
Abstract
Interleukin-17A (IL-17), a potent proinflammatory cytokine, has been shown to participate in cardiac electrical disorders. Diabetes mellitus is an independent risk factor for ventricular arrhythmia. In this study, we investigated the role of IL-17 in ventricular arrhythmia of diabetic mice. Diabetes was induced in both wild-type and IL-17 knockout mice by intraperitoneal injection of streptozotocin (STZ). High-frequency electrical stimuli were delivered into the right ventricle to induce ventricular arrhythmias. We showed that the occurrence rate of ventricular tachycardia was significantly increased in diabetic mice, which was attenuated by IL-17 knockout. We conducted optical mapping on perfused mouse hearts and found that cardiac conduction velocity (CV) was significantly decreased, and action potential duration (APD) was prolonged in diabetic mice, which were mitigated by IL-17 knockout. We performed whole-cell patch clamp recordings from isolated ventricular myocytes, and found that the densities of Ito, INa and ICa,L were reduced, the APDs at 50% and 90% repolarization were increased, and early afterdepolarization (EAD) was markedly increased in diabetic mice. These alterations were alleviated by the knockout of IL-17. Moreover, knockout of IL-17 alleviated the downregulation of Nav1.5 (the pore forming subunit of INa), Cav1.2 (the main component subunit of ICa,L) and KChIP2 (potassium voltage-gated channel interacting protein 2, the regulatory subunit of Ito) in the hearts of diabetic mice. The expression of NF-κB was significantly upregulated in the hearts of diabetic mice, which was suppressed by IL-17 knockout. In neonatal mouse ventricular myocytes, knockdown of NF-κB significantly increased the expression of Nav1.5, Cav1.2 and KChIP2. These results imply that IL-17 may represent a potential target for the development of agents against diabetes-related ventricular arrhythmias.
Similar content being viewed by others

Introduction
Cardiac complications are the major causes of mortality and morbidity in patients with diabetes. Sudden cardiac death is one of the most common causes of death in patients with diabetes [1]. Diabetes is considered an independent risk factor for ventricular arrhythmias and sudden cardiac death [2, 3]. A host of studies have indicated that diabetes induces electrical remodeling, structural changes, metabolic disorders, etc., which contribute to increased susceptibility to atrial fibrillation and ventricular arrhythmias [4].
Electrical remodeling has been shown to promote the development of ventricular arrhythmias in the diabetic heart [5,6,7,8]. Inflammatory cytokines are pivotal regulators of cardiac electrical remodeling [9]. They are also involved in diabetes-related electrical remodeling. A recent study showed that increased IL-1β secretion from macrophages causes prolongation of the action potential duration (APD) and induces a decrease in potassium current and an increase in calcium sparks in the cardiomyocytes of diabetic mice; moreover, an IL-1 receptor antagonist inhibits the occurrence of arrhythmia in diabetic mice [10]. Considering that there are many different cytokines, our current understanding of the involvement of these factors in diabetes-related arrhythmia remains quite limited.
Interleukin-17A (IL-17), a potent proinflammatory cytokine, has been implicated in the development of a number of inflammatory reactions [11, 12]. It is also a key regulator of cardiac diseases [13, 14]. The expression of IL-17 is increased in the context of several cardiac diseases, including atrial fibrillation [15], myocardial ischemia/reperfusion injury [13], and dilated cardiomyopathy [16]. IL-17 leads to adverse postmyocardial infarction remodeling by inducing murine cardiomyocyte apoptosis through the p38 MAPK-p53-Bax signaling pathway [17]. Liao et al. found that IL-17 exacerbates cardiac ischemia/reperfusion injury by promoting neutrophil infiltration [13]. We observed that IL-17 expression was upregulated in the hearts of diabetic model mice and that deletion of IL-17 mitigated cardiac fibrosis and improved cardiac function in a manner mediated by the lncRNA-AK081284/TGFβ1 signaling pathway [18]. Despite the fact that the participation of IL-17 in cardiac diseases has been extensively studied, its role in cardiac electrical remodeling and arrhythmia in diabetes remains unknown.
Therefore, in the current study, we explored the influence of IL-17 deletion on cardiac electrical remodeling and susceptibility to ventricular arrhythmia in diabetic mice. We found that knockout of IL-17 prevented diabetes-induced electrical changes in sodium, calcium and potassium currents and reduced the occurrence of ventricular arrhythmia.
Materials and methods
Experimental animals
All animal handling procedures were approved by the Institutional Animal Care and Use Committee of Harbin Medical University, China. All animal studies were performed in accordance with the regulations of the Institutional Animal Care and Use Committee of Harbin Medical University. Adult male C57BL/6J mice were purchased from Charles River Laboratories (Beijing, China). Interleukin-17A knockout mice were provided by Prof. Zhi-nan Yin (Jinan University, China). Mice were kept at a constant temperature (23 °C) on a standard light/dark cycle (12 h/12 h) and provided free access to standard chow and water.
Induction of diabetes
Eight-week-old wild-type mice (WT) and interleukin-17 knockout mice (IL-17 KO) were intraperitoneally injected with streptozotocin (STZ, 50 mg·kg−1) dissolved in citric acid buffer (pH = 4.5, 4 °C) to establish a diabetes model. Age-matched control mice were administered citrate buffer. Mice with fasting blood glucose levels over 11 mmol· L−1 were considered diabetic. The mice were subjected to further experiments 3 months after diabetes induction.
High-frequency electrical stimulation
Mice were anesthetized by injection of avertin (0.2 g· kg−1, ip) and fixed in the supine position. The right jugular vein was opened, and an 8-electrode catheter (1.1F, Octapolar EP catheter; NY, Ithaca, USA) was inserted deep into the right ventricle. S2 and S3 extrastimuli with 2 ms decrements following a train of S1 stimuli (10 consecutive electrical pulses) were applied to induce ventricular arrhythmia by using an automated stimulator interfaced with a data acquisition system (GY6000; HeNanHuaNan Medical Science & Technology Ltd., Henan, China). S1 stimuli of three cycle lengths (90 ms, 80 ms and 70 ms) were employed. The occurrence of rapid irregular ventricular rhythm lasting for three beats or more was regarded as successful induction of ventricular arrhythmias.
Ventricular cardiomyocyte isolation
The mice were injected with heparin sodium (400 IU· kg−1) and sacrificed by cervical dislocation. The heart was quickly removed, cannulated and perfused with Tyrode’s solution (in mmol·L−1: 150 NaCl, 5.4 KCl, 5 HEPES, 1.2 MgCl2•6H2O, 2 NaH2PO4, and 10 glucose; pH adjusted to 7.35 with NaOH). A solution containing 1 mg ·mL−1 type II collagenase (Invitrogen, Carlsbad, USA) and 0.75 mg· mL−1 bovine serum albumin V (Solarbio, Beijing, China) was then used to digest the heart. When the tissue turned soft, the ventricle was cut off and immersed in Tyrode’s solution comprising 200 µM CaCl2 and 1% bovine serum albumin at room temperature. Single ventricular myocytes were obtained by gently agitating the digested tissue.
Whole-cell patch clamp experiment
A whole-cell patch clamp equipment (an Axopatch 700B amplifier) was used to record the APD, potassium currents, sodium currents and the L-type calcium current. Borosilicate glass electrodes with pipette resistances of 2–4 MΩ were prepared by using a Brown-Flamming puller (model P-97, Sutter Instrument Co., Novato, CA., UAS) and a microforge (F-83, Narishige). The action potentials were recorded with pipette solution (in mmol·L−1: 130 K-glutamate, 1 MgCl2•6H2O, 5 NaCl, 15 KCl, 5 Mg-ATP, 1 CaCl2, 5 EGTA, and 10 HEPES; pH adjusted to 7.20 with KOH) and external solution (normal Ca2+-containing Tyrode’s solution) at 37 °C. Sodium currents were recorded in external solution (in mmol·L−1: 10 NaCl, 1.2 MgCl2•6H2O, 1.8 CaCl2, 5 CsCl, 125 TEACl, 20 HEPES, 10 glucose, 0.02 nifedipine, and 3 4-AP; pH adjusted to 7.30 with CsOH) and pipette solution (in mmol·L−1: 5 NaCl, 35 L-aspartic acid, 30 TEACl, 11 HEPES, 5 Mg-ATP, 10 EGTA, and 125 CsOH; pH adjusted to 7.20 with CsOH) at room temperature. Potassium currents were recorded in external solution (in mmol·L−1: 138 NaCl, 5.4 KCl, 10 HEPES, 1.0 MgCl2•6H2O, 10 glucose, 1.8 CaCl2, and 0.02 nifedipine; pH adjusted to 7.30 with NaOH) and pipette solution (in mmol·L−1: 130 KOH, 130 L-glutamic acid, 1 MgCl2•6H2O, 5 NaCl, 15 KCl, 5 Mg-ATP, 1 CaCl2, 5 EGTA, 10 HEPES; pH adjusted to 7.20 with KOH) at 37 °C. The L-type calcium current (ICa,L) was recorded in external solution (in mmol·L−1: 120 tetraethylammonium (TEA), 10 HEPES, 1 MgCl2•6H2O, 10 CsCl, 10 glucose, and 1.8 CaCl2; pH was adjusted to 7.30 with CsOH) and pipette solution (in mmol·L−1: 120 CsCl, 40 CsOH, 1 MgCl2•6H2O, 11 EGTA, 5 Mg-ATP, and 10 HEPES; pH adjusted to 7.20 with CsOH) at 37 °C. Whole-cell currents were sampled at 10 kHz and filtered at 2 kHz. The current amplitude data for each cell were normalized to the cell capacitance (current density, pA/pF) data, and the current voltage relationship (I-V curve) was plotted. Voltage-dependent activation and steady-state inactivation profiles were fitted to the Boltzmann equation.
Optical mapping
Cardiac conduction velocity (CV) was measured as previously described [19, 20]. Mice were injected with heparin sodium (400 IU· kg−1) and sacrificed by cervical dislocation. The hearts were isolated and Langendorff perfused with Tyrode’s solution (in mmol·L−1: 128.2 NaCl, 1.3 CaCl2, 4.7 KCl, 1.85 MgCl2•6H2O, 1.19 NaH2PO4, 20 NaHCO3, and 11 glucose; pH adjusted to 7.35 with NaOH) at 37 °C. The perfusate was oxygenated by bubbling with O2/CO2 (95%/5%) gas. After 10 min of stabilization, the beating of the heart was stopped by perfusion with the electric-mechanical uncoupler blebbistatin (50 nM, Selleckchem, Houston, TX, USA) and paced at a frequency of 10 Hz at the apex. The heart was then loaded with 30 μL (1.25 mg· mL−1) voltage-sensitive dye (RH 237 [N-(4-sulfobutyl)-4-(6-(4-(dibutylamino)phenyl)hexatrienyl)pyridinium, inner salt]; AAT Bioquest Inc., USA). The dye was excited at 710 nm using a monochromatic light-emitting device after 15 min. Images were acquired with a MiCAM05 CMOS camera (Sci Media, USA) at a rate of 2000 frames/s with a filter setting of 1 kHz. Cardiac CV was calculated from the gradient of the scalar field of 17 ms isochronal activation maps along the septal apex-base axis. The time for a cardiac excitation wave to travel a certain distance was measured, and the CV was calculated by the equation CV = distance/conduction time.
Isolation of neonatal mouse ventricular myocytes (NMVMs)
Neonatal mouse ventricular myocytes were isolated from 1- to 3-day-old neonatal mice as described in our previous study [19]. Briefly, neonatal mouse ventricles were cut into 1–2 mm3 pieces after the hearts were rapidly removed, and the cells were dissociated in 0.25% trypsin at 37 °C for 10 min. Heart tissues were trypsinized until the tissues disappeared, and cell suspensions were collected by centrifugation at 1500 × g for 5 min. The collected cells were then resuspended in DMEM (Biological Industries, Kibbutz Beit Haemek, Israel) supplemented with 10% fetal bovine serum (Biological Industries, Kibbutz Beit Haemek, Israel) and penicillin (100 U·mL−1)/streptomycin (100 U·mL−1; Beyotime, Shanghai, China) and cultured at 37 °C in 5% CO2 and 95% O2 in a humidified incubator. After allowing 90 min for fibroblast adherence, the cell suspension was plated in DMEM in a 6-well plate at a density of 3 × 105 cells per well.
Quantitative real-time RT-PCR (qPCR)
Total RNA was extracted from the hearts of mice or NMVMs using TRIzol (Invitrogen, Camarillo, CA, USA) reagent according to the manufacturer’s instructions. The RNA samples were reverse-transcribed according to the manufacturer’s instructions (Reverse Transcription System, Promega, USA). Real-time PCR was performed on an ABI 7500 Fast Real-Time PCR system (Applied Biosystems, USA) by using the SYBR Green PCR Master Mix Kit (Applied Biosystems, USA) to quantify the expression of target genes. The relative RNA expression of the genes was calculated using the comparative cycle threshold (Ct) method (2-ΔΔCt). β-Actin was used as an internal control. The results are expressed as fold changes, which were determined by normalizing the data to the values of the control subjects.
Western blot analysis
Heart tissue and primary cultured cardiomyocytes were ground in lysis buffer (RIPA: PI=100:1; Beyotime, China), lysed for 30 min on ice, and then centrifuged for 20 min (4 °C, 13,500 r/min). Protein samples (80 μg) were separated by SDS-PAGE and transferred onto nitrocellulose membranes (PALL, USA). The membranes were blotted with primary antibody overnight at 4 °C. After washing, the membranes were incubated with fluorescent secondary antibodies. The primary antibodies included rabbit anti-mouse Nav1.5 (Alomone Labs, Israel), Kv4.2 (Alomone Labs, Israel), Kv4.3 (Alomone Labs, Israel), KChIP2 (Boster, China), Cav1.2 (Alomone Labs, Israel), and NF-κB (CST, USA). β-Actin was used as an internal control.
Statistical analysis
The data are expressed as the mean ± SEM. Statistical comparisons of more than two groups were performed using analysis of variance (ANOVA) followed by Tukey’s test. Student’s t test was used for comparisons between two groups. GraphPad Prism 7.0 software was used for the calculations. A two-tailed P < 0.05 was considered to indicate a statistically significant difference.
Results
Knockout of IL-17 reduced the susceptibility diabetic mice to ventricular arrhythmia
Our previous study showed that IL-17 expression was significantly upregulated in diabetic mice [18]. Diabetes mellitus is often associated with ventricular arrhythmias, which prompted us to evaluate the potential influence of IL-17 on ventricular arrhythmias in a diabetes model. We established a mouse model of STZ-induced diabetes. The blood glucose levels of wild-type mice (WT+DM) and IL-17 knockout mice (IL-17 KO+DM) were significantly increased at 12 weeks after STZ injection (Fig. 1a). The induction rate, number of average ventricular arrhythmia episodes and duration of ventricular arrhythmia were increased in the WT+DM mice compared with WT controls, while knockout of IL-17 reduced the occurrence of ventricular arrhythmias (Fig. 1b–e). These results suggested that IL-17 knockout is protective against diabetes-induced ventricular arrhythmias.

a Fasting blood glucose level. *P < 0.05 vs. WT or IL-17 KO mice; n = 11–27 mice. b Representative traces of pacing-induced ventricular tachycardia (VT). c Induction rate of VT. *P < 0.05 vs. WT mice; #P < 0.05 vs. IL-17 KO mice; $P < 0.05 vs. WT+DM mice; n = 8–10 mice. d Average VT episodes. *P < 0.05 vs. WT mice; #P < 0.05 vs. IL-17 KO mice; $P < 0.05 vs. WT + DM mice; n = 8–10 mice. e Duration of VT. *P < 0.05 vs. WT mice; #P < 0.05 vs. IL-17 KO mice; $P < 0.05 vs. WT + DM mice; n = 8–10 mice. WT wild-type, IL-17 KO IL-17 knockout, WT+DM wild-type+diabetes mellitus, IL-17 KO+DM IL-17 knockout+diabetes mellitus.
Knockout of IL-17 prevented APD prolongation and CV reduction in the hearts of diabetic mice
To explore the potential mechanism underlying the influence of IL-17 knockout on the susceptibility of diabetic mice to ventricular arrhythmia, we performed optical mapping on perfused mouse hearts to measure the ventricular CV and APD. No significant change in the CV was observed between WT and IL-17 KO mice at different pacing cycle lengths (PCLs) under normal conditions. Upon induction of diabetes, the ventricular CV was significantly slowed in WT mice and was restored by knockout of IL-17 (Fig. 2a, b). The APD50 of normal IL-17 KO mice was shorter than that of control WT mice at PCLs of 200 ms and 150 ms but not 120 ms and 100 ms (Fig. 2c–e). Induction of diabetes led to a significant prolongation of the APD50 and APD80 in WT mice, which were significantly shortened by IL-17 knockout (Fig. 2c–f).

a Representative images showing the conduction velocity (CV). b Statistical analysis of the CV under various pacing cycle lengths (PCLs). c, d Representative surface images and traces of the APD by optical mapping recording of perfused hearts. e, f Statistical analysis of the APD50 and APD80 of perfused hearts. *P < 0.05 vs. WT mice; #P < 0.05 vs. WT + DM mice; n = 8–10 mice. WT wild-type, IL-17 KO IL-17 knockout, WT+DM wild-type+diabetes mellitus, IL-17 KO+DM IL-17 knockout+diabetes mellitus.
Effects of IL-17 knockout on APD and EAD in ventricular myocytes isolated from diabetic mice
The APDs of isolated ventricular myocytes were recorded by the whole-cell patch clamp technique. Knockout of IL-17 shortened the APD50 but not APD90 in control mice (Fig. 3a–c). The APD50 and APD90 were significantly prolonged in diabetic mice compared with control mice, and these changes were prevented by knockout of IL-17 (Fig. 3a–c). The action potential overshoot (OS) was increased in IL-17 KO mice compared with WT mice under both normal conditions and in the context of diabetes (Fig. 3d). There was no significant change in the resting membrane potential (RMP) among the groups (Fig. 3e). In addition, ventricular myocytes from diabetic WT mice were more likely to induce early afterdepolarization (EAD) than those from diabetic IL-17 KO mice (Fig. 3f).

a Representative traces of the APDs of single ventricular myocytes. b, c Statistical analysis of the APD50 and APD90 of single ventricular myocytes determined by the patch-clamp technique. n = 9–18 cells. d, e Statistical analysis of the overshoot (OS) and resting membrane potential (RMP). n = 11–18 cells. f Representative traces and the rate of occurrence of EAD in single ventricular myocytes determined by patch-clamp recording. n = 18–19 cells. *P < 0.05 vs. WT mice; #P < 0.05 vs. WT + DM mice. WT wild-type, IL-17 KO IL-17 knockout, WT+DM wild-type+diabetes mellitus, IL-17 KO+DM IL-17 knockout + diabetes mellitus.
Knockout of IL-17 increased the I to in ventricular myocytes in diabetic mice
The whole-cell patch clamp technique was used to record the transient outward potassium current (Ito)and steady-state outward potassium current (Iss) (Fig. 4). The results showed that knockout of IL-17 increased Ito under normal conditions. The density of Ito was decreased in the cardiomyocytes of diabetic mice, and this decrease was prevented by IL-17 knockout (Fig. 4a, b). There was no evident change in the steady-state outward potassium current (Iss) or cell membrane capacitance (Fig. 4c, d). The protein level of Kv4.2/Kv4.3 and mRNA level of potassium voltage-gated channel subfamily D member 2/potassium voltage-gated channel subfamily D member 3 (KCND2/KCND3) did not change after knockout of IL-17, while the protein expression of KChIP2 and the mRNA level of potassium voltage-gated channel interacting protein 2 (KCNIP2) was significantly upregulated in IL-17 knockout mice (Fig. 4e–g).

a Representative traces of the Ito. b Current density-voltage (I–V) relationship of the Ito. n = 8–11 cells. c Current density-voltage (I–V) relationship of the Iss. n = 9–16 cells. d Mean membrane capacitance of the Ito and Iss n = 9–16 cells. e The protein and mRNA levels of Kv4.2. n = 7. f The protein and mRNA levels of Kv4.3. n = 7. g The protein and mRNA levels of KChIP2. n = 5. *P < 0.05 vs. WT mice; #P < 0.05 vs. WT+DM mice. WT wild-type, IL-17 KO IL-17 knockout, WT+DM wild-type+diabetes mellitus, IL-17 KO+DM IL-17 knockout+diabetes mellitus.
Knockout of IL-17 increased the peak Na+ current (I Na) in the ventricular myocytes of diabetic mice
Consistent with a previous study [21], the density of the peak INa was reduced in the ventricular cardiomyocytes of mice with STZ-induced diabetes (Fig. 5a, b). Knockout of IL-17 increased the peak INa under control condition and prevented the reduction in peak INa in the diabetes model, while no significant changes in the kinetics of peak INa or cell membrane capacitance were observed (Fig. 5c–f). Knockout of IL-17 increased the protein level of Nav1.5 and the mRNA level of sodium voltage-gated channel alpha subunit 5 (SCN5A) in control mice and inhibited the downregulation of this protein and mRNA in diabetic mice (Fig. 5g, h).

a Representative traces of the sodium current. b Current density-voltage (I–V) relationship of the sodium current. n = 11–14 cells. c Activation-Vm relationship of the sodium current. n = 11–14 cells. d Steady-state inactivation-Vm relationship of the sodium current. n = 11–14 cells. e Recovery from inactivation of the sodium current. n = 11–14 cells. f Mean membrane capacitance of the INa. n = 11–14 cells. g The protein level of Nav1.5. n = 8. h The level of SCN5A determined by RT-PCR. n = 6–8 hearts. *P < 0.05 vs. WT mice; #P < 0.05 vs. WT + DM mice. WT wild-type, IL-17 KO IL-17 knockout, WT+DM wild-type+diabetes mellitus, IL-17 KO+DM IL-17 knockout+diabetes mellitus.
Knockout of IL-17 increased the L-type calcium current (I Ca,L) in the ventricular myocytes of diabetic mice
Knockout of IL-17 increased the density of the ICa,L under normal conditions and in the context of STZ-induced diabetes (Fig. 6a, b), but no significant changes in the kinetics of the ICa,L or cell membrane capacitance were observed (Fig. 6c–f). Knockout of IL-17 increased the protein level of Cav1.2 and mRNA level of calcium voltage-gated channel subunit alpha 1 C (CACNA1C) in control mice and inhibited the downregulation of Cav1.2 expression in diabetic mice (Fig. 6g, h).

a Representative traces of the L-type calcium current. b Current density-voltage (I-V) relationship of the L-type calcium current. n = 9–18 cells. c Activation-Vm relationship of the L-type calcium current. n = 9–18 cells. d Steady-state inactivation-Vm relationship of the L-type calcium current. n = 9–18 cells. e Recovery from inactivation of the L-type calcium current. n = 9–18 cells. f Mean membrane capacitance of the ICa,L. n = 9–18 cells. g The protein level of Cav1.2. n = 4. h The level of CACNA1C determined by RT-PCR. n = 6–10 hearts. *P < 0.05 vs. WT mice; #P< 0.05 vs. WT + DM mice. WT wild-type, IL-17 KO IL-17 knockout, WT+DM wild-type+diabetes mellitus, IL-17 KO+DM IL-17 knockout+diabetes mellitus.
Knockout of IL-17 inhibited the upregulation of NF-κB expression in the hearts of diabetic mice
NF-κB is an important downstream molecule in the IL-17 signaling pathway, which has been shown to regulate the transcription of SCN5A, CACNA1C and KCNIP2 [22,23,24]. In this study, we found that knockout of IL-17 downregulated the protein expression of NF-κB and reversed the upregulation of NF-κB expression induced by diabetes (Fig. 7a). Moreover, we confirmed that NF-κB exerts regulatory effects on SCN5A, CACNA1C and KCNIP2. Knockdown of NF-κB with siRNA (si-NF-κB) in neonatal mouse ventricular myocytes (NMVMs) significantly increased the protein and mRNA levels of Nav1.5, Cav1.2 and KChIP2 but had no influence on the expression of Kv4.2/KCND2 or Kv4.3/KCND3 (Fig. 7b, c). These results suggested that the regulation of Nav1.5, Cav1.2 and KChIP2 expression by IL-17 is mediated by NF-κB. These data indicated that the effects of IL-17 knockout on ion channel remodeling during diabetes are mediated by NF-κB.

a The protein level of NF-κB in the hearts of mice determined by Western blotting. *P < 0.05 vs. WT mice; #P < 0.05 vs. WT+DM mice. n = 8. WT wild-type, IL-17 KO IL-17 knockout, WT+DM wild-type+diabetes mellitus, IL-17 KO+DM IL-17 knockout+diabetes mellitus. b Protein levels of NF-κB and ion channels in neonatal mouse ventricular myocytes (NMVMs) after silencing of NF-κB. n = 4–5. c The mRNA level of ion channels in NMVMs after knockdown of NF-κB. *P < 0.05 vs. NC. n = 7–11. NC negative control, si-NF-κB siRNA for NF-κB.
Discussion
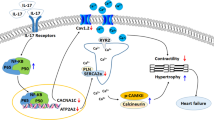
In this study, we demonstrated that knockout of IL-17 reduced the susceptibility of diabetic mice to ventricular arrhythmia (Fig. 8). An electrophysiological study showed that knockout of IL-17 prevented the reduction in the Ito, INa and ICa,L in the ventricular myocytes of diabetic mice and thus restored the APD and CV. Mechanistically, NF-κB-mediated downregulation of KChIP2, Cav1.2 and Nav1.5 expression underlies the effects of IL-17 knockout on ionic remodeling and the occurrence of ventricular arrhythmia in diabetic mice.

Knockout of IL-17 downregulates the expression of NF-κB, which suppresses the expression of KCNIP2, which encodes potassium voltage-gated channel interacting protein 2; CACNA1C, which encodes the pore-forming subunit of the voltage-gated L-type calcium channel Cav1.2; and SCN5A, which encodes the pore-forming subunit of the voltage-gated sodium channel Nav1.5. Decreased expression of KChIP2 and Nav1.5 prolonged the APD and slowed the conduction velocity, which increased susceptibility to ventricular arrhythmias. KCNIP2, potassium voltage-gated channel interacting protein 2; CACNA1C, calcium voltage-gated channel subunit alpha 1 C; KCND2, potassium voltage-gated channel subfamily D member 2; KCND3, potassium voltage-gated channel subfamily D member 3; SCN5A, sodium voltage-gated channel alpha subunit 5; WT+DM, wild-type+diabetes mellitus; IL-17 KO+DM, IL-17 knockout+diabetes mellitus.
Patients with diabetes are at a high risk of developing cardiac arrhythmias. A host of studies have verified that the susceptibility of diabetic animals to ventricular arrhythmia is increased [25, 26]. Consistently, in this study, we found that the induction rate of ventricular arrhythmias was increased in mice with STZ-induced diabetes mellitus. Diabetes is often associated with the occurrence of many inflammatory reactions and the upregulation of the expression of inflammatory factors, which have been implicated in the development of cardiac arrhythmia [27]. Serum IL-17A levels are increased in patients with diabetes [28]. Our previous studies showed that interleukin-17A expression is significantly upregulated in the sera and heart tissues of diabetic mice [18]. We showed here that knockout of IL-17 reduced the induction rate of ventricular tachycardia, occurrence of ventricular tachycardia events and duration of ventricular tachycardia in a diabetes model. Chang et al. also demonstrated that neutralizing IL-17 with an antibody reduces ventricular arrhythmia in mice with ischemic heart failure [29]. These findings indicate that inhibition of IL-17 may be a therapeutic strategy for the prevention of diabetes-induced ventricular arrhythmia.
Prolongation of APD is an important cause of ventricular arrhythmias, which can trigger early afterdepolarization (EAD). Meo et al. discovered that STZ-treated animals exhibit prolongation of the QT interval, ventricular myocytes with increased APD and susceptibility to ventricular arrhythmias, which is attributed to a reduction in the expression of voltage-gated potassium channels [30]. Sato et al. found that type 2 diabetes induces prolongation of APD due to a reduction in the transient outward K+ current and downregulation of Kv4.2 and KChIP2 expression [7]. Our study suggested that APD prolongation in the diabetes model depends on a decrease in the transient outward potassium current (Ito) and that knockout of IL-17 increases the Ito and thereby prevents diabetes-induced APD prolongation. Although a reduction in the ICa,L tends to shorten APD, the fact that APD is prolonged in the cardiomyocytes of diabetic mice indicates that the contribution of the potassium current increase to APD overcame that of the reduction in the ICa,L. We observed that APD prolongation was accompanied by an increase in the number of episodes of EAD in ventricular myocytes isolated from diabetic mice, which was dramatically inhibited by the deletion of IL-17. The Ito is propagated by channels comprised of voltage-gated α pore-forming subunits (Kv4.3 in humans and Kv4.2 and Kv4.3 in rodents) and auxiliary subunit KChIP2 [24, 31]. KChIP2 is a K+ channel-interacting protein 2 that prominently increases the amplitude of current by facilitating voltage-gated potassium channel assembly and stabilizing the cell surface channel complex [32]. KChIP2 expression is decreased during cardiac hypertrophy and heart failure [33]. In our study, neither diabetes induction nor IL-17 knockout changed the expression of Kv4.2/Kv4.3, while IL-17 knockout prevented the downregulation of KChIP2 expression in diabetic mice. Tatsuya Sato et al. found that type 2 diabetes induces Kv4.2 protein downregulation in OLETF rats only in Endo myocytes, although KChIP2 protein levels in both Endo and Epi myocytes are lower in OLETF rats than in LETO rats [7]. Nishiyama et al. showed that Kv4.2 mRNA expression, but not Kv4.3 mRNA expression, is downregulated in the ventricles of diabetic rats [34]. Chang et al. also demonstrated that inhibition of IL-17 prevents the prolongation of APD, although under different pathological conditions, they provided no ionic evidence [29]. To the best of our knowledge, we explored the influence of IL-17 on cellular ionic remodeling for the first time.
Impaired cardiac CV is critical for the development of cardiac arrhythmias. In a number of diabetic models, slower CV, greater QRS prolongation, and greater propensity for ventricular fibrillation have been observed [21, 35]. Shimoni Y et al. reported that in a rat model of STZ-induced type 1 diabetes, cardiac impulse propagation is impaired in both male and female animals [36]. We found here that the ventricular CV was slowed in diabetic mice and was restored to normal levels by deletion of IL-17. The amplitude of the INa affects myocardial action potential upstroke velocity and hence CV [37]. Thus, we found that the INa and the levels of Nav1.5, the pore-forming subunit of sodium channels, were reduced in diabetic mice and recovered by IL-17 knockout. Consistently, the cardiac CV is reduced due to a reduction in the Na+ current density in a rabbit model of alloxan-induced diabetes [21]. It has also been reported that the level of Nav1.5 exhibits an decreasing tendency, although not statistically significant.
NF-κB signaling is important in heart development, and its activation has been suggested to contribute to the development of cardiac diseases [38, 39]. The NF-κB pathway is a key downstream pathway of IL-17A, which is involved in regulating many inflammatory genes [40,41,42]. NF-κB has also been reported to regulate the expression of KCNIP2, CACNA1C and SCN5A. Liu et al demonstrated that the NF-κB protein interacts with the KChIP2 gene and suppresses its transcription [23]. Narayanan et al. reported that mitochondria control functional Cav1.2 expression in smooth muscle cells in the cerebral arteries by NF-κB [43]. Shang LL et al. showed that NF-κB can bind to the promoter region of the SCN5A gene and reduce the mRNA level of SCN5A [22]. We found that the expression of NF-κB was increased in the hearts of diabetic mice; the expression of KChIP2, Cav1.2 and Nav1.5 was decreased; and these changes were reversed by IL-17 knockout. Knockdown of NF-κB increased the expression of KChIP2, Cav1.2 and Nav1.5 at both the protein and mRNA levels. These data are consistent with the concept that NF-κB negatively regulates KChIP2, Cav1.2 and Nav1.5, which implies that IL-17 can regulate cardiac electrical remodeling in diabetic mice through the NF-κB signaling pathway.
Conclusion
Knockout of IL-17 reduced the susceptibility of the diabetic heart to ventricular arrhythmia by preventing the remodeling of KChIP2 and SCN5A through the NF-κB pathway. IL-17 represents a novel therapeutic target for the intervention of diabetes-related ventricular arrhythmias.
References
Weidner K, Behnes M, Schupp T, Rusnak J, Reiser L, Bollow A, et al. Type 2 diabetes is independently associated with all-cause mortality secondary to ventricular tachyarrhythmias. Cardiovasc Diabetol. 2018;17:125.
Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Europace 2015;17:v319.
Jouven X, Lemaître RN, Rea TD, Sotoodehnia N, Empana J. Siscovick D S. Diabetes, glucose level, and risk of sudden cardiac death. Eur Heart J. 2005;26:2142–7.
Grisanti LA. Diabetes and arrhythmias: pathophysology, mechanisms and therapeutic outcomes. Front Physiol. 2018;9:1669.
Pacher U, Nanasi K. Electrophysiological changes in rat ventricular and atrial myocardium at different stages of experimental diabetes. Acta Physiol (Oxf). 1999;166:7–13.
Lu Z, Jiang YP, Wu CYC, Ballou LM, Liu S, Carpenter ES, et al. Increased persistent sodium current due to decreased PI3K signaling contributes to QT prolongation in the diabetic heart. Diabetes. 2013;62:4257–65.
Sato T, Kobayashi T, Kuno A, Miki T, Tanno M, Kouzu H, et al. Type 2 diabetes induces subendocardium-predominant reduction in transient outward K+ current with downregulation of Kv4.2 and KChIP2. Am J Physiol-Heart C. 2014;306:H1054–65.
Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–6.
El Khoury N, Mathieu S, Fiset C. Interleukin-1β reduces L-type Ca2+ current through protein kinase Cϵ activation in mouse heart. J Biol Chem. 2014;289:21896–908.
Monnerat G, Alarcón ML, Vasconcellos LR, Hochman-Mendez C, Brasil G, Bassani RA, et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat Commun. 2016;7:13344.
Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity 2011;34:149–62.
Song X, Qian Y. IL-17 family cytokines mediated signaling in the pathogenesis of inflammatory diseases. Cell Signal. 2013;25:2335–47.
Liao Y, Xia N, Zhou S, Tang T, Yan X, Lv B, et al. Interleukin-17A contributes to myocardial ischemia/reperfusion injury by regulating cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll Cardiol. 2012;59:420–9.
Robert M, Miossec P. Effects of interleukin 17 on the cardiovascular system. Autoimmun Rev. 2017;16:984–91.
Fu X, Zhao N, Dong Q, Du L, Chen X, Wu Q, et al. Interleukin-17A contributes to the development of post-operative atrial fibrillation by regulating inflammation and fibrosis in rats with sterile pericarditis. Int J Mol Med. 2015;36:83–92.
Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106:1646–55.
Zhou SF, Yuan J, Liao MY, Xia N, Tang TT, Li JJ, et al. IL-17A promotes ventricular remodeling after myocardial infarction. J Mol Med (Berl). 2014;92:1105–16.
Zhang Y, Zhang Y, Li T, Wang J, Jiang Y, Zhao Y, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol. 2018;115:64–72.
Zhang Y, Sun L, Xuan L, Pan Z, Hu X, Liu H, et al. Long non-coding RNA CCRR controls cardiac conduction via regulating intercellular coupling. Nat Commun. 2018;9:4176.
Lang D, Sulkin M, Lou Q, Efimov IR. Optical mapping of action potentials and calcium transients in the mouse heart. J Vis Exp. 2011;55:e3275.
Stables CL, Musa H, Mitra A, Bhushal S, Deo M, Guerrero-Serna G, et al. Reduced Na+ current density underlies impaired propagation in the diabetic rabbit ventricle. J Mol Cell Cardiol. 2014;69:24–31.
Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, et al. NF-kappaB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. 2008;294:C372–9.
Liu W, Wang G, Zhang C, Ding W, Cheng W, Luo Y, et al. MG53, a novel regulator of KChIP2 and Ito,f, plays a critical role in electrophysiological remodeling in cardiac hypertrophy. Circulation. 2019;139:2142–56.
Panama BK, Latour-Villamil D, Farman GP, Zhao D, Bolz S, Kirshenbaum LA, et al. Nuclear factor κB downregulates the transient outward potassium current Ito,f through control of KChIP2 expression. Circ Res. 2011;108:537–43.
Rolim N, Skårdal K, Høydal M, Sousa MML, Malmo V, Kaurstad G, et al. Aerobic interval training reduces inducible ventricular arrhythmias in diabetic mice after myocardial infarction. Basic Res Cardiol. 2015;110:44.
Rajab M, Jin H, Welzig CM, Albano A, Aronovitz M, Zhang Y, et al. Increased inducibility of ventricular tachycardia and decreased heart rate variability in a mouse model for type 1 diabetes: effect of pravastatin. Am J Physiol-Heart C. 2013;305:H1807–16.
Lakin R, Polidovitch N, Yang S, Guzman C, Gao X, Wauchop M, et al. Inhibition of soluble TNFα prevents adverse atrial remodeling and atrial arrhythmia susceptibility induced in mice by endurance exercise. J Mol Cell Cardiol. 2019;129:165–73.
Arababadi MK, Nosratabadi R, Hassanshahi G, Yaghini N, Pooladvand V, Shamsizadeh A, et al. Nephropathic complication of type-2 diabetes is following pattern of autoimmune diseases?. Diabetes Res Clin Pr. 2010;87:33–7.
Chang S, Hsiao Y, Tsai Y, Lin S, Liu S, Lin Y, et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J Mol Cell Cardiol. 2018;122:69–79.
Meo M, Meste O, Signore S, Sorrentino A, Cannata A, Zhou Y, et al. Reduction in Kv current enhances the temporal dispersion of the action potential in diabetic myocytes: insights from a novel repolarization algorithm. J Am Heart Assoc. 2016;5:e003078.
Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol. 2010;48:12–25.
Foeger NC, Marionneau C, Nerbonne JM. Co-assembly of Kv4 α subunits with K+ channel-interacting protein 2 stabilizes protein expression and promotes surface retention of channel complexes. J Biol Chem. 2010;285:33413–22.
Nattel S, Maguy A, Le Bouter S, Yeh Y. Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev. 2007;87:425–56.
Nishiyama A, Ishii DN, Backx PH, Pulford BE, Birks BR, Tamkun MM. Altered K+ channel gene expression in diabetic rat ventricle: isoform switching between Kv4.2 and Kv1.4. Am J Physiol-Heart C. 2001;281:H1800–7.
Axelsen LN, Calloe K, Braunstein TH, Riemann M, Hofgaard JP, Liang B, et al. Diet-induced pre-diabetes slows cardiac conductance and promotes arrhythmogenesis. Cardiovasc Diabetol. 2015;14:87.
Shimoni Y, Emmett T, Schmidt R, Nygren A, Kargacin G. Sex-dependent impairment of cardiac action potential conduction in type 1 diabetic rats. Am J Physiol-Heart C. 2009;296:H1442–50.
Bohne LJ, Johnson D, Rose RA, Wilton SB, Gillis AM. The association between diabetes mellitus and atrial fibrillation: clinical and mechanistic insights. Front Physiol. 2019;10:135.
Cominacini L, Anselmi M, Garbin U, Fratta Pasini A, Stranieri C, Fusaro M, et al. Enhanced plasma levels of oxidized low-density lipoprotein increase circulating nuclear factor-kappa B activation in patients with unstable angina. J Am Coll Cardiol. 2005;46:799–806.
Timmers L, van Keulen JK, Hoefer IE, Meijs MFL, van Middelaar B, den Ouden K, et al. Targeted deletion of nuclear factor κB p50 enhances cardiac remodeling and dysfunction following myocardial infarction. Circ Res. 2009;104:699–706.
Shembade N, Harhaj EW. IKKi: a novel regulator of Act1, IL-17 signaling and pulmonary inflammation. Cell Mol Immunol. 2011;8:447–9.
Mcdaniel DK, Eden K, Ringel VM, Allen IC. Emerging roles for noncanonical NF-κB signaling in the modulation of inflammatory bowel disease pathobiology. Inflamm Bowel Dis. 2016;22:2265–79.
Baldwin AS. The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–81.
Narayanan D, Xi Q, Pfeffer LM, Jaggar JH. Mitochondria control functional CaV 1.2 expression in smooth muscle cells of cerebral arteries. Circ Res. 2010;107:631–41.
Acknowledgements
This work was supported by National Key R&D Program of China (2017YFC1307404 to ZP), National Natural Science Foundation of China (82070344, 81870295 to ZP 81861128022 to BY), Heilongjiang Touyan Innovation Team Program and CAMS Innovation Fund for Medical Sciences (CIFMS), 2019-I2M-5-078 (to BY).
Author information
Authors and Affiliations
Contributions
DSL and GLX performed experiments, analyzed data, and prepared the paper. JMY, CZL, RXZ, TT, ZL, KWS, YG, XNL, and JW helped perform experiments and collect data. YJL and ZWP designed the project, oversaw the experiments and prepared the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Li, Ds., Xue, Gl., Yang, Jm. et al. Knockout of interleukin-17A diminishes ventricular arrhythmia susceptibility in diabetic mice via inhibiting NF-κB-mediated electrical remodeling. Acta Pharmacol Sin 43, 307–315 (2022). https://doi.org/10.1038/s41401-021-00659-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-021-00659-8
