
Abstract
Endothelin-1 (ET-1), an endogenous vasoactive peptide, has been found to play an important role in peripheral pain signaling. Acid-sensing ion channels (ASICs) are key sensors for extracellular protons and contribute to pain caused by tissue acidosis. It remains unclear whether an interaction exists between ET-1 and ASICs in primary sensory neurons. In this study, we reported that ET-1 enhanced the activity of ASICs in rat dorsal root ganglia (DRG) neurons. In whole-cell voltage-clamp recording, ASIC currents were evoked by brief local application of pH 6.0 external solution in the presence of TRPV1 channel blocker AMG9810. Pre-application with ET-1 (1−100 nM) dose-dependently increased the proton-evoked ASIC currents with an EC50 value of 7.42 ± 0.21 nM. Pre-application with ET-1 (30 nM) shifted the concentration–response curve of proton upwards with a maximal current response increase of 61.11% ± 4.33%. We showed that ET-1 enhanced ASIC currents through endothelin-A receptor (ETAR), but not endothelin-B receptor (ETBR) in both DRG neurons and CHO cells co-expressing ASIC3 and ETAR. ET-1 enhancement was inhibited by blockade of G-protein or protein kinase C signaling. In current-clamp recording, pre-application with ET-1 (30 nM) significantly increased acid-evoked firing in rat DRG neurons. Finally, we showed that pharmacological blockade of ASICs by amiloride or APETx2 significantly alleviated ET-1-induced flinching and mechanical hyperalgesia in rats. These results suggest that ET-1 sensitizes ASICs in primary sensory neurons via ETAR and PKC signaling pathway, which may contribute to peripheral ET-1-induced nociceptive behavior in rats.
Similar content being viewed by others


Introduction
Endothelin-1 (ET-1) is an endogenous vasoactive peptide. It is derived from endothelial cells and is also produced by other cell types, including inflammatory cells, tumor cells, and neurons [1,2,3,4]. ET-1 exerts its biological effects by binding to two specific G-protein-coupled receptors, endothelin-A receptor (ETAR) and endothelin-B receptor (ETBR) [5, 6]. In the peripheral nervous system, ETAR is expressed in nociceptive primary sensory neurons, including the dorsal root ganglion (DRG) neurons, whereas ETBR is mainly found in satellite glial cells [7]. Numerous studies have demonstrated that ET-1 plays an important role in peripheral pain signaling. In humans, exogenous ET-1 causes tactile allodynia and severe pain [8]. In rodents, an intraplantar injection of ET-1 produces mechanical and thermal hyperalgesia and spontaneous pain-like behaviors [9,10,11,12]. Paw withdrawal thresholds to mechanical stimuli and heat are significantly lower in conditional ET-1 knockout mice [13]. Peripheral ET-1 acts on nociceptors through its cognate receptors, which subsequently modify pain-related ion channels to amplify signal generation [14]. In DRG neurons, ET-1 increases neuronal excitation by hyperpolarizing tetrodotoxin-resistant (TTX-R) Na+ channels and suppressing the delayed outward rectifier current (IK) via ETAR [15, 16]. In contrast, ET-1 decreases the excitability of DRG neurons through the activation of ETBR [17]. ET-1 potentiates TRPV1-mediated currents by an ETAR-mediated and protein kinase C (PKC)-dependent mechanism in both the expression system and sensory neurons [18, 19]. ET-1-induced pain-like behavior is attenuated in TRPV1-deficient mice [20]. TRPV1 also contributes to ET-1-induced thermal and mechanical hyperalgesia [10, 21]. ET-1 sensitizes TRPA1 in primary sensory neurons via an ETAR and protein kinase A (PKA) pathway [22]. TRPA1 is involved in ET-1-induced mechanical hyperalgesia and spontaneous pain-like behavior [22, 23]. However, ET-1-induced nociceptive responses are not completely inhibited in TRPV1-deficient mice or by TRPV1 and TRPA1 antagonists, suggesting that other mechanisms are also involved.
Acid-sensing ion channels (ASICs), such as pH sensors, are also pain-related ion channels. ASICs are expressed in both DRG cell bodies and sensory terminals, where they contribute to proton-evoked pain signaling [24, 25]. ASICs, rather than TRPV1, are shown to mainly mediate pain sensation induced by a moderate (up to pH 6.0) peripheral pH [26,27,28,29]. ASICs were found to be the major player in pain associated with tissue acidosis, such as inflammation, tissue damage, ischemia, and tumors [30,31,32]. Thus, ASICs represent a novel therapeutic target for pain [32,33,34].
ET-1 and protons activate their cognate receptors expressed in nociceptors and separately contribute to the nociceptive process. Since both endothelin receptors and ASICs are distributed in DRG neurons, we hypothesized that there may be an interaction between endothelin receptors and ASICs in the same DRG neuron. Herein, we showed that ET-1 enhanced the electrophysiological activity of ASICs in DRG neurons through ETAR, which may contribute to ET-1-induced spontaneous flinching and mechanical hyperalgesia in rats.
Materials and methods
Isolation of DRG neurons
All experimental protocols were approved by the animal research ethics committee of Hubei University of Science and Technology. All procedures were performed to minimize the suffering of animals. Male Sprague-Dawley rats (5- to 6-week-old) were sacrificed. The DRGs were removed and minced with fine spring scissors. The ganglion fragments were placed in a flask containing 5 mL of Dulbecco's modified Eagle's medium (DMEM, Sigma). DMEM contained trypsin (type II-S, Sigma) 0.5 mg/mL, collagenase (type I-A, Sigma) 1.0 mg/mL and DNase (type IV, Sigma) 0.1 mg/mL, and were incubated at 35 °C in a shaking water bath for 25–30 min. Soybean trypsin inhibitor (type II-S, Sigma) 1.25 mg/mL was then added to stop trypsin digestion.
Electrophysiological recordings
Electrophysiological experiments were carried out as described previously [35]. Whole-cell patch clamp and voltage-clamp recordings were carried out at room temperature (22–25 °C) using a MultiClamp-700B amplifier and Digidata-1440A A/D converter (Axon Instruments, CA, USA). Dissociated neurons were placed into a 35-mm Petri dish and were bathed in an external solution containing (mM): NaCl 150, KCl 5, CaCl2 2.5, MgCl2 2, HEPES 10, and d-glucose 10, and the pH and osmolarity were adjusted to 7.4 with NaOH and to 330 mOsm/L with sucrose, respectively. Cells were kept for at least 60 min in normal external solution before the start of electrophysiological experiments. The neurons selected for electrophysiological experiments were 15–35 μm in diameter, which are thought to be nociceptive neurons. Recording pipettes were pulled using a Sutter P-97 puller (Sutter Instruments, CA, USA). The micropipettes were filled with internal solution containing (mM): KCl 140, MgCl2 2.5, HEPES 10, EGTA 11 and ATP 5; the pH of the solution was adjusted to 7.2 with KOH and its osmolarity was adjusted to 310 mOsm/L with sucrose. The resistance of the recording pipette was in the range of 3–6 MΩ. To establish a whole-cell configuration, a small patch of membrane underneath the tip of the pipette was aspirated to form a giga seal, and then, negative pressure was applied to rupture it. The series resistance was compensated for by 70%–80%. The capacitance compensation was also adjusted before recording the membrane currents. The membrane voltage was maintained at −60 mV in all voltage-clamp experiments. Current-clamp recordings were obtained by switching to current-clamp mode after a stable whole-cell configuration was formed in voltage-clamp mode. Only cells with a stable resting membrane potential (more negative than −50 mV) were used in the study. Signals were sampled at 10 to 50 kHz and filtered at 2 to 10 kHz, and the data were stored in a compatible PC computer for offline analysis using pCLAMP 10 acquisition software (Axon Instruments, CA, USA).
Drug application
Drugs were obtained from Sigma Chemical Co. (St. Louis, MO, USA), including hydrochloric acid, ET-1, BQ-123, BQ-778, amiloride, APETx2, capsaicin, and AMG9810. Different pH values were configured with hydrochloric acid and external solution. Working ET-1 and other drugs were freshly prepared in normal external solution and held in a series of independent reservoirs. The pipette tips connecting the reservoirs were positioned ∼30 μm away from the recorded neurons. The application of each drug was driven by gravity and controlled by the corresponding valve. In some experiments where GDP-β-S (Sigma) or GF109203X (RBI) was applied for intracellular dialysis through recording patch pipettes, they were dissolved in the internal solution before use. To ensure that the cell interior was perfused with the dialysis drug, there was at least a 30 min interval between the establishment of whole-cell access and current measurement. To functionally characterize ASIC activity, we used AMG9810 (5 μM) to block TRPV1 in the extracellular solution [36].
Cell culture and transfection
ASIC3 and ETAR complementary DNA (cDNAs) were used for heterologous expression in CHO cells as described previously [37]. In brief, CHO cells were cultured at 37 °C in a humidified atmosphere of 5% CO2 and 95% O2 and passaged twice a week. Transient transfection of CHO cells was performed using a HilyMax liposome transfection reagent (Dojindo Laboratories). CHO cells were maintained in F-12 nutrient mixture (added 1.176 g of NaHCO3/L medium) supplemented with 10% fetal bovine serum and 1% gluta-MAXTM-1 (100 ×; Invitrogen). When ASIC3 and ETAR cDNA was cotransfected, the ratio was maintained at 1:1. All plasmids contained, in addition to the desired ASIC3 cDNA, the coding sequence for enhanced green fluorescent protein to aid in the identification of transfected cells. Electrophysiological measurements were performed 24–48 h after transfection.
Animal behavioral assay
Rats were placed in a 30 × 30 × 30 cm Plexiglas chamber and allowed to habituate for at least 30 min before nociceptive behavioral experiments. A double-blind experiment was carried out. To assess ET-1-induced flinching, 49 rats were divided randomly into seven groups, with seven rats in each group. Separate groups of rats were coded and administered drugs. ET-1 (10 ng/paw) or vehicle (phosphate-buffered saline) was injected into the hind paw of rats using 100 μL syringe and 30-gauge needle in a volume of 50 μL, with BQ-123 (1 µg/paw), BQ-788 (100 ng/paw), amiloride (100 µM, 50 µL/paw) or APETx2 (10 μM, 50 µL/paw) co-injected with ET-1. The other experimenters measured nociceptive behavior. Flinching (time spent licking, biting, and lifting the injected paw) was counted for 50 min starting immediately after the injection. To assess ET-1-induced mechanical hyperalgesia, 42 rats were divided randomly into 6 groups, with seven rats in each group. Separate groups of rats were coded and administered drugs as above. Paw withdrawal thresholds (PWTs) were determined thereafter, at 0, 0.5, and 2.5 h after the injection. Paw withdrawal thresholds (PWTs) were tested using the up and down method [38]. A series of von Frey filaments (Stoelting, Wood Dale, IL) were used to vertically press against the injected hind paw until the filaments were bent for 6 s. Quick withdrawal, licking or paw flinching were counted as positive reactions. The test was carried out many times in each rat, and the interval between tests was at least 5 min. All rats were tested only in one behavioral experiment, either for flinching or mechanical hyperalgesia.
Data analysis
The data were statistically compared using Student's t-test or analysis of variance (ANOVA), followed by Bonferroni’s post hoc test. Statistical analysis of concentration–response data was performed using the nonlinear curve-fitting program ALLFIT. The data are expressed as the mean ± S.E.M.
Results
Potentiation of ASIC currents by ET-1 in rat DRG neurons
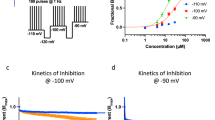
In the present study, AMG9810 (5 μM) was added to the external solution to block proton-induced TRPV1 activation [36]. As shown in Fig. 1a, a perfusion of pH 6.0 external solution to DRG neurons for 5 s caused a rapid inward current (IpH6.0). This IpH6.0 could be completely blocked by both 10 μM amiloride, a broad-spectrum ASIC channel blocker, and 2 μM APETx2, an ASIC3 blocker. However, capsaicin (50 nM) failed to evoke any membrane currents in the presence of AMG9810. Thus, these proton-induced currents were considered to be pure ASIC currents after AMG9810 blocked proton-induced TRPV1 activation.

a Representative current traces were produced by the application of a pH 6.0 acidic solution for 5 s on a DRG neuron. The proton-gated current (IpH6.0) could be blocked by 10 μM amiloride, a broad-spectrum ASIC channel blocker. This current was also blocked by 2 μM APETx2, an ASIC3 blocker. The proton-gated currents were recorded in the presence of AMG9810 (5 μM) to block proton-induced TRPV1 activation. Under such conditions, capsaicin (50 nM) failed to evoke any membrane currents. The membrane potential was clamped at –60 mV. b The sequential current traces illustrate that the IpH6.0 amplitude was enhanced by different concentrations of ET-1 in a representative DRG neuron. ET-1 was preapplied to external solution for 2 min. c The graph shows ET-1 concentration-dependently increased the IpH6.0 with an EC50 of 7.42 ± 0.21 nM. Each point represents the mean ± S.E.M of 8–10 cells.
In some DRG neurons sensitive to acid stimuli (62.50%, 10/16), we observed that the pre-application of ET-1 for 2 min increased the peak amplitude of the ASIC currents. As shown in Fig. 1b, c, the IpH6.0 was enhanced by ET-1 pretreatment, and the potentiation of the IpH6.0 was dependent upon the concentration of ET-1. Fig. 1b shows that the peak amplitude of the IpH6.0 increased as the concentration of pretreated ET-1 increased from 1 to 100 nM in a representative DRG neuron. The potentiation of the IpH6.0 was reversible after the washout of ET-1. Fig. 1c shows the concentration–response curve for ET-1 with an EC50 (half-maximal effective concentration) value of 7.42 ± 0.21 nM. The results indicated that ET-1 increased ASIC currents in rat DRG neurons in a concentration-dependent manner.
We then investigated whether the ET-1 potentiation of ASIC currents was dependent upon pH values. ASIC currents were measured by applying a range of different low pH values in the absence and presence of ET-1 (30 nM). Fig. 2a shows that the peak amplitudes of IpH7.0, IpH6.0, and IpH5.0 increased after the pre-application of ET-1 for 2 min. Fig. 2b shows the effect of ET-1 (30 nM) on the concentration–response curve to protons. First, the pre-application of ET-1 shifted the concentration–response curve to protons upwards, as indicated by an increase of 61.11% ± 4.33% in the maximal current response to protons in the presence of ET-1. Second, neither the Hill coefficient nor slope of the curve was significantly changed by ET-1 (pH: n = 1.89 ± 0.37; ET-1 + pH: n = 1.86 ± 0.42; P > 0.1, Bonferroni’s post hoc test). Third, the pH0.5 (pH for half-maximal activation) values of both curves also showed no significant difference (pH: pH0.5 = 5.81 ± 0.11; ET-1 + pH: pH0.5 = 5.94 ± 0.16; P > 0.1, Bonferroni’s post hoc test). We therefore concluded that the potentiation of the ASIC current by ET-1 was not due to a change in the apparent affinity of protons for ASICs.

a Sequential currents were evoked by different low pH values in the absence and presence of ET-1 (30 nM) pretreatment. b Proton concentration–response curves for ASIC activation in the absence (white circle) and presence (black circle) of extracellular 30 nM ET-1. The pre-application of ET-1 shifted the concentration–response curve for protons upwards. Each point represents the mean ± S.E.M. of 7–10 neurons. All peak current values were normalized to the peak current maximally activated by pH 5.0 applied alone in the absence of ET-1 (marked with an asterisk). The figure shows averaged data fitted with the Hill equation.
Involvement of ETAR and PKC signaling in the ET-1 potentiation of ASIC currents
To verify whether the ET-1 potentiation of ASIC currents was mediated by ET receptors, we observed the effects of the selective ETAR antagonist BQ-123 and the selective ETBR antagonist BQ-788 on the ET-1 enhancement of ASIC currents. As shown in Fig. 3a, b, the amplitude of the IpH6.0 increased 59.39 % ± 9.47% after ET-1 (30 nM) pretreatment alone. However, the amplitude of the IpH6.0 increased only 8.97% ± 4.13% when both BQ-123 (100 nM) and ET-1 (30 nM) were coapplied to DRG neurons (P < 0.01, compared with ET-1 alone, Bonferroni’s post hoc test, n = 8; Fig. 3a, b). In contrast, ET-1 (30 nM) caused an increase of 54.48% ± 6.19% on the IpH6.0 after BQ-788 (100 nM) was coapplied with ET-1 (P > 0.1, compared with ET-1 alone, Bonferroni’s post hoc test, n = 8; Fig. 3a, b). These results indicated that ETAR, but not ETBR, is involved in the ET-1 potentiation of ASIC currents in rat DRG neurons.

The current traces in a and the bar graph in b show that the IpH6.0 was potentiated by ET-1 (30 nM) preapplied alone for 2 min, and the ET-1 potentiation of the IpH6.0 was blocked by the coapplication of the ETAR-specific antagonist BQ-123 (100 nM), but not by the ETBR-specific antagonist BQ-788 (100 nM). Statistical tests were performed using Bonferroni’s post hoc test, and significance is shown as follows: **P < 0.01, compared with control column, ##P < 0.01, compared with ET-1 alone column. n = 8 in each column. The current traces in c and the bar graph in d show the effects of ET-1 (30 nM) on the IpH6.0 with recording pipettes filled with normal internal solution, the nonhydrolyzable GDP analog GDP-β-S (500 μM), or the PKC inhibitor GF109203X (2 μM) containing internal solution. Intracellular dialysis of GDP-β-S or GF109203X almost completely abolished the potentiation of the IpH6.0 by ET-1. **P < 0.01, post hoc Bonferroni’s test, compared with normal internal solution. n = 8 in each column.
We further explored the signaling pathway downstream of ETAR in the ET-1 potentiation of ASIC currents using an intracellular dialysis technique. ETAR is most commonly associated with the Gq/11 G-protein subtype, which activates PLC and leads to a cascade of events, including the activation of PKC [5, 39]. Thus, we internally applied GDP-β-S (a nonhydrolyzable GDP analog) or GF109203X (a selective PKC inhibitor) to DRG cells through recording patch pipettes. Unlike the increase of 59.39% ± 9.47% on the IpH6.0 produced under the normal internal solution condition, the pre-application of ET-1 (30 nM) for 2 min produced only increases of 6.54% ± 2.85% and 12.47% ± 3.06% on the IpH6.0 when GDP-β-S (500 μM) or GF109203X (2 μM), respectively, was included in the pipette solution (P < 0.01, compared with normal internal solution, post hoc Bonferroni’s test, n = 8; Fig. 3c, d). These results indicated that the potentiation of ASIC currents by ET-1 was dependent upon G-protein and PKC signaling.
To further verify whether ET-1 could enhance acid-evoked currents mediated by ASIC3, ASIC3 was coexpressed with ETAR in CHO cells. We observed that a rapid reduction in the extracellular pH from 7.4 to 6.0 for 5 s also caused a rapid inward current. The acid-evoked currents were ASIC3 currents, since they were blocked by 2 μM APETx2, an ASIC3 blocker. Similar to that observed in DRG neurons, the ASIC3 currents were enhanced by the pre-application of ET-1 (30 nM) in CHO cells co-expressing ASIC3 and ETAR (Fig. 4a, b). The ET-1 potentiation of ASIC3 currents was also inhibited after pharmacological blockade of ETAR by BQ-123 (Fig. 4a, b). In contrast, ET-1 had no effect on ASIC3 currents at a concentration of 30 nM in CHO cells expressing ASIC3 alone, but not expressing ETAR (Fig. 4c, d).

The current traces in a and bar graphs in b show that the IpH6.0 was enhanced by ET-1 (30 nM) preapplied alone for 2 min in CHO cells co-expressing ASIC3 and ETAR. This enhancing effect was inhibited by the coapplication of ET-1 and BQ-123 (100 nM), but not by the coapplication of ET-1 and BQ-788 (100 nM). Statistical tests were performed using Bonferroni’s post hoc test, and significance is shown as follows: **P < 0.01. n = 6 in each column. The current traces in c and bar graphs in d show that ET-1 had no effect on the IpH6.0 in CHO cells expressing ASIC3 alone, but not expressing ETAR. Currents were normalized to the control (100%, white column). n = 5 in each column. The proton-gated current in CHO cells could be blocked by 2 μM APETx2, an ASIC3 inhibitor.
Enhancement of proton-evoked action potentials by ET-1 in rat DRG neurons
Under current-clamp conditions, a stimulus of pH 6.0 could trigger bursts of action potentials (APs) in DRG neurons in the presence of 5 μM AMG9810 to block proton-induced TRPV1 activation (Fig. 5a). Similar to that observed under voltage-clamp conditions, ET-1 also increased the number of APs evoked by acidic stimuli in DRG neurons (Fig. 5a, b). As shown in Fig. 5b, the number of APs evoked by a stimuli of pH 6.0 increased in seven DRG neurons with ET-1 (30 nM) pretreatment for 2 min (P < 0.01, paired t-test, n = 7, Fig. 5b). However, pretreatment with vehicle had no effect on the number of APs evoked by stimuli of pH 6.0 (P > 0. 1, paired t-test; n = 7, Fig. 5c). These results indicated that ET-1 exerted an enhancing effect on acid-evoked action potentials in rat DRG neurons.

a Under current-clamp conditions, action potentials were evoked by a pH 6.0 acid stimulus in two DRG neurons with ET-1 (30 nM) or vehicle pretreatment for 2 min. AMG9810 (5 μM) was used to block proton-induced TRPV1 activation. b, c The graphs show the pre-application of ET-1 (30 nM), but not vehicle, increased the number of action potentials by pH 6.0 acid perfusions. **P < 0.01, paired t-test, n = 7 cells.
Attenuation of ET-1-induced flinching and mechanical hyperalgesia by the pharmacological blockade of ETAR, ETBR, or ASICs in rats
We finally examined the contribution of ASICs to ET-1-induced nocifensive behavior in vivo. Rats displayed an obvious flinching response after ET-1 (10 ng/paw) was injected subcutaneously into the ipsilateral hind paw compared with rats receiving vehicle injection only (Fig. 6a). The ET-1-induced flinching was almost completely blocked by subcutaneous cotreatment with BQ-123 (100 ng/paw), but not with BQ-788 (100 ng/paw). As shown in Fig. 6a, when ASICs blocker amiloride (100 µM, 50 µL/paw) or ASIC3 blocker rAPETx2 (10 μM, 50 µL/paw) was coadministered subcutaneously with ET-1 (10 ng/paw), it significantly attenuated hind paw flinching (P < 0.05, Bonferroni’s post hoc test, compared with ET-1 alone group, n = 7 rats). Rats injected with ET-1 into the hind paw (10 ng/paw) also displayed obvious mechanical hyperalgesia, measured 0.5 and 2.5 h after injection (Fig. 6b, c). ET-1-induced mechanical hyperalgesia disappeared 24 h after injection. Fig. 6b shows that ET-1-induced mechanical hyperalgesia was partially attenuated by subcutaneous cotreatment with BQ-123 (100 ng/paw) or BQ-788 (100 ng/paw). Furthermore, the intraplantar application of both BQ-123 (100 ng/paw) and BQ-788 (100 ng/paw) together almost completely blocked the mechanical hyperalgesia induced by ET-1 (Fig. 6b). As shown in Fig. 6c, amiloride (100 µM, 50 µL/paw) or APETx2 (10 μM, 50 µL/paw) also significantly attenuated the mechanical hyperalgesia induced by ET-1 (P < 0.05 and 0.01, Bonferroni’s post hoc test, compared with ET-1 alone group, n = 7 rats). The injection of these antagonists alone at the same dosage used above did not produce any effects on the number of flinches or the PWT (data not shown). Together, these results demonstrated that ASICs mediated, at least partially, ET-1-induced flinching and mechanical hyperalgesia.

a ET-1-induced flinching was prevented by the ETAR-specific antagonist BQ-123, but not by the ETBR-specific antagonist BQ-788. When the ASIC blocker amiloride or APETx2 was subcutaneously administered together with ET-1, ET-1 induced an attenuated flinching response. b ET-1-induced mechanical hyperalgesia was attenuated by BQ-123 or BQ-788 alone and was blocked by the combined application of BQ-123 and BQ-788. c ET-1-induced mechanical hyperalgesia was also attenuated after the pharmacological blockade of ASICs by amiloride or APETx2. Flinching and mechanical hyperalgesia were induced by the injection of ET-1 subcutaneously into the rat plantar hind paw. Spontaneous flinching was evaluated by the time spent licking/lifting the injected paw (in seconds). The paw withdrawal threshold (PWT, in g) of rats was measured by von Frey hair test before injection (0 h) and 0.5, 2.5, 24 h after injection. BQ-123 (100 ng/paw) and/or BQ-788 (100 ng/paw), amiloride (100 µM, 50 µL/paw), or APETx2 (10 μM, 50 µL/paw) were co-injected with ET-1 (10 ng/paw) into the hind paw of rats. n = 7 rats in each group. *P < 0.05, **P < 0.01, ***P < 0.001, Bonferroni’s post hoc test, compared with the ET-1 alone group.
Discussion
We demonstrated here that ET-1 sensitized ASICs through ETAR. ET-1 increased ASIC-medicated currents and action potentials in rat primary sensory neurons, which may contribute to ET-1-induced spontaneous flinching and mechanical hyperalgesia in rats.
Low pH-evoked currents are mediated by proton-gated ion channels, including ASICs and TRPV1 [40]. In the present study, the activation of TRPV1 channels was completely blocked by AMG9810, so capsaicin failed to evoke any membrane currents. We believe that ASICs mediated these low pH-evoked currents, which could be completely blocked by ASIC channel blockers; these evoked currents were, therefore, considered to be ASIC currents, although precise ASIC subunits need to be identified. To date, at least seven ASIC subunits have been identified in mammals. There are six ASIC subunits (ASIC1a and b, ASIC2a and b, ASIC3, and ASIC4) expressed in both DRG cell bodies and sensory terminals [24, 25]. These ASIC subunits exist as homomeric or heteromeric channels in DRG neurons. The ASIC3 subunit is the most abundant in DRG and has emerged as a critical pH sensor [26, 32].
The current data demonstrated that ET-1 can enhance the electrophysiological activity of ASICs in rat DRG neurons. ET-1 increased not only ASIC-mediated currents in voltage-clamp experiments but also the number of action potentials evoked by extracellular acid stimuli in current-clamp experiments. Obviously, the two results corroborated each other. ET-1 shifted the proton concentration–response curve upward without changing the pH0.5. Thus, the sensitization of ASICs by ET-1 did not change the apparent affinity of protons for ASICs.
We found that ET-1 sensitized ASICs in rat DRG neurons through ETAR, but not through ETBR, because the ET-1 potentiation of ASIC currents was completely blocked by the selective ETAR antagonist BQ-123 and was not affected by the selective ETBR antagonist BQ-788. In CHO cells co-expressing ASIC3 and ETAR, ET-1 also sensitized ASIC3 through ETAR. Together, ETAR, but not ETBR, appeared to contribute to the ET-1 potentiation of ASIC currents. The results were consistent with the expression characteristics of ETAR and ETBR in primary sensory neurons. ETAR is present in a subset of small- and medium-sized DRG neurons and their axons, whereas ETBR is mainly expressed by satellite glial cells and nonmyelinating Schwann cells, but not neurons [7]. It has been found that ETAR is mainly expressed in isolectin B4-negative (IB4−) DRG neurons [41]. ASICs are also more prevalent in IB4− DRG neurons than in IB4+ neurons [42]. Thus, the ET-1 potentiation of ASICs may mainly occur in peptidergic and IB4- DRG neurons. ETAR belongs to a subfamily of GPCRs and is coupled to multiple second messenger systems. Although ETAR couples to Gs in other tissues, ET-1 does not increase intracellular cAMP levels in isolated DRG neurons [43]. ETAR also couples to Gq/11 to activate PLCβ, which, in turn, produces IP3 and DAG, followed by intracellular Ca2+ release and the activation of PKC [5, 39, 44]. ET-1 can trigger intracellular calcium mobilization through ETAR in DRG neurons [45]. ETAR, but not ETBR, activates PKC in peptidergic, IB4− sensory neurons [41]. In the present study, the lack of ET-1 potentiation in DRG neurons treated with GDP-β-S or the PKC inhibitor GF109203X indicated that a G-protein- and PKC-dependent pathway was involved in the sensitization of ASICs by ET-1. It has been shown that PKC is involved in the ET-1-mediated enhancement of capsaicin-induced Ca2+ increases in sensory neurons. ET-1 potentiates TRPV1 currents via the ETAR-mediated activation of PKC in both DRG neurons and the trigeminal ganglion neurons [18, 19]. The stimulation of ETAR by ET-1 inhibits inward rectifier K+ channels via the PKC pathway [46]. However, ET-1 sensitizes TRPA1 via the ETAR and PKA pathways in primary sensory neurons [22]. Our previous studies show that ASICs are modulated by intracellular PKC signaling [35, 37].
Initial studies show that exogenous ET-1 elicits spontaneous pain and hyperalgesia. In the present study, rats displayed flinching responses and mechanical hyperalgesia after ET-1 was injected subcutaneously into the ipsilateral hind paw. ET-1-induced flinching was almost completely blocked by subcutaneous cotreatment with BQ-123, but not BQ-788, suggesting this behavior is mediated by ETAR, but not ETBR. A combined intraplantar injection of BQ-123 and BQ-788 completely blocked ET-1-induced mechanical hyperalgesia, suggesting it is mediated by both ETAR and ETBR. Previous studies have shown that a subcutaneous injection of ET-1 into the rat plantar hind paw induces flinching behavior and spike responses in nociceptors through ETAR [9, 47]. It has been shown that ETAR activation contributes to ET-1-induced pain-like behavior and thermal hyperalgesia [9, 10, 48]. In contrast, ET-1-induced mechanical hyperalgesia requires both ETAR and ETBR activation [11, 21, 49]. ET-1 has been implicated in inflammation, so we cannot rule out ET-1-elicited nociceptive behavior that may be due to the induction of inflammatory responses after hind paw injection. However, some studies also show that ET-1 elicits nociceptive behavior due to ET-1’s direct action on primary afferents, since this nociceptive behavior is mediated by ETAR located immunocytochemically on primary afferents [7, 9]. In this work, we used cell bodies of DRG neurons as a simple and accessible model to examine the characteristics of the membrane of peripheral terminals. The sensitization of ASICs by ET-1 may also occur in peripheral terminals. The pharmacological blockade of ASICs significantly alleviated ET-1-induced flinching and mechanical hyperalgesia in rats, suggesting that ASICs mediated, at least partially, ET-1-induced nociceptive behavior. It is possible that ET-1 sensitizes ASICs on primary afferents, which underlies ET-1-induced nociceptive behavior.
During tissue damage and inflammation, ET-1 is generated and released in a variety of cells, such as endothelial cells, monocytes and macrophages, which results in an increase of local ET-1 levels [3, 4]. DRG neurons are also an important source of ET-1 [1]. ET-1 can excite these sensory neurons in a paracrine or autocrine manner. ET-1 levels are higher within a cancerous microenvironment, and the administration of an ETAR antagonist alleviates cancer pain [3, 5, 50, 51]. Thus, an endogenous concentration of ET-1 in diseased tissue is required to cause pain. The present tested concentration of ET-1 (30 nM) falls within the endogenous concentration of ET-1 ranges reported [52]. ET-1 is implicated in the pathogenesis of a variety of types of pain, such as inflammatory, neuropathic, and tumoral pain [5, 14, 53]. During tissue damage and inflammation, protons are released from damaged cells and the degranulation of mast cells. These released protons result in a drop in the local extracellular pH. For instance, the local extracellular pH drops to approximately 5.4 during acute inflammation, which is low enough to activate ASICs and cause pain [30, 54]. An acidic pH is commonly associated with cancerous microenvironments, contributing to cancer-related pain via the activation of ASICs and TRPV1 [55, 56]. Under inflammatory and cancer states, once both ET-1 and protons are locally released together in diseased tissue, and they activate their cognate receptors expressed in nociceptors to initiate and/or sensitize nociceptive processes. Furthermore, the present study showed that released ET-1 could sensitize ASICs through ETAR. ET-1 and protons may potentially work synergistically to produce more intense pain and mechanical hyperalgesia.
Conclusion
In summary, the major finding of this study was that ET-1 enhanced the electrophysiological activity of ASICs in DRG neurons via the intracellular PKC signaling pathway, which may contribute to ET-1-induced nociceptive behavior in rats. Our results demonstrated that ASICs act as novel targets of ET-1, which may provide a strategy for the treatment of pain associated with ET-1, such as inflammatory pain and cancer-related pain.
References
Giaid A, Gibson SJ, Ibrahim BN, Legon S, Bloom SR, Yanagisawa M, et al. Endothelin 1, an endothelium-derived peptide, is expressed in neurons of the human spinal cord and dorsal root ganglia. Proc Natl Acad Sci USA. 1989;86:7634–8.
Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–5.
Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, et al. Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med. 1995;1:944–9.
Ehrenreich H, Anderson RW, Fox CH, Rieckmann P, Hoffman GS, Travis WD, et al. Endothelins, peptides with potent vasoactive properties, are produced by human macrophages. J Exp Med. 1990;172:1741–8.
Smith TP, Haymond T, Smith SN, Sweitzer SM. Evidence for the endothelin system as an emerging therapeutic target for the treatment of chronic pain. J Pain Res. 2014;7:531–45.
Davenport AP, Hyndman KA, Dhaun N, Southan C, Kohan DE, Pollock JS, et al. Endothelin. Pharmacol Rev. 2016;68:357–418.
Pomonis JD, Rogers SD, Peters CM, Ghilardi JR, Mantyh PW. Expression and localization of endothelin receptors: implications for the involvement of peripheral glia in nociception. J Neurosci. 2001;21:999–1006.
Dahlof B, Gustafsson D, Hedner T, Jern S, Hansson L. Regional haemodynamic effects of endothelin-1 in rat and man: unexpected adverse reaction. J Hypertens. 1990;8:811–7.
Gokin AP, Fareed MU, Pan HL, Hans G, Strichartz GR, Davar G. Local injection of endothelin-1 produces pain-like behavior and excitation of nociceptors in rats. J Neurosci. 2001;21:5358–66.
Kawamata T, Ji W, Yamamoto J, Niiyama Y, Furuse S, Namiki A. Contribution of transient receptor potential vanilloid subfamily 1 to endothelin-1-induced thermal hyperalgesia. Neuroscience. 2008;154:1067–76.
Motta EM, Chichorro JG, D'Orleans-Juste P, Rae GA. Roles of endothelin ETA and ETB receptors in nociception and chemical, thermal and mechanical hyperalgesia induced by endothelin-1 in the rat hindpaw. Peptides. 2009;30:918–25.
Motta EM, Calixto JB, Rae GA. Mechanical hyperalgesia induced by endothelin-1 in rats is mediated via phospholipase C, protein kinase C, and MAP kinases. Exp Biol Med (Maywood). 2006;231:1141–5.
Hasue F, Kuwaki T, Kisanuki YY, Yanagisawa M, Moriya H, Fukuda Y, et al. Increased sensitivity to acute and persistent pain in neuron-specific endothelin-1 knockout mice. Neuroscience. 2005;130:349–58.
Barr TP, Kam S, Khodorova A, Montmayeur JP, Strichartz GR. New perspectives on the endothelin axis in pain. Pharmacol Res. 2011;63:532–40.
Feng B, Strichartz G. Endothelin-1 raises excitability and reduces potassium currents in sensory neurons. Brain Res Bull. 2009;79:345–50.
Zhou Z, Davar G, Strichartz G. Endothelin-1 (ET-1) selectively enhances the activation gating of slowly inactivating tetrodotoxin-resistant sodium currents in rat sensory neurons: a mechanism for the pain-inducing actions of ET-1. J Neurosci. 2002;22:6325–30.
Mule NK, Singh JN, Shah KU, Gulati A, Sharma SS. Endothelin-1 decreases excitability of the dorsal root ganglion neurons via ETB receptor. Mol Neurobiol. 2018;55:4297–310.
Plant TD, Zollner C, Kepura F, Mousa SS, Eichhorst J, Schaefer M, et al. Endothelin potentiates TRPV1 via ETA receptor-mediated activation of protein kinase C. Mol Pain. 2007;3:35.
Yamamoto T, Ono K, Hitomi S, Harano N, Sago T, Yoshida M, et al. Endothelin receptor-mediated responses in trigeminal ganglion neurons. J Dent Res. 2013;92:335–9.
Kawamata T, Ji W, Yamamoto J, Niiyama Y, Furuse S, Omote K, et al. Involvement of transient receptor potential vanilloid subfamily 1 in endothelin-1-induced pain-like behavior. Neuroreport. 2009;20:233–7.
Balonov K, Khodorova A, Strichartz GR. Tactile allodynia initiated by local subcutaneous endothelin-1 is prolonged by activation of TRPV-1 receptors. Exp Biol Med (Maywood). 2006;231:1165–70.
Zheng X, Tai Y, He D, Liu B, Wang C, Shao X, et al. ETAR and protein kinase A pathway mediate ET-1 sensitization of TRPA1 channel: A molecular mechanism of ET-1-induced mechanical hyperalgesia. Mol Pain. 2019;15:1744806919842473.
Liang J, Bi H, Ji W. Involvement of TRPA1 in ET-1-induced pain-like behavior in mice. Neuroreport. 2010;21:201–5.
Alvarez de la Rosa D, Zhang P, Shao D, White F, Canessa CM. Functional implications of the localization and activity of acid-sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci USA. 2002;99:2326–31.
Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ, et al. Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons. Proc Natl Acad Sci USA. 2002;99:2338–43.
Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V, et al. ASIC3, a sensor of acidic and primary inflammatory pain. EMBO J. 2008;27:3047–55.
Lingueglia E. Acid-sensing ion channels in sensory perception. J Biol Chem. 2007;282:17325–9.
Ugawa S, Ueda T, Ishida Y, Nishigaki M, Shibata Y, Shimada S. Amiloride-blockable acid-sensing ion channels are leading acid sensors expressed in human nociceptors. J Clin Invest. 2002;110:1185–90.
Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nat Rev Neurosci. 2013;14:461–71.
Reeh PW, Steen KH. Tissue acidosis in nociception and pain. Prog Brain Res. 1996;113:143–51.
Deval E, Lingueglia E. Acid-Sensing Ion Channels and nociception in the peripheral and central nervous systems. Neuropharmacology. 2015;94:49–57.
Lee CH, Chen CC. Roles of ASICs in nociception and proprioception. Adv Exp Med Biol. 2018;1099:37–47.
Li WG, Xu TL. Acid-sensing ion channels: a novel therapeutic target for pain and anxiety. Curr Pharm Des. 2015;21:885–94.
Dussor G. ASICs as therapeutic targets for migraine. Neuropharmacology. 2015;94:64–71.
Qiu F, Qiu CY, Liu YQ, Wu D, Li JD, Hu WP. Potentiation of acid-sensing ion channel activity by the activation of 5-HT(2) receptors in rat dorsal root ganglion neurons. Neuropharmacology. 2012;63:494–500.
Gavva NR, Tamir R, Qu Y, Klionsky L, Zhang TJ, Immke D. et al. AMG 9810 [(E)-3-(4-t-butylphenyl)-N-(2,3-dihydrobenzo[b][1,4] dioxin-6-yl)acrylamide], a novel vanilloid receptor 1 (TRPV1) antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2005;313:474–84.
Wu J, Liu TT, Zhou YM, Qiu CY, Ren P, Jiao M, et al. Sensitization of ASIC3 by proteinase-activated receptor 2 signaling contributes to acidosis-induced nociception. J Neuroinflammation. 2017;14:150.
Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63.
Masaki T, Miwa S, Sawamura T, Ninomiya H, Okamoto Y. Subcellular mechanisms of endothelin action in vascular system. Eur J Pharmacol. 1999;375:133–8.
Kweon HJ, Yu SY, Kim DI, Suh BC. Differential regulation of proton-sensitive ion channels by phospholipids: a comparative study between ASICs and TRPV1. PLoS ONE. 2015;10:e0122014.
Vellani V, Prandini M, Giacomoni C, Pavesi G, Ravegnani L, Magherini PC. Functional endothelin receptors are selectively expressed in isolectin B4-negative sensory neurons and are upregulated in isolectin B4-positive neurons by neurturin and glia-derived neurotropic factor. Brain Res. 2011;1381:31–7.
Liu M, Willmott NJ, Michael GJ, Priestley JV. Differential pH and capsaicin responses of Griffonia simplicifolia IB4 (IB4)-positive and IB4-negative small sensory neurons. Neuroscience. 2004;127:659–72.
Dymshitz J, Vasko MR. Endothelin-1 enhances capsaicin-induced peptide release and cGMP accumulation in cultures of rat sensory neurons. Neurosci Lett. 1994;167:128–32.
Zhang WM, Yip KP, Lin MJ, Shimoda LA, Li WH, Sham JS. ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am J Physiol Lung Cell Mol Physiol. 2003;285:L680–90.
Yamamoto H, Kawamata T, Ninomiya T, Omote K, Namiki A. Endothelin-1 enhances capsaicin-evoked intracellular Ca2+ response via activation of endothelin a receptor in a protein kinase Cepsilon-dependent manner in dorsal root ganglion neurons. Neuroscience. 2006;137:949–60.
Park WS, Han J, Kim N, Youm JB, Joo H, Kim HK, et al. Endothelin-1 inhibits inward rectifier K+ channels in rabbit coronary arterial smooth muscle cells through protein kinase C. J Cardiovasc Pharmacol. 2005;46:681–9.
McKelvy AD, Mark TR, Sweitzer SM. Age- and sex-specific nociceptive response to endothelin-1. J Pain. 2007;8:657–66.
Piovezan AP, D'Orleans-Juste P, Souza GE, Rae GA. Endothelin-1-induced ET(A) receptor-mediated nociception, hyperalgesia and oedema in the mouse hind-paw: modulation by simultaneous ET(B) receptor activation. Br J Pharmacol. 2000;129:961–8.
da Cunha JM, Rae GA, Ferreira SH, Cunha Fde Q. Endothelins induce ETB receptor-mediated mechanical hypernociception in rat hindpaw: roles of cAMP and protein kinase C. Eur J Pharmacol. 2004;501:87–94.
Pickering V, Jay Gupta R, Quang P, Jordan RC, Schmidt BL. Effect of peripheral endothelin-1 concentration on carcinoma-induced pain in mice. Eur J Pain. 2008;12:293–300.
Yuyama H, Koakutsu A, Fujiyasu N, Tanahashi M, Fujimori A, Sato S, et al. Effects of selective endothelin ET(A) receptor antagonists on endothelin-1-induced potentiation of cancer pain. Eur J Pharmacol. 2004;492:177–82.
van Papendorp CL, Cameron IT, Davenport AP, King A, Barker PJ, Huskisson NS, et al. Localization and endogenous concentration of endothelin-like immunoreactivity in human placenta. J Endocrinol. 1991;131:507–11.
Hans G, Deseure K, Adriaensen H. Endothelin-1-induced pain and hyperalgesia: a review of pathophysiology, clinical manifestations and future therapeutic options. Neuropeptides. 2008;42:119–32.
Kweon HJ, Suh BC. Acid-sensing ion channels (ASICs): therapeutic targets for neurological diseases and their regulation. BMB Rep. 2013;46:295–304.
Lam DK. Emerging factors in the progression of cancer-related pain. Pain Manag. 2016;6:487–96.
Nagae M, Hiraga T, Yoneda T. Acidic microenvironment created by osteoclasts causes bone pain associated with tumor colonization. J Bone Miner Metab. 2007;25:99–104.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81671101 and No. 31471062).
Author information
Authors and Affiliations
Contributions
WPH designed this research. LW, TTL, YJ, SW, and CYQ performed the experiments. LW and TTL participated in data analysis. WPH, LW, and TTL wrote the paper. All authors contributed substantially to this research and reviewed this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Wu, L., Liu, Tt., Jin, Y. et al. Endothelin-1 enhances acid-sensing ion channel currents in rat primary sensory neurons. Acta Pharmacol Sin 41, 1049–1057 (2020). https://doi.org/10.1038/s41401-019-0348-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0348-z
