
Abstract
β-amyloid (Aβ) is one of the inducing factors of astrocytes activation and neuroinflammation, and it is also a crucial factor for the development of Alzheimer’s disease (AD). Icariside II (ICS II) is an active component isolated from a traditional Chinese herb Epimedium, which has shown to attnuate lipopolysaccharide (LPS)-induced neuroinflammation through regulation of NF-κB signaling pathway. In this study we investigated the effects of ICS II on LPS-induced astrocytes activation and Aβ accumulation. Primary rat astrocytes were pretreated with ICS II (5, 10, and 20 μM) or dexamethasone (DXMS, 1 μM) for 1 h, thereafter, treated with LPS for another 24 h. We found that ICS II pretreatment dose dependently mitigated the levels of tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2) in the astrocytes. Moreover, ICS II not only exerted the inhibitory effect on LPS-induced IκB-α degradation and NF-κB activation, but also decreased the levels of Aβ1–40, Aβ1–42, amyloid precursor protein (APP) and beta secretase 1 (BACE1) in the astrocytes. Interestingly, molecular docking revealed that ICS II might directly bind to BACE1. It is concluded that ICS II has potential value as a new therapeutic agent to treat neuroinflammation-related diseases, such as AD.
Similar content being viewed by others


Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by beta-amyloid (Aβ) peptide fibrils, which are extracellular depositions of a particular protein and are accompanied by extensive neuroinflammation [1,2,3]. A number of studies have reported that inflammation, which may precede amyloid deposition, exerts vital effects in the pathogenesis of AD [4, 5]. Moreover, inflammatory mediators increase the expression of amyloid precursor protein (APP) and the formation of Aβ and upregulate beta-secretase activity [6]. Therefore, antiinflammatory drugs may prevent or treat AD by inhibiting neuroinflammation, thereby reducing the production or deposition of Aβ [7,8,9,10]. However, there are still no ideal antiinflammatory drugs to prevent or treat AD.
Astrocytes and microglia are important components of homeostasis in the brain [11]. When the brain is exposed to undesirable environmental conditions, both astrocytes and microglia, which are crucial perpetrators of inflammation and potential neuronal dysfunction [12, 13], acquire special “response” or “activation” phenotypes [14, 15]. In AD, the interaction between microglia and astrocytes may be of great significance for the development of neurodegenerative disease. In particular, astrocytes represent the most plentiful cell type in the brain and play an imperative role in maintaining the homeostasis of the central nervous system [16,17,18]. Under physiological conditions, astrocytes play a role in supporting and separating nerve cells [19,20,21,22]. Nevertheless, under neuroinflammatory conditions, the excessive activation of astrocytes is involved in the inflammatory response through its ability to release multiple molecules and further lead to neurodegenerative diseases [23,24,25].
Epimedium is a traditional Chinese herb used for the treatment of cardiovascular diseases, osteoporosis, and sexual and neurological disorders [26]. Icariside II (ICS II) is known as one of the major active pharmaceutical ingredients of Epimedium, and it has been indicated to have an extensive range of pharmacological effects, including antiinflammatory [27, 28], anticancer [29, 30], antioxidative [31, 32], and antiaging activities [33]. Our previous study found that ICS II attenuates streptozotocin- or Aβ25-35-induced cognitive deficits in rats by increasing the number of surviving neurons in the hippocampus [28] or inhibiting neuronal apoptosis and reducing PDE5 protein expression [34]. In our previous in vivo studies, we proved that ICS II exerts beneficial effects on lipopolysaccharide (LPS)-induced neuroinflammation by regulating the TLR4/MyD88/NF-κB signaling pathway in rats and inhibiting LPS-induced astrocyte overactivation [35]. However, whether ICS II can suppress neuroinflammatory responses in vitro remains unclear. Thus, in the current study, we investigated the effects of ICS II on lipopolysaccharide (LPS)-induced neuroinflammation in primary astrocytes and the underlying molecular mechanism.
Materials and methods
ICS II (purity ≥ 98%) was obtained from Nanjing Zelang Medical Technology Corporation Ltd (Nanjing, China). DMEM/F12 was obtained from HyClone (Logan, UT, USA), and fetal bovine serum (FBS) was purchased from Gibco (Thermo Fisher Scientific, MA, USA). Dexamethasone (DXMS) and LPS (L 2630) (Escherichia coli O111:B4) were purchased from Sigma-Aldrich (St Louis, MO, USA). DXMS and LPS were dissolved in normal saline at concentrations of 1 mM and 1 mg/mL, respectively, and ICS II was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM. Anticyclooxygenase-2 (COX-2) (#ab15191), anti-nitric oxide synthase (iNOS) (#ab15323), antinuclear factor-κB (NF-κB) (p65) (#ab16502), anti-p-NF-κB (p65) (#ab86299), anti-IκB-α (#ab7217), anti-IKK-α (#ab32041), anti-p-IKK-α (#ab38515), anti-IKK-β (#ab124957), anti-p-IKK-β (#ab59195), and anti-β-site APP cleaving enzyme (BACE1) (#ab108394) antibodies were purchased from Abcam (Cambridge, UK). Anti-APP (#AB60097b) was purchased from BBI Life Sciences Corporation (Shanghai, China). Tumor necrosis factor-alpha (TNF-α) enzyme-linked immunosorbent assay (ELISA) kits (an interleukin-1 beta (IL-1β) ELISA kit, an Aβ1–40 ELISA assay kit (JL10226), and an Aβ1–42 ELISA kit (JL10958)) were purchased from Shanghai Jianglai Biotechnology (Shanghai, China). A nitric oxide (NO) detection kit (S0021) was purchased from Beyotime Biotechnology (Shanghai, China).
Astrocyte culture and drug treatment
Sprague-Dawley rats (250 ± 50 g) were housed under a 12-h-light/dark cycle at a humidity of 50% ± 5% and a temperature of 24 °C. All animals were given free access to water and food. Animal experiments were performed in compliance with the State Committee of Science and Technology of the People’s Republic of China Order No. 2 of November 14, 1988, and the protocol was approved by the Experimental Animal Ethics Committee of the Zunyi Medical University. Primary astrocytes were extracted from neonatal rat brains as previously described [36]. Primary astrocytes were isolated from the cerebral cortex of 24-h-old neonatal rats. Briefly, suckling rats were repeatedly disinfected with 75% alcohol three times, and then the brains were removed, and the brain tissues were separated. Then, the cerebral cortex was collected under low temperatures, and the meninges and blood vessels were slightly removed. The cerebral cortex was fully dissociated by the addition of 10 mL of trypsin, the flask was treated with polylysine, and the suspension was spread in a culture flask at a cell density of 3 × 105 cells/mL. The differential adherence method was used to remove other types of cells in the tissue, and then astrocytes were cultured in DMEM/F12 containing 10% FBS. The DMEM/F12 was changed every 3 days until the 13th day. The primary cells were shaken at 37 °C at a constant temperature to remove other glial cells, such as microglia and oligodendrocytes. Thereafter, the astrocytes were fixed with 4% paraformaldehyde, incubated with an anti-GFAP antibody (1:500, Abcam), and visualized using an Alexa Fluor-conjugated secondary antibody (1:1000, Abcam). More than 95% of the cultured astrocytes were identified by GFAP staining, and the astrocytes were pretreated with different concentrations of ICS II or DXMS (1 μM) for 1 h. Thereafter, they were treated with LPS for another 24 h. Since DXMS is an effective and classical antiinflammatory drug, it was selected as a positive control drug to evaluate the antiinflammatory effect of ICS II, and 1 μM was chosen as the desired concentration of DXMS according to a previous study [37].
Measurement of cell viability
Cell viability was assessed using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay as described in our previous study [38]. In brief, astrocytes were seeded in a 96-well plate at 1 × 105 cells/well and treated as described above. Then, 20 μL of MTT (5 mg/mL) was added to FBS-free medium and cultured for another 4 h. The MTT was removed, and the cells were lysed with 150 μL of DMSO in each well. The optical density was measured at 490 nm using a microplate reader. The results of the treatments were expressed as a percentage of the control.
ELISA assay
In brief, astrocytes were inoculated into 96-well plates at a density of 1 × 105 cells/well and pretreated with ICS II (5, 10, 20 μM) for 1 h. LPS (1 mg/mL) and ICS II were added to the plates for 24 h. Thereafter, the culture medium was collected and centrifuged for 10 min at 16 000 × g. The supernatants were then collected and used to measure TNF-α, IL-1β, Aβ1–40, and Aβ1-42 levels using ELISA kits. All data were obtained from at least three independent experiments.
Measurement of nitric oxide (NO)
Astrocytes were inoculated in 96-well plates at a density of 1 × 105 cells/well and pretreated with ICS II (5, 10, 20 μM) for 1 h. LPS (1 mg/mL) and ICS II were added to the plates for 24 h. The accumulation of nitrite in the supernatant was evaluated using the Griess reaction. Each 50 μL of supernatant was reacted with an equal volume of Griess reagent and cultured for 15 min at room temperature. The absorbance was detected in a microplate absorbance reader at a wavelength of 540 nm, and a series of known concentrations of sodium nitrite was utilized as a standard.
Western blot analysis
Western blot analysis was performed as described in our previous study [38]. Briefly, astrocytes were treated as mentioned above, homogenized with protein extraction solution, and lysed for 40 min on ice. The lysate was centrifuged for 15 min at 14 000 × g. Equal amounts of protein (30 μg) were separated on a 10% SDS-polyacrylamide gel, transferred onto a polyvinylidene difluoride membrane and incubated with the following specific antibodies: anti-iNOS (1:1000), anti-COX-2 (1:2000), anti-p65 (1:2000), anti-p-p65 (1:1000), IκB-α (1:2000), anti-IKK-α (1:2000), anti-p-IKK-α (1:1000), anti-IKK-β (1:2000), anti-p-IKK-β (1:1000), anti-APP (1:1000), anti-BACE1 (1:1000), and anti-β-actin (1:2000). Then, the blots were subjected to the corresponding horseradish peroxidase-conjugated anti-rabbit or mouse immunoglobulin G. Thereafter, immunoreactive proteins were measured using an enhanced chemiluminescence Western blotting detection system.
Molecular docking
Molecular docking was performed using Autodock 4.2 and AutodockTools software to observe the affinity of ICS II and BACE1. The pdb format file for the BACE1 protein was obtained from the Protein Data Bank (http://www.rcsb.org) database with a PDB ID of 2ZHV. The molecular docking of the ICS II and BACE1 proteins was performed using Autodock 4.2.
Statistical analysis
All data were analyzed by SPSS 17.0 statistics software and are expressed as the mean ± SD. The data were analyzed using one-way ANOVA followed by the post hoc least significant difference. All data were obtained from at least three independent experiments. P < 0.05 was considered significant.
Results
Immunofluorescence identification of astrocytes
We extracted the cultured cells and identified them by staining for GFAP, which is a specific marker of astrocytes [39]. The results showed that astrocytes made up 95% of the cultured cells (Fig. 1a). Furthermore, the results showed that ICS II (1.56, 3.12, 6.25, 12.5, 25, or 50 μM) for 24, 48, or 72 h had no effect on the astrocytes; however, ICS II (100, 200, or 400 μM) for 24, 48, or 72 h was cytotoxic to the astrocytes. Since the concentration of DMSO used to dissolve ICS II in the experiment did not exceed 0.1%, it was assumed that the concentration of DMSO itself was not toxic to the cells and that the toxicity to the cells therefore resulted from the high concentration of ICS II. Thus, a concentration of ICS below 50 μM was considered to be the safe concentration range (Fig. 1b–d). Thereafter, 5, 10, and 20 μM ICS II were used in subsequent experiments, and DXMS (1 μM) was used as a positive control agent.

Identification of astrocytes and the determination of the safe concentration range of ICS II. a Identification of cellular immunofluorescence (scale bar = 50 μm, x200). b The safe concentration range of ICS II within 24 h (n = 5). c The safe concentration range of ICS II within 48 h (n = 5). d The safe concentration range of ICS II within 72 h (n = 5). *P < 0.05, **P < 0.01 versus control group
Effect of ICS II on the LPS-induced production of TNF-α and IL-1β
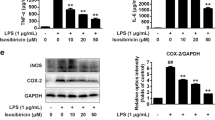
The effect of ICS II on the LPS-induced production of TNF-α and IL-1β in astrocytes was determined using an ELISA assay. The results showed that ICS II (5, 10, or 20 μM) slightly mitigated the production of TNF-α (Fig. 2a) and IL-1β (Fig. 2b). Following LPS stimulation, the production of TNF-α and IL-1β was substantially elevated compared with that in the control group, while ICS II markedly alleviated LPS-induced TNF-α and IL-1β overproduction in a concentration-dependent manner.

ICS II suppressed inflammatory factors in astrocytes after LPS insult. Astrocytes were pretreated with or without ICS II (5, 10, and 20 μM) or DXMS (1 μM) for 1 h followed by LPS (1 μg/mL) for another 24 h. a TNF-α level (n = 5). b IL-1β level (n = 5). c Production of NO (n = 5). d Representative Western blot bands for iNOS and COX-2 proteins. e Quantitation of iNOS protein (n = 3). f Quantitation of COX-2 protein (n = 3). **P < 0.01 versus control group; #P < 0.05, ##P < 0.01 versus LPS group
Effect of ICS II on LPS-induced NO production and iNOS and COX-2 expression in astrocytes
The effect of ICS II on LPS-induced NO production and iNOS and COX-2 expression in astrocytes was determined using the Griess reaction and Western blot analysis, respectively. The results showed that ICS II decreased LPS-induced NO production in astrocytes (Fig. 2c). Moreover, iNOS and COX-2 expression levels were dramatically increased after LPS insult. However, ICS II (5, 10, and 20 μM) induced a concentration-dependent decrease in the expression of iNOS and COX-2 in astrocytes compared with that in the LPS group (Fig. 2d–f).
Effect of ICS II on LPS-induced NF-κB (p65) translocation and the degradation of IKK-α, IKK-β, and IκB-α
The results suggested that ICS II not only suppressed the LPS-induced phosphorylation of p65 (Fig. 3a, b) but also prevented the nuclear translocation of p65 (Fig. 3c–f). Furthermore, ICS II also suppressed the LPS-induced degradation of IκB-α (Fig. 4a, b), IKK-α, and IKK-β (Fig. 4c, d). These results indicate that ICS II mitigates the LPS-induced activation of NF-κB via inhibiting IκB-α phosphorylation and the translocation of p65 to the nucleus.

ICS II inhibited the LPS-induced phosphorylation of NF-κB in astrocytes. a Representative Western blot bands for p-NF-κB and NF-κB. b Quantitation of p-NF-κB levels (n = 3). c Representative Western blot bands for NF-κB (nuclear) and NF-κB (cytoplasmic) proteins. d Quantitation of NF-κB (nucleus) protein (n = 3). e Quantitation of NF-κB (cytoplasmic) protein (n = 3). f Quantitation of NF-κB (nuclear)/NF-κB (cytoplasmic) protein (n = 3). *P < 0.05, **P < 0.01 versus control group; #P < 0.05, ##P < 0.01 versus LPS group

ICS II suppressed the LPS-induced activation of the NF-κB pathway in astrocytes. a Representative Western blot bands for IκB-α, p-IKK-α, IKK-α, p-IKK-β, and IKK-β proteins. b Quantitation of IκB-α protein (n = 3). c Quantitation of p-IKK-α/IKK-α protein (n = 3). d Quantitation of p-IKK-β/IKK-β protein (n = 3). **P < 0.01 versus control group; #P < 0.05, ##P < 0.01 versus LPS group
Effect of ICS II on LPS-induced amyloidogenesis in astrocytes
The effect of inflammation on amyloid formation in vitro was also investigated because neuroinflammation can cause amyloid production, whereas the aberrant activation of astrocytes is a major source of neuroinflammation. Astrocytes provide both mechanical and metabolic support to neurons, regulating the environment in which they function. To determine the relationship between neuroinflammation and amyloidogenesis, we investigated whether the antiinflammatory effect of ICS II can result in antiamyloidogenesis. As shown in Fig. 5a, b, when the cells were unstimulated, they expressed low protein levels of APP and BACE1, whereas the protein expression of APP and BACE1 increased in response to LPS after 24 h. In addition, ICS II also decreased LPS-induced Aβ1–40 and Aβ1–42 secretion into the culture media of astrocytes (Fig. 5c, d). In astrocytes, we also found that ICS II inhibited the LPS-induced expression of APP and BACE1, as well as Aβ1–40 and Aβ1–42 levels in a concentration-dependent manner. These results further indicate that the amyloidogenic pathway can be promoted by neuroinflammatory stimulation and that the antiinflammatory effect of ICS II can result in antiamyloidogenesis.

Effects of ICS II on the expression of APP and BACE1 and the levels of Aβ1–40 and Aβ1–42. a Quantitation of APP expression (n = 3). b Quantitation of BACE1 expression (n = 3). c Aβ1–40 level (n = 5). d Aβ1–42 level (n = 5). **P < 0.01 versus control group; #P < 0.05, ##P < 0.01 versus LPS group
ICS II binds and inhibits BACE1
Interestingly, mock molecular docking was used to verify whether ICS II binds to BACE1 protein, and the results showed that the binding energy of ICS II and BACE1 was −5.78 kcal/mol, which confirmed that ICS II can bind to BACE1, as the standard threshold for a molecule to bind to a protein is thought to be greater than or equal to −5 kcal/mol (Fig. 6a, b). We further investigated the supposed binding modes and interactions within the amino acid pocket, including Gly13, Gly230, Gly11, Thr232, Tyr71, Gly74, Lys107, Asp106, and Phe108 (Fig. 6c, d). Taken together, these findings show that ICS II may directly affect BACE1 and thus exert neuroprotective effects.

ICS II binds and inhibits BACE1. a Visual of the binding sites between ICS II and BACE1. b Visual of the binding surface between ICS II and BACE1. c Crystal structure of ICS II (green) displaying BACE1 (yellow and purple) bound to the docking pocket. d Amino acid residues
Discussion
The present study revealed that (1) ICS II protects against LPS-induced inflammation in primary-cultured astrocytes; (2) the inhibitory effect of ICS II is due to regulation of the IKK/IκB/NF-κB signaling pathway; and (3) ICS II decreases Aβ1–40 and Aβ1–42 levels by downregulating APP and BACE1 expression (Fig. 7).

A schematic showing the underlying mechanisms by which ICS II protects primary-cultured astrocytes against LPS-induced inflammation. ICS II inhibits proinflammatory mediators via regulating the IKK/IκB/NF-κB signaling pathway
LPS is an effective component of gram-negative bacilli endotoxin, which can cause a series of inflammatory reactions in the body and is widely applied to establish animal or cellular inflammatory models. Moreover, astrocytes represent the most abundant cell type in the central nervous system, and they exert a variety of physiological functions through close association with neurons and other brain structures. Similar to microglia, the immune and inflammatory properties of astrocytes, which can promote the secretion of various neuroinflammatory factors, such as TNF-α and IL-1β, are activated by LPS. Accumulating evidence has demonstrated that the production of multiple neuroinflammatory factors, including TNF-α, IL-1β, and iNOS, can activate the NF-κB pathway, resulting in neuroinflammation [40]. Moreover, the excessive production of neuroinflammatory factors leads to neuronal damage through the activation of glial cells. Notably, astrocytes play a key role in central nervous system inflammation by producing cytokines, such as TNF-α, IL-1β, and NO, leading to neuronal injury [41]. Therefore, the suppression of astrocyte activation may be an effective treatment for neuroinflammation-related diseases [42]. Our findings showed that LPS induces an increase in proinflammatory factors, which is consistent with the results of previous studies [43, 44]. ICS II significantly inhibited the LPS-induced accumulation of TNF-α, IL-1β, NO, iNOS, and COX-2 in primary culture astrocytes. In addition, these effects were not due to the cytotoxic effect of ICS II, as evidenced by the observation that there was no effect on astrocytes at concentrations up to 50 μM. Moreover, ICS II also inhibited iNOS and COX-2 protein expression in astrocytes. These findings indicate that ICS II exerts inhibitory effects on inflammation.
Notably, NF-κB not only regulates a variety of inflammatory factors, such as IL-1β and TNF-α but also plays a key role in mediating COX-2 and iNOS expression [45]. NF-κB (p50/p65) heterodimers exist in the cytoplasm of resting cells. However, under stimulation by LPS, the phosphorylation of IKK was induced, which subsequently phosphorylates the IκB protein, resulting in the release of NF-κB (p50/p65) heterodimers. Then, NF-κB-p65 translocates from the cytoplasm to the nucleus, thereby promoting the release of proinflammatory factors, including TNF-α and IL-6 [43, 44]. Consistent with these studies, the present study showed that LPS induces the phosphorylation of NF-κB-p65, IκB, and IKK. However, ICS II abolished these changes, suggesting that ICS II exerts potent antiinflammatory effects, at least partly through modulating the IKK/IκB/NF-κB pathway.
APP is a membrane-intrinsic protein expressed in a variety of tissues and is closely related to AD. APP can be cleaved by α, β, and γ proteases, and the continuous action of β-protease and γ-protease can cause APP to be cleaved to produce Aβ [46]. However, Aβ can cause the formation of senile plaques in the brain and the apoptosis of neuronal cells, thereby causing AD. Most importantly, mounting evidence has shown that neuroinflammation is related to the accumulation of Aβ in the brains of AD patients [47, 48]. In addition, APP is first cleaved by β-secretase at its β-cleavage site and is then proteolyzed by the second enzyme, γ-secretase, to produce Aβ1–42 and Aβ1–40. In particular, the Aβ1–40 and Aβ1–42 peptides are involved in the amyloidogenic pathway; Aβ1–40 is the most plentiful species, and Aβ1–42 shows the strongest neurotoxicity [49, 50]. Of note, BACE1 is an important rate-limiting enzyme in the APP–Aβ pathway and plays an important role in the production of Aβ. BACE1 was overexpressed in the brains of animal models of AD [51]. In contrast, in the brains of BACE1 knockout mice, the levels of Aβ and sAPPβ are decreased [52]. In addition, activated astrocytes that release mediators can induce APP and BACE1 expression, thereby stimulating Aβ production [53,54,55,56]. Notably, NF-κB mediates the accumulation of APP and the expression of BACE1 to increase Aβ production [57, 58]. In this study, LPS not only augmented the production of Aβ1–40 and Aβ1–42 but also upregulated APP and BACE1, and these findings are consistent with those of previous studies [36, 59]. However, ICS II reversed these changes, revealing that ICS II exerts inhibitory effects on neuroinflammation via suppressing the NF-κB/BACE1 signaling pathway, and this was also evidenced by molecular docking.
Conclusion
In conclusion, the current study revealed that ICS II exerts inhibitory effects on LPS-induced inflammation in astrocytes through the IKK/IκB/NF-κB/BACE1 signaling pathway, and thus ICS II may be a promising therapeutic agent for neuroinflammatory diseases, including AD.
References
Monson NL, Ireland SJ, Ligocki AJ, Chen D, Rounds WH, Li M, et al. Elevated CNS inflammation in patients with preclinical Alzheimer’s disease. J Cereb Blood Flow Metab. 2014;34:30–3.
Abbott A. Is ‘friendly fire’ in the brain provoking Alzheimer’s disease? Nature. 2018;556:426–8.
Birch AM, Katsouri L, Sastre M. Modulation of inflammation in transgenic models of Alzheimer’s disease. J Neuroinflamm. 2014;11:25.
Holmes C. Review: systemic inflammation and Alzheimer’s disease. Neuropathol Appl Neurobiol. 2013;39:51–68.
Nazem A, Sankowski R, Bacher M, Al-Abed Y. Rodent models of neuroinflammation for Alzheimer’s disease. J Neuroinflamm. 2015;12:74.
Zhan X, Cox C, Ander BP, Liu D, Stamova B, Jin LW, et al. Inflammation combined with ischemia produces myelin injury and plaque-like aggregates of myelin, amyloid-beta and AbetaPP in adult rat brain. J Alzheimers Dis. 2015;46:507–23.
McGeer PL, Rogers J, McGeer EG. Inflammation, antiinflammatory agents, and Alzheimer’s Disease: the last 22 years. J Alzheimers Dis. 2016;54:853–7.
Leoutsakos JM, Muthen BO, Breitner JC, Lyketsos CG, ADAPT Research Team. Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: findings from the randomized controlled Alzheimer’s Disease Anti-inflammatory Prevention Trial. Int J Geriatr Psychiatry. 2012;27:364–74.
Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F, et al. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796–804.
Zhang C, Wang Y, Wang D, Zhang J. Zhang F. NSAID exposure and risk of Alzheimer’s disease: an updated meta-analysis from cohort studies. Front Aging Neurosci. 2018;10:83.
Wekerle H. Brain inflammatory cascade controlled by gut-derived molecules. Nature. 2018;557:642–3.
Bouvier DS, Murai KK. Synergistic actions of microglia and astrocytes in the progression of Alzheimer’s disease. J Alzheimers Dis. 2015;45:1001–14.
Nott A, Glass CK. Immune memory in the brain. Nature. 2018;556:312–3.
Renner NA, Sansing HA, Inglis FM, Mehra S, Kaushal D, Lackner AA, et al. Transient acidification and subsequent proinflammatory cytokine stimulation of astrocytes induce distinct activation phenotypes. J Cell Physiol. 2013;228:1284–94.
Awada R, Saulnier-Blache JS, Grès S, Bourdon E, Rondeau P, Parimisetty A, et al. Autotaxin downregulates LPS-induced microglia activation and pro-inflammatory cytokines production. J Cell Biochem. 2014;115:2123–32.
Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nat Rev Neurosci. 2013;14:311–21.
Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504:394–400.
Liddelow S, Barres B. SnapShot: astrocytes in health and disease. Cell. 2015;162:1170–1170.e1.
Verkhratsky A, Parpura V. Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol Dis. 2016;85:254–61.
Adamsky A, Kol A, Kreisel T, Doron A, Ozeri-Engelhard N, Melcer T, et al. Astrocytic activation generates de novo neuronal potentiation and memory enhancement. Cell. 2018;174:59–71.e14.
Covelo A, Araque A. Stimulating astrocytes to remember. Cell. 2018;174:12–3.
Henneberger C. Does rapid and physiological astrocyte-neuron signalling amplify epileptic activity? J Physiol. 2017;595:1917–27.
Ben Haim L, Carrillo-de Sauvage MA, Ceyzériat K, Escartin C. Elusive roles for reactive astrocytes in neurodegenerative diseases. Front Cell Neurosci. 2015;9:278.
Rossi D. Astrocyte physiopathology: at the crossroads of intercellular networking, inflammation and cell death. Prog Neurobiol. 2015;130:86–120.
Zhao J, O’Connor T, Vassar R. The contribution of activated astrocytes to Abeta production: implications for Alzheimer’s disease pathogenesis. J Neuroinflamm. 2011;8:150.
Wu H, Lien EJ, Lien LL. Chemical and pharmacological investigations of Epimedium species: a survey. Prog Drug Res. 2003;60:1–57.
Deng Y, Long L, Wang K, Zhou J, Zeng L, He L, et al. Icariside II, a broad-spectrum anti-cancer agent, reverses beta-amyloid-induced cognitive impairment through reducing inflammation and apoptosis in rats. Front Pharmacol. 2017;8:39.
Yin C, Deng Y, Gao J, Li X, Liu Y, Gong Q. Icariside II, a novel phosphodiesterase-5 inhibitor, attenuates streptozotocin-induced cognitive deficits in rats. Neuroscience. 2016;328:69–79.
Yang L, Wang Y, Guo H, Guo M. Synergistic anti-cancer effects of icariin and temozolomide in glioblastoma. Cell Biochem Biophys. 2015;71:1379–85.
Li J, Jiang K, Zhao F. Icariin regulates the proliferation and apoptosis of human ovarian cancer cells through microRNA-21 by targeting PTEN, RECK and Bcl-2. Oncol Rep. 2015;33:2829–36.
Gao J, Deng Y, Yin C, Liu Y, Zhang W, Shi J, et al. Icariside II, a novel phosphodiesterase 5 inhibitor, protects against H2 O2 -induced PC12 cells death by inhibiting mitochondria-mediated autophagy. J Cell Mol Med. 2017;21:375–86.
Feng L, Gao J, Liu Y, Shi J, Gong Q. Icariside II alleviates oxygen-glucose deprivation and reoxygenation-induced PC12 cell oxidative injury by activating Nrf2/SIRT3 signaling pathway. Biomed Pharmacother. 2018;103:9–17.
Zhang SQ, Cai WJ, Huang JH, Wu B, Xia SJ, Chen XL, et al. Icariin, a natural flavonol glycoside, extends healthspan in mice. Exp Gerontol. 2015;69:226–35.
Liu S, Li X, Gao J, Liu Y, Shi J, Gong Q. Icariside II, a phosphodiesterase-5 inhibitor, attenuates beta-amyloid-induced cognitive deficits via BDNF/TrkB/CREB signaling. Cell Physiol Biochem. 2018;49:985.
Zhou J, Deng Y, Li F, Yin C, Shi J, Gong Q. Icariside II attenuates lipopolysaccharide-induced neuroinflammation through inhibiting TLR4/MyD88/NF-kappaB pathway in rats. Biomed Pharmacother. 2019;111:315–24.
Song SY, Jung YY, Hwang CJ, Lee HP, Sok CH, Kim JH, et al. Inhibitory effect of ent-Sauchinone on amyloidogenesis via inhibition of STAT3-mediated NF-kappaB activation in cultured astrocytes and microglial BV-2 cells. J Neuroinflamm. 2014;11:118.
Korhonen R. Dexamethasone inhibits inducible nitric-oxide synthase expression and nitric oxide production by destabilizing mRNA in lipopolysaccharide-treated macrophages. Mol Pharmacol. 2002;62:698–704.
Gao J, Liu S, Xu F, Liu Y, Lv C, Deng Y, et al. Trilobatin protects against oxidative injury in neuronal PC12 cells through regulating mitochondrial ROS homeostasis mediated by AMPK/Nrf2/Sirt3 signaling pathway. Front Mol Neurosci. 2018;11:267.
Brenner M. Role of GFAP in CNS injuries. Neurosci Lett. 2014;565:7–13.
Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Sci World J. 2012;2012:756357.
Lu Y, Jiang BC, Cao DL, Zhang ZJ, Zhang X, Ji RR, et al. TRAF6 upregulation in spinal astrocytes maintains neuropathic pain by integrating TNF-alpha and IL-1beta signaling. Pain. 2014;155:2618–29.
Morales I, Guzmán-Martínez L, Cerda-Troncoso C, Farías GA, Maccioni RB. Neuroinflammation in the pathogenesis of Alzheimer’s disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci. 2014;8:112.
Zeng KW, Yu Q, Liao LX, Song FJ, Lv HN, Jiang Y, et al. Anti-neuroinflammatory effect of MC13, a novel coumarin compound from condiment murraya, through inhibiting lipopolysaccharide-induced TRAF6-TAK1-NF-kappaB, P38/ERK MAPKS and Jak2-Stat1/Stat3 pathways. J Cell Biochem. 2015;116:1286–99.
Fan X, Zhang Y, Dong H, Wang B, Ji H, Liu X. Trilobatin attenuates the LPS-mediated inflammatory response by suppressing the NF-kappaB signaling pathway. Food Chem. 2015;166:609–15.
Shu Z, Yang B, Zhao H, Xu B, Jiao W, Wang Q, et al. Tangeretin exerts anti-neuroinflammatory effects via NF-kappaB modulation in lipopolysaccharide-stimulated microglial cells. Int Immunopharmacol. 2014;19:275–82.
Wang X, Zhou X, Li G, Zhang Y, Wu Y, Song W. Modifications and trafficking of APP in the pathogenesis of Alzheimer’s Disease. Front Mol Neurosci. 2017;10:294.
Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–45.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42.
Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA. 1994;91:12243–7.
Schreiner B, Hedskog L, Wiehager B, Ankarcrona M. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis. 2015;43:369–74.
Du Y, Qu J, Zhang W, Bai M, Zhou Q, Zhang Z, et al. Morin reverses neuropathological and cognitive impairments in APPswe/PS1dE9 mice by targeting multiple pathogenic mechanisms. Neuropharmacology. 2016;108:1–13.
Sankaranarayanan S, Price EA, Wu G, Crouthamel MC, Shi XP, Tugusheva K, et al. In vivo beta-secretase 1 inhibition leads to brain Abeta lowering and increased alpha-secretase processing of amyloid precursor protein without effect on neuregulin-1. J Pharmacol Exp Ther. 2008;324:957–69.
Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–15.
Kim EK, Moon JC, Lee JM, Jeong MS, Oh C, Ahn SM, et al. Large-scale production of soluble recombinant amyloid-beta peptide 1-42 using cold-inducible expression system. Protein Expr Purif. 2012;86:53–7.
Albuquerque MS, Mahar I, Davoli MA, Chabot JG, Mechawar N, Quirion R, et al. Regional and sub-regional differences in hippocampal GABAergic neuronal vulnerability in the TgCRND8 mouse model of Alzheimer’s disease. Front Aging Neurosci. 2015;7:30.
Dai X, Hou W, Sun Y, Gao Z, Zhu S, Jiang Z. Chitosan oligosaccharides inhibit/disaggregate fibrils and attenuate amyloid beta-mediated neurotoxicity. Int J Mol Sci. 2015;16:10526–36.
Sambamurti K, Kinsey R, Maloney B, Ge YW, Lahiri DK. Gene structure and organization of the human beta-secretase (BACE) promoter. FASEB J. 2004;18:1034–6.
Chen CH, Zhou W, Liu S, Deng Y, Cai F, Tone M, et al. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int J Neuropsychopharmacol. 2012;15:77–90.
Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflamm. 2008;5:37.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 81560585), the Program for Excellent Young Talents of Zunyi Medical University (Grant No. 15zy-002), the Zunyi Medical University Masters Startup Fund (F-758), The “Hundred” Level of High-Level Innovative Talents in Guizhou Province (Grant No. QKHRCPT 20165684), the Education Department of Guizhou Province of China (Grant No. GNYL (2017) 006, YLXKJS-YS-06), Shijingshan’s Tutor Studio of Pharmacology[GZS-2016(07)] and the Program for Changjiang Scholars and Innovative Research Team in University, China (Grant No. IRT_17R113), and the 2011 Collaborative Innovation Center of Guizhou Traditional Chinese Medicine and Ethnic Medicine (No. Qianjiaokeyanfa [2012] 311).
Author information
Authors and Affiliations
Contributions
QHG and JSS designed the experimental protocols. YZ carried out all the studies except the Western blot analysis, which was performed by CL, LHL, and YD. YZ wrote the manuscript with help from QHG, CYY, and JMG.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Zheng, Y., Deng, Y., Gao, Jm. et al. Icariside II inhibits lipopolysaccharide-induced inflammation and amyloid production in rat astrocytes by regulating IKK/IκB/NF-κB/BACE1 signaling pathway. Acta Pharmacol Sin 41, 154–162 (2020). https://doi.org/10.1038/s41401-019-0300-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0300-2
Keywords
This article is cited by
-
Knockdown and inhibition of hippocampal GPR17 attenuates lipopolysaccharide-induced cognitive impairment in mice
Journal of Neuroinflammation (2023)
-
Effects of genus Epimedium in the treatment of osteoarthritis and relevant signaling pathways
Chinese Medicine (2023)
-
Pharmacological mechanism and therapeutic efficacy of Icariside II in the treatment of acute ischemic stroke: a systematic review and network pharmacological analysis
BMC Complementary Medicine and Therapies (2022)
-
Brucine-Induced Neurotoxicity by Targeting Caspase 3: Involvement of PPARγ/NF-κB/Apoptosis Signaling Pathway
Neurotoxicity Research (2022)
