n = 10, ANOVA, F(3, 36) = 36.10, P < 0.01; post hoc LSD, t = 5.77, P < 0.01, Fig. 6c). Two-way repeated measures ANOVA revealed a main effect of trials (n = 12 and 29, F(5,35) = 43.575, P < 0.01), with post hoc tests confirming a significant increase in the freezing rate in trials 2 and 3 (P < 0.05, P < 0.01, Fig. 6d). After extinction, the freezing rate decreased significantly in the Ext + NAC group (49% ± 4% in the Ext + vehicle group, 35% ± 3% in the Ext + NAC group, n = 12–29 in each group, ANOVA, F(3, 78) = 85.21, P < 0.01; post hoc LSD, t = 2.17, P < 0.05 Ext + vehicle vs. Ext + NAC, Fig. 6e). During extinction, we examined the effect of DTT infusion into the BLA on fear extinction to confirm the role of GSH in the BLA in contrast to other brain areas (Fig. 6f). Extinction training was performed 3 days after DTT infusion (the first infusion was 24 h after fear conditioning, and the last infusion was half an hour before fear extinction). DTT did not affect GSH levels (Fig. 6g), but it mimicked the redox function of GSH. As shown in Fig. 6h, two-way repeated measures ANOVA identified a main effect of trials (F(5,18) = 21.848, P< 0.01), with post hoc tests confirming a significant increase in freezing rate in trials 3 and 4 (P < 0.05, P < 0.01). The freezing rate on extinction recall day decreased significantly in the DTT group (44% ± 7% in aCSF group vs. 32% ± 6% in DTT group, n = 12, t = 2.56, P < 0.05, Fig. 6i).

a Experimental protocols of NAC/vehicle treatment. NAC or vehicle was added to the drinking water of rats for 3 days. b,c GSH and GSSG contents were detected after fear conditioning (n = 10, *P < 0.05, **P < 0.01 vs. naïve group; #P < 0.05, ##P < 0.01 vs. Cond group). d Rats in each group received auditory fear extinction in context B after 3 days of treatment with NAC/vehicle water (n = 12 and 29, two-way ANOVA F(5,35) = 43.575, P < 0.01, post hoc LSD test *P < 0.05, **P < 0.01). e The percentage of freezing during 4 CS-alone trials (n = 12–29, $P < 0.05 Ext + NAC vs. Ext+Vehicle). f Experimental protocols of rats with DTT or aCSF intra-BLA infusion. g DTT intra-BLA infusion reversed the change in GSH content in the BLA. The GSH assay was performed 30 min after extinction recall. h Rats of each group received auditory fear extinction in context B after a 3-day treatment with NAC/vehicle water (n = 12, two-way ANOVA, F(5,18) = 21.848, P < 0.01, post hoc LSD test *P < 0.05, **P < 0.01). i Fear memory was assessed 24 h after the extinction session by placing the animals in context B and measuring conditional freezing during tone presentation (n = 12, *P < 0.05). All data are mean ± SEM. One-way or two-way repeated measures ANOVA was used for analysis as appropriate, and the LSD method was used as a post hoc test.
To exclude the possibility that nociception is altered by redox status, pain thresholds during electric foot shock stimulation were examined. There were no differences in vocalization or jump response to gradually increased intensity of electric shock among rats in the control, BSO-treated or diamide-treated groups (n = 8, Fig. S7a), indicating that the impairment of fear memory is not due to pain sensitivity alterations. Similarly, no significant difference was observed between rats in the control, DTT-treated or NAC-treated groups (n = 8, Fig. S7b).
Discussion
Emerging evidence has revealed that oxidative stress may be involved in the pathophysiology of psychiatric diseases; however, much less is known about the mechanisms coupling oxidative stress with aberrant behavior. In the present study, we demonstrated that impaired GSH homeostasis in the BLA, a brain region that is crucial in the regulation of fear memory, blocks the extinction of fear memory by triggering redox-dependent NMDAR hypofunction. Importantly, pharmacologically reestablishing GSH homeostasis facilitated fear extinction, which may represent a novel strategy for therapy in panic disorder and other psychiatric diseases (Fig. 7). Further support for this hypothesis comes from a finding of our comprehensive meta-analysis of published data indicating that oxidative biomarkers, such as MDA, were significantly elevated in the blood of patients with fear-related anxiety disorders. Our data highlight the therapeutic and preventive value of GSH supplementation in fear-related psychiatric diseases.

Hypothesis for the role of redox homeostasis underlying individual differences in learned fear inhibition.
Fear conditioning is a compelling platform for investigating fear-related behavior. In this paradigm, a neutral CS, such as a tone, acquires the capacity to induce fear responses after being associated with an aversive US, such as a foot shock. Fear extinction, a process wherein the CS is repeatedly presented in the absence of the US to diminish conditioned fear responses, has proven valuable as a translational assay for studying the mechanisms underlying cognitive behavioral therapy of fear-related diseases [2, 3]. We trained rats with 1.2 mA electronic stimuli on the fear training day. Approximately one-third of the rats exhibited a freezing rate >50% after extinction, which was consistent with previous studies [7] and defined as extinction deficits. We focused on fear-related oxidative stress in the BLA because most available evidence points to the BLA as a major site of subsequent plasticity changes related to fear memory and extinction [25, 58, 61,62,63]. Control over fear behavior relies on the activation of specific prefrontal projections to the BLA, which may excite more principal neurons in the BLA and evoke fear responses. The principal neurons of the BLA are glutamatergic, and their excitation may trigger glutamate-dependent ROS generation via various pathways [64,65,66]. In our study, a similar increase in MDA levels in experimental extinction deficits and clinical fear-related anxiety disorders indicates that redox status may represent a potential mechanism underlying the individual differences in the development of uncontrollable fear in responses to traumatic stressors. Interestingly, although all fear-exposed rats displayed elevated oxidative stress in the BLA, after extinction learning, increases in the levels of oxidative biomarkers, such as GSH, GSSG, GPx and total thiols, were reversed in the extinction rats but not in the extinction-resistant rats. Levels of the main endogenous antioxidant, GSH, decreased with the freezing rate. Therefore, we hypothesized that the recovery of redox homeostasis in the BLA may be essential for fear extinction. This point was confirmed by the phenomenon in which compounds that mimic the impairment of GSH homeostasis in extinction-resistant rats, such as BSO, an inhibitor of γ-glutamylcysteine synthetase, or diamide, a selective thiol-oxidizing agent, blocked fear extinction. Thus, impairment of GSH homeostasis may underlie the mechanism of resistance to fear extinction. It has been reported that restoring GSH levels may represent a therapeutic strategy for diseases such as Alzheimer’s disease and cancer [25, 59]. In the present study, pharmacological reestablishment of GSH homeostasis in the BLA by DTT injection and NAC supplementation facilitated fear extinction in rats, raising the possibility of reestablishing GSH homeostasis in the therapy and prevention of mental illnesses.
NMDARs in the BLA have been identified as crucial targets for facilitating fear extinction. In recent years, augmenting NMDARs with different pharmacological agents, including D-cycloserine, BDNF, pituitary adenylate cyclase-activating polypeptide, CGRP and leptin, to erase fear has drawn increasing interest from researchers seeking to explore the therapeutic drugs against fear-related disorders [20,21,22, 30, 31, 66, 67]. In parallel to a substantial number of pharmacological findings, much less is known about the alteration of NMDARs underlying fear-related psychiatric diseases. It has been demonstrated that learning-induced changes in NMDAR subunit composition may serve as a mechanism for maintaining fear memory and preventing fear destabilization [48, 68,69,70]. In fear conditioning, very little is known about the role of endogenous changes in NMDAR subunits. It has long been recognized that NMDARs are sensitive to redox status. Converging lines of evidence suggest that oxidative stress inhibits NMDAR function via direct modifications, whereas thiol reduction augments NMDAR activity [71,72,73]. Our previous studies revealed that reversal of redox status re-established hippocampal synaptic plasticity in aged animals [34]. In this study, we found that NMDARs in the BLA were sensitive to redox status, which was consistent with previous work in other brain regions. Fear training may induce redox-dependent modifications of NMDARs in the BLA, including NR1 and NR2A, which mediate the hypofunction of NMDARs, as indicated by impaired NMDAR-dependent LTP. The findings that DTT regenerated NMDAR-dependent LTP in the BLA of extinction-deficit rats confirmed that redox status plays a critical role in NMDAR hypofunction.
Links between synaptic plasticity in the LA and fear learning are well established. Fear conditioning induces LTP-like changes in thalamo-LA synaptic transmission [23,24,25], which conveys specific CS information to the LA in discriminative fear learning [26]. Both fear conditioning- and LTP-induced plasticity share a common molecular mechanism. Therefore, the molecular basis of NMDAR-dependent LTP is vital for generating auditory fear conditioning. It should be noted that auditory fear conditioning occludes LTP at the thalamo-LA pathway [27, 28]. Memory-encoding synaptic circuits, such as the LA, are reversibly modified after fear conditioning and become less plastic, consistent with fear-occluded LTP induction [12,13,14]. These alterations may hinder the remaining capacity of new information storage. Extinction is a learning process that depends on parallel NMDA-dependent learning that competes with the first process. Thus, reversal of fear-triggered modifications in synaptic plasticity of the BLA may facilitate extinction. Our previous work identified extinction-promoting agents by inducing chemical LTP with agents such as calcitonin gene-related peptide and leptin or facilitating LTP depotentiation in the BLA [29, 30]. In this study, we observed that fear conditioning reversibly inhibited LTP in the thalamo-LA pathway. In the extinction rats, LTP in the thalamo-LA pathway was recovered. However, in the extinction-deficit rats, sustained inhibition of LTP was observed, which might be due to the persistent redox modifications of NMDARs. In contrast, DTT reversed NMDAR-dependent LTP in the BLA of extinction-resistant rats and facilitated fear extinction. These results indicate that an intrinsic linkage may exist between these two parallel findings. However, more evidence is required to confirm these findings.
LTD or the depotentiation of fear-induced LTP in the BLA are also thought to be a process central to the extinction of fear memories [29, 74]. We found that, like LTP, extinction regenerated NMDAR-dependent LTD in the BLA, but in Ext-deficient rats, sustained inhibition of NMDAR-dependent LTD was observed. DTT rescued LTD from 94% ± 5% in the Ext deficit group to 76% ± 4%, indicating that a redox-dependent mechanism exists. Although NMDA hypofunction may be involved in deficits in LTD, the mechanisms may be multifactorial because both mGluRs and the endocannabinoid system play key roles in the induction of LTD [74, 75]. Considering the beneficial effects of depotentiation of fear-modified LTP on extinction, the effect of reductants on LTD may contribute to the extinction-promoting activity.
In the last two decades, some advances in translational research have diminished the gaps between basic neuroscience and clinical psychiatry in the treatment of fear-related anxiety disorders. For instance, animal studies have revealed that NMDARs are important in fear extinction, and based on this, it was observed in the clinic that the NMDAR partial agonist D-cycloserine increased the effect of exposure therapy in psychiatric patients [76, 77]. Our findings from animal experiments highlight a possible relationship between redox status and fear extinction. Despite accumulating clinical data about the alteration of oxidative biomarkers in psychiatric diseases, many conflicting reports exist, especially in anxiety disorders, such as panic disorder, obsessive-compulsive disorder, posttraumatic stress disorder, social phobia, specific phobias and generalized anxiety disorder. We performed a translational study using published data to analyze various oxidative biomarkers in fear-related anxiety disorders. Seventeen studies researched the association between MDA content and anxiety disorders, including 645 patients and 638 HCs in total. We found that oxidative biomarkers were significantly elevated in the blood of patients with fear-related disorders, including social phobia, panic disorders, PTSD or general anxiety disorder, but not obsessive-compulsive disorder. However, only a few studies have focused on oxidative biomarkers in the blood of patients with fear-related anxiety, which needs further investigation.
Several questions remain to be verified in subsequent studies. One interesting observation in our experiments was that the oxidative stress level, which was elevated in the BLA in response to fear stimulation, could be reversed by extinction. The exact mechanism by which this occurs requires further investigation. In the BLA, both interneurons and principal neurons are involved in the control of anxiety. The question is whether the oxidative stress triggered by fear conditioning occurs in the principal neurons. Although NMDARs have been identified as redox-modified targets, other targets underlying fear conditions may also be oxidized. This should be further evaluated by a redox-coupled proteomic assay.
In summary, the current results, together with our previous findings, provide a new explanation for the role of oxidative stress in fear-related mental illness and present evidence that redox regulation of NMDAR function may represent a pharmacological strategy for the prevention and therapy of fear-related mental illness.
References
Parsons RG, Ressler KJ. Implications of memory modulation for post-traumatic stress and fear disorders. Nat Neurosci. 2013;16:146–53.
Bukalo O, Pinard CR, Holmes A. Mechanisms to medicines: elucidating neural and molecular substrates of fear extinction to identify novel treatments for anxiety disorders. Br J Pharmacol. 2014;171:4690–718.
Milad MR, Quirk GJ. Fear Extinction as a model for translational neuroscience: ten years of progress. Annu Rev Psychol. 2012;63:129–51.
Kessler RC, Chiu WT, Demler O, Walters EE. Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:617.
Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the national comorbidity survey replication. Arch Gen Psychiatry. 2005;62:593.
Sharko AC, Fadel JR, Kaigler KF, Wilson MA. Activation of orexin/hypocretin neurons is associated with individual differences in cued fear extinction. Physiol Behav. 2017;178:93–102.
Russo AS, Parsons RG. Acoustic startle response in rats predicts inter-individual variation in fear extinction. Neurobiol Learn Mem. 2017;139:157–64.
Furini C, Myskiw J, Izquierdo I. The learning of fear extinction. Neurosci Biobehav Rev. 2014;47:670–83.
Bouton ME. Conditioning, remembering, and forgetting. J Exp Psychol Anim Behav Process. 1994;20:219–31.
Huynh TN, Santini E, Mojica E, Fink AE, Hall BS, Fetcho RN, et al. Activation of a novel p70 S6 kinase 1-dependent intracellular cascade in the basolateral nucleus of the amygdala is required for the acquisition of extinction memory. Mol Psychiatr. 2018;23:1394–401.
Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–85.
Schroeder BW, Shinnick-Gallagher P. Fear learning induces persistent facilitation of amygdala synaptic transmission. Eur J Neurosci. 2005;22:1775–83.
Rosenkranz JA, Moore H, Grace AA. The prefrontal cortex regulates lateral amygdala neuronal plasticity and responses to previously conditioned stimuli. J Neurosci. 2003;23:11054–64.
Tsvetkov E, Carlezon WA, Benes FM, Kandel ER, Bolshakov VY. Fear conditioning occludes LTP-induced presynaptic enhancement of synaptic transmission in the cortical pathway to the lateral amygdala. Neuron. 2002;34:289–300.
Davis M. NMDA receptors and fear extinction: implications for cognitive behavioral therapy. Dialogues Clin Neurosci. 2011;13:463–74.
Shen H, Igarashi H, Imamura N, Matsuki N, Nomura H. N-methyl-D-aspartate receptors and protein synthesis are necessary for reinstatement of conditioned fear. NeuroReport. 2013;24:763–7.
Myers KM, Carlezon WA, Davis M. Glutamate receptors in extinction and extinction-based therapies for psychiatric illness. Neuropsychopharmacology. 2010;36:274–93.
Zimmerman JM, Maren S. NMDA receptor antagonism in the basolateral but not central amygdala blocks the extinction of Pavlovian fear conditioning in rats. Eur J Neurosci. 2010;31:1664–70.
Sotres-Bayon F, Bush DEA, LeDoux JE. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology. 2007;32:1929–40.
Peters J, Dieppa-Perea LM, Melendez LM, Quirk GJ. Induction of fear extinction with hippocampal-infralimbic BDNF. Science. 2010;328:1288–90.
Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–51.
Klass A, Glaubitz B, Tegenthoff M, Lissek S. d-Cycloserine facilitates extinction learning and enhances extinction-related brain activation. Neurobiol Learn Mem. 2017;144:235–47.
Cho JH, Bayazitov IT, Meloni EG, Myers KM, Carlezon WA, Zakharenko SS, et al. Coactivation of thalamic and cortical pathways induces input timing–dependent plasticity in amygdala. Nat Neurosci. 2011;15:113–22.
McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–11.
Rogan MT, Stäubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–7.
Kim WB, Cho J. Encoding of discriminative fear memory by input-specific LTP in the amygdala. Neuron. 2017;95:1129.
Hong I, Kim J, Song B, Park K, Shin K, Eom KD, et al. Fear conditioning occludes late-phase long-term potentiation at thalamic input synapses onto the lateral amygdala in rat brain slices. Neurosci Lett. 2012;506:121–5.
Huang CC, Chen CC, Liang YC, Hsu KS. Long-term potentiation at excitatory synaptic inputs to the intercalated cell masses of the amygdala. Int J Neuropsychopharmacol. 2014;17:1233–42.
Hong I, Song B, Lee S, Kim J, Kim J, Choi S. Extinction of cued fear memory involves a distinct form of depotentiation at cortical input synapses onto the lateral amygdala. Eur J Neurosci. 2009;30:2089–99.
Wu X, Zhang JT, Liu J, Yang S, Chen T, Chen JG, et al. Calcitonin gene-related peptide erases the fear memory and facilitates long-term potentiation in the central nucleus of the amygdala in rats. J Neurochem. 2015;135:787–98.
Wang W, Liu SL, Li K, Chen Y, Jiang B, Li YK, et al. Leptin: a potential anxiolytic by facilitation of fear extinction. CNS Neurosci Ther. 2015;21:425–34.
Kumar A, Yegla B, Foster TC. Redox signaling in neurotransmission and cognition during aging. Antioxid Redox Signal. 2018;28:1724–45.
Ishii T, Takanashi Y, Sugita K, Miyazawa M, Yanagihara R, Yasuda K, et al. Endogenous reactive oxygen species cause astrocyte defects and neuronal dysfunctions in the hippocampus: a new model for aging brain. Aging Cell. 2017;16:39–51.
Yang YJ, Wu PF, Long LH, Yu DF, Wu WN, Hu ZL, et al. Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell. 2010;9:709–21.
Haxaire C, Turpin FR, Potier B, Kervern M, Sinet PM, Barbanel G, et al. Reversal of age-related oxidative stress prevents hippocampal synaptic plasticity deficits by protecting d-serine-dependent NMDA receptor activation. Aging Cell. 2012;11:336–44.
Sugihara I, Robillard JM, Gordon GR, Choi HB, Christie BR, MacVicar BA. Glutathione restores the mechanism of synaptic plasticity in aged mice to that of the adult. PLoS ONE. 2011;6:e20676.
Hovatta I, Tennant RS, Helton R, Marr RA, Singer O, Redwine JM, et al. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature. 2005;438:662–6.
Nagahara N, Nagano M, Ito T, Shimamura K, Akimoto T, Suzuki H. Antioxidant enzyme, 3-mercaptopyruvate sulfurtransferase-knockout mice exhibit increased anxiety-like behaviors: a model for human mercaptolactate-cysteine disulfiduria. Sci Rep. 2013;3:1986. https://doi.org/10.1038/srep01986.
Mazereeuw G, Herrmann N, Andreazza AC, Khan MM, Lanctôt KL. A meta-analysis of lipid peroxidation markers in major depression. Neuropsychiatr Dis Treat. 2015;11:2479–91.
Ibi M, Liu J, Arakawa N, Kitaoka S, Kawaji A, Matsuda KI, et al. Depressive-like behaviors are regulated by NOX1/NADPH oxidase by redox modification of NMDA receptor 1. J Neurosci. 2017;37:4200–12.
Steullet P, Cabungcal JH, Coyle J, Didriksen M, Gill K, Grace AA, et al. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol Psychiatry. 2017;22:936–43.
Baumann PS, Griffa A, Fournier M, Golay P, Ferrari C, Alameda L, et al. Impaired fornix–hippocampus integrity is linked to peripheral glutathione peroxidase in early psychosis. Transl Psychiatry. 2016; 6: e859-e.
Jang EY, Ryu YH, Lee BH, Chang SC, Yeo MJ, Kim SH, et al. Involvement of reactive oxygen species in cocaine-taking behaviors in rats. Addict Biol. 2015;20:663–75.
Hardingham GE, Do KQ. Linking early-life NMDAR hypofunction and oxidative stress in schizophrenia pathogenesis. Nat Rev Neurosci. 2016;17:125–34.
Kuloglu M, Atmaca M, Tezcan E, Ustundag B, Bulut S. Antioxidant enzyme and malondialdehyde levels in patients with panic disorder. Neuropsychobiology. 2002;46:186–9.
Atmaca M, Tezcan E, Kuloglu M, Ustundag B, Tunckol H. Antioxidant enzyme and malondialdehyde values in social phobia before and after citalopram treatment. Eur Arch Psychiatry Clin Neurosci. 2004;254:231–5.
Kuloglu M, Atmaca M, Tezcan E, Gecici O, Tunckol H, Ustundag B. Antioxidant enzyme activities and malondialdehyde levels in patients with obsessive-compulsive disorder. Neuropsychobiology. 2002;46:27–32.
Quinlan EM, Lebel D, Brosh I, Barkai E. A molecular mechanism for stabilization of learning-induced synaptic modifications. Neuron. 2004;41:185–92.
Wang CM, Yang YJ, Zhang JT, Liu J, Guan XL, Li MX, et al. Regulation of emotional memory by hydrogen sulfide: role of GluN2B-containing NMDA receptor in the amygdala. J Neurochem. 2015;132:124–34.
Fan H, Wu PF, Zhang L, Hu ZL, Wang W, Guan XL, et al. Methionine sulfoxide reductase A negatively controls microglia-mediated neuroinflammationviaInhibiting ROS/MAPKs/NF-κB signaling pathways through a catalytic antioxidant function. Antioxid Redox Signal. 2015;22:832–47.
Luo Y, Wu PF, Zhou J, Xiao W, He JG, Guan XL, et al. Aggravation of seizure-like events by hydrogen sulfide: involvement of multiple targets that control neuronal excitability. CNS Neurosci Ther. 2014;20:411–9.
Sartori SB, Maurer V, Murphy C, Schmuckermair C, Muigg P, Neumann ID, et al. Combined neuropeptide S and D-cycloserine augmentation prevents the return of fear in extinction-impaired rodents: advantage of dual versus single drug approaches. Int J Neuropsychopharmacol. 2016;19:pyv128.
Whittle N, Hauschild M, Lubec G, Holmes A, Singewald N. Rescue of impaired fear extinction and normalization of cortico-amygdala circuit dysfunction in a genetic mouse model by dietary Zinc restriction. J Neurosci. 2010;30:13586–96.
Habermann KJ, Grünewald L, van Wijk S, Fulda S. Targeting redox homeostasis in rhabdomyosarcoma cells: GSH-depleting agents enhance auranofin-induced cell death. Cell Death Dis. 2017;8:e3067.
Arrick BA, Griffith OW, Cerami A. Inhibition of glutathione synthesis as a chemotherapeutic strategy for trypanosomiasis. J Exp Med. 1981;153:720–5.
Obin M, Shang F, Gong X, Handelman G, Blumberg J, Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J. 1998;12:561–9.
Ochi T. Mechanism for the changes in levels of glutathione upon exposure of cultured mammalian cells to tertiary-butylhydroperoxide and diamide. Arch Toxicol. 1993;67:401–10.
Dalton GL, Wu DC, Wang YT, Floresco SB. Phillips AG. NMDA GluN2A and GluN2B receptors play separate roles in the induction of LTP and LTD in the amygdala and in the acquisition and extinction of conditioned fear. Neuropharmacology. 2012;62:797–806.
das Neves Duarte JM, Kulak A, Gholam-Razaee MM, Cuenod M, Gruetter R, Do KQ. N-Acetylcysteine normalizes neurochemical changes in the glutathione-deficient schizophrenia mouse model during development. Biol Psychiatry. 2012;71:1006–14.
Joshi D, Mittal DK, Shukla S, Srivastav AK, Srivastav SK. Methylmercury toxicity: amelioration by selenium and water-soluble chelators as N-acetyl cysteine and dithiothreitol. Cell Biochem Funct. 2014;32:351–60.
Knapska E, Macias M, Mikosz M, Nowak A, Owczarek D, Wawrzyniak M, et al. Functional anatomy of neural circuits regulating fear and extinction. Proc Natl Acad Sci USA. 2012;109:17093–8.
Pape HC, Pare D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol Rev. 2010;90:419–63.
Zhang C, Yi F, Xia M, Boini KM, Zhu Q, Laperle LA, et al. NMDA receptor-mediated activation of NADPH oxidase and glomerulosclerosis in hyperhomocysteinemic rats. Antioxid Redox Signal. 2010;13:975–86.
Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–27. 5 Pt 1
Lobysheva NV, Selin AA, Vangeli IM, Byvshev IM, Yaguzhinsky LS, Nartsissov YR. Glutamate induces H2O2 synthesis in nonsynaptic brain mitochondria. Free Radic Biol Med. 2013;65:428–35.
Tell F, Shin R-M, Higuchi M, Suhara T. Nitric oxide signaling exerts bidirectional effects on plasticity inductions in amygdala. PLoS ONE. 2013;8:e74668.
Peter C, Braidy N, Zarka M, Welch J, Bridge W. Therapeutic approaches to modulating glutathione levels as a pharmacological strategy in Alzheimer’s disease. Curr Alzheimer Res. 2015;12:298–313.
Whitt JD, Keeton AB, Gary BD, Sklar LA, Sodani K, Chen ZS, et al. Sulindac sulfide selectively increases sensitivity of ABCC1 expressing tumor cells to doxorubicin and glutathione depletion. J Biomed Res. 2016;30:120–33.
Amaral OB, Roesler R. Targeting the NMDA receptor for fear-related disorders. Recent Pat CNS Drug Discov. 2008;3:166–78.
Zinebi F, Xie JG, Liu J, Russell RT, Gallagher JP, McKernan MG, et al. NMDA currents and receptor protein are downregulated in the amygdala during maintenance of fear memory. J Neurosci. 2003;23:10283–91.
Aizenman E, Lipton SA, Loring RH. Selective modulation of NMDA responses by reduction and oxidation. Neuron. 1989;2:1257–63.
Kohr G, Eckardt S, Luddens H, Monyer H, Seeburg PH. NMDA receptor channels: subunit-specific potentiation by reducing agents. Neuron. 1994;12:1031–40.
Sucher NJ, Lipton SA. Redox modulatory site of the NMDA receptor-channel complex: regulation by oxidized glutathione. J Neurosci Res. 1991;30:582–91.
Bennett MR, Arnold J, Hatton SN, Lagopoulos J. Regulation of fear extinction by long-term depression: the roles of endocannabinoids and brain derived neurotrophic factor. Behav Brain Res. 2017;319:148–64.
Wang SJ, Gean PW. Long-term depression of excitatory synaptic transmission in the rat amygdala. J Neurosci. 1999;19:10656–63.
Rothbaum BO, Price M, Jovanovic T, Norrholm SD, Gerardi M, Dunlop B, et al. A randomized, double-blind evaluation of d-cycloserine or alprazolam combined with virtual reality exposure therapy for posttraumatic stress disorder in Iraq and Afghanistan war veterans. Am J Psychiatry. 2014;171:640–8.
Difede J, Cukor J, Wyka K, Olden M, Hoffman H, Lee FS, et al. D-Cycloserine augmentation of exposure therapy for post-traumatic stress disorder: a pilot randomized clinical trial. Neuropsychopharmacology. 2013;39:1052–8.
Acknowledgements
This work was supported by grants from the Foundation for Innovative Research Groups of NSFC (No. 81721005 to JGC and FW), the National Basic Research Program of China (973 Program, No. 2014CB744601 to FW and No. 2013CB531303 to JGC), National Natural Science Foundation of China (No. 81773712 to PFW, No. 81803503 to XLG, No. 81471377 to FW, No. 81473198 to JGC), Science Fund for Creative Research Groups of the Natural Science Foundation of Hubei Province (2015CFA020) and PCSIRT (No. IRT13016) to JGC.
Author information
Authors and Affiliations
Contributions
PFW conceived the study, designed the experiments, performed redox state assay and meta-analysis, created the figure and wrote the paper. XLG performed electrophysiological recording, behavioral experiments, redox state assay, meta-analysis and statistical analysis. FW and JGC supervised the project, designed the experiments, revised the paper and supported funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Wu, Pf., Guan, Xl., Wang, F. et al. N-acetylcysteine facilitates extinction of cued fear memory in rats via reestablishing basolateral amygdala glutathione homeostasis. Acta Pharmacol Sin 43, 260–272 (2022). https://doi.org/10.1038/s41401-021-00661-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00661-0
