
Abstract
c-Maf is a critical oncogenic transcription factor that contributes to myelomagenesis. Our previous studies demonstrated that the deubiquitinase USP5 stabilizes c-Maf and promotes myeloma cell proliferation and survival; therefore, the USP5/c-Maf axis could be a potential target for myeloma therapy. As a concept of principle, the present study established a USP5/c-Maf-based luciferase system that was used to screen an FDA-approved drug library. It was found that mebendazole, a typical anthelmintic drug, preferentially induced apoptosis in c-Maf-expressing myeloma cells. Moreover, oral administration of mebendazole delayed the growth of human myeloma xenografts in nude mice but did not show overt toxicity. Further studies showed that the selective antimyeloma activity of mebendazole was associated with the inhibition of the USP5/c-Maf axis. Mebendazole downregulated USP5 expression and disrupted the interaction between USP5 and c-Maf, thus leading to increased levels of c-Maf ubiquitination and subsequent c-Maf degradation. Mebendazole inhibited c-Maf transcriptional activity, as confirmed by both luciferase assays and expression measurements of c-Maf downstream genes. In summary, this study identified mebendazole as a USP5/c-Maf inhibitor that could be developed as a novel antimyeloma agent.
Similar content being viewed by others


Introduction
Multiple myeloma (MM) is a hematological malignancy derived from plasma cells that produce antibodies [1]. MM features gene mutations and chromosomal aberrations, including deletions, inversions, interchanges, and translocations. These genetic abnormalities lead to discrepant responses of MM patients to individual therapeutic agents [2]. There are at least six types of chromosomal translocations, among which t(14:16) results in the overexpression of the transcription factor gene c-Maf [2, 3]. c-Maf modulates the transcription of several key genes, including cyclin D2 (CCND2), integrin B7 (ITGB7), AMPK-related protein kinase 5 (ARK5), and C-C chemokine receptor 1 (CCR1), by binding to its specific recognition element on the upstream regulatory region of these genes [4]. Among these c-Maf-modulated genes, CCND2 is a key factor in cell cycle progression and thus promotes MM cell proliferation, while ITGB7 mediates MM cell adhesion to bone marrow stromal cells and facilitates MM cell migration and invasion, thereby conferring chemoresistance [5]. Therefore, c-Maf promotes MM cell proliferation and survival. The expression level of the c-Maf protein is negatively associated with the overall survival of MM patients [2, 6]. Therefore, targeting c-Maf degradation is proposed as a promising therapeutic approach for MM [4, 6]. Our previous studies show that c-Maf can be degraded through the ubiquitin-proteasome pathway [7], while the deubiquitinase USP5 abolishes K48-linked polyubiquitination, thus stabilizing the c-Maf protein and promoting MM cell survival [8]. In contrast, knockdown of USP5 leads to c-Maf degradation and MM cell apoptosis [8]. Therefore, targeting the USP5/c-Maf axis may be a potential therapy for MM.
In the present study, we established a USP5/c-Maf luciferase system and applied it to screen a library composed of 1700 small molecule chemical drugs approved by the US Food and Drug Administration. From this screen, we identified mebendazole, an agent widely used for the treatment of human gastrointestinal parasitic infections [9], selectively induces MM cell apoptosis by targeting USP5/c-Maf signaling.
Materials and methods
Cell culture
Human embryonic kidney cells (HEK293T) were maintained in Dulbecco’s modified Eagle’s medium. MM cell lines, including RPMI-8226, LP1, and U266, were cultured in Iscove’s modified Dulbecco’s medium. All media were supplemented with 10% fetal bovine serum, glutamine and antibiotics, as described previously [10].
Plasmids
The c-Maf and USP5 plasmids were constructed as described previously [8, 10]. A nucleotide sequence containing six tandem Maf recognition elements (MAREs) was chemically synthesized by GeneWiz, Inc. (Suzhou, Jiangsu, China) and inserted into a pGL4 vector to generate the MARE.Luci reporter plasmid (pMARE.Luci) [8, 10].
Chemicals and antibodies
Monoclonal anti-HA (1:10000, M180–3), anti-Myc (1:10000, M192–3), and anti-Flag (1:10000, M185–3L) were obtained from MBL Beijing Biotech Co., Ltd. (Beijing, China). Antibodies against USP5 (1:1000, 10473–1-AP) and GAPDH (1:5000, 60004–1-Ig) were purchased from Proteintech Group, Inc. (Wuhan, Hubei, China). The anti-c-Maf antibody was obtained from Santa Cruz Biotechnology, Inc. (1:1000, sc-7866) (Santa Cruz, CA, USA) and Proteintech (1:1000, 55013–1-AP). An anti-Ub antibody was obtained from Santa Cruz Biotechnology, Inc. (1:1000, sc-271289). The US FDA-approved drug library and mebendazole were purchased from TargetMol Corp. (Boston, MA, USA). HRP-labeled goat anti-mouse and goat anti-rabbit IgG (H + L) antibodies were purchased from Beyotime Institute of Biotechnology (Nantong, China).
Gene transfection
One day before transfection, HEK293T cells were seeded in six-well plates. When grown to 50% confluence, cells were subjected to gene delivery using polyethyleneimine (PEI) as described previously [10].
Screening of the FDA-approved drug library
HEK293T cells were cotransfected with the c-Maf, USP5, and pMARE.Luci plasmids. Forty-eight hours later, cells were isolated and reseeded into 96-well plates at a density of 1 × 105 cells/well for 24 h before each drug (final concentration: 5 μM) was added and were then incubated for another 24 h. Cells were then collected for the luciferase activity assay, which was performed according to the manufacturer’s instructions, as described previously [8]. The drugs that decreased the luciferase activity by more than 50% were selected for further evaluation as candidate USP5/c-Maf axis inhibitors [10].
Immunoblot (IB) assays
After cell lysates were prepared, protein concentrations were determined by a BCA assay (Beyotime). Equal amounts of protein (30 µg) were separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes. After normalization, the blots were probed with appropriate antibodies, as described previously [8].
Immunoprecipitation (IP)
After being treated with mebendazole for 24 h, LP1 and RPMI-8226 cells were harvested to prepare whole cell lysates, and the IP assay was conducted as described previously [10]. After IP, proteins were resuspended in RIPA buffer and used for an IB assay [10].
Apoptosis detection via flow cytometric analysis
Cells were treated with mebendazole for 24 h followed by staining in dark for 5 min with Annexin V-FITC and propidium iodide (PI, Multi Sciences Biotech Co., Ltd., Hangzhou, China). Cells were then subjected to analysis on a BD® flow cytometer as described previously [11].
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted using Trizol (Sangon Biotech, Shanghai, China). Total RNA (2 µg) was reverse transcribed using an EasyScipt First-Strand cDNA Synthesis kit (Vazyme Biotech Co., Ltd., Nanjing, China) according to the manufacturer’s instructions. PCR amplification was carried out using the following primers: USP5, 5’-CGG ATT TGA CCT TAG CG-3’ (forward) and 5’-CTG CCA TCG AAG TAG CG-3’ (reverse); c-Maf, 5’-AAA AAG GAA CCG GTG GAG AC-3’ (forward) and 5’-GGT AGC CGG TCA TCC AGT AG-3’ (reverse); and GAPDH, 5’-AAT CCC ATC ACC ATC TTC C-3’ (forward) and 5’-CAT CAC GCC ACA GTT TCC-3’ (reverse). The primers used for CCND2, ITGB7, and ARK5 were described previously [11]. PCR products were visualized by GoldView® staining (TransGen Biotech Co., Ltd., Beijing, China) following electrophoresis on 2% agarose gels.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The mRNA expression levels of USP5, c-Maf, CCND2, ITGB7 and ARK5 were measured by qRT-PCR with the following specific primers: USP5, 5’-GCT GCT GTC AGT ATT ACC GAC-3’ (forward) and 5’-AAA GCC CAG AAA CGT GTT CAT A-3’ (reverse); c-Maf, 5‘-CTG CCG CTT CAA GAG GGT GCA GC-3’ (forward) and 5‘-GAT CTC CTG CTT GAG GTG GTC-3’ (reverse); CCND2, 5‘-TTA CCT GGA CCG TTT CTT GG-3’ (forward) and 5‘-GAG GCT TGA TGG AGT TGT CG-3’ (reverse); ITGB7, 5‘-GAA TCA ACC AGA CGG TGA CTT TCT-3’ (forward) and 5‘-GCC CGG AGC CTC AGG A-3’ (reverse); ARK5, 5‘-GGG AGC TGT ACG ATT ACA TCA G-3’ (forward) and 5‘-ACA CCG TTC TTG TGA CAA TAG TG-3’ (reverse); and GAPDH, 5’-GCA CCG TCA AGG CTG AGA AC-3’ (forward) and 5’-TGG TGA AGA CGC CAG TGG A-3’ (reverse). RT-PCR amplification was carried out with 200 ng of cDNA in a 20-µL reaction mixture containing 0.5 µL of primers and 10 µL of SYBR green mix (Beyotime). Data quantification was carried out by the 2–ΔΔCt method.
Myeloma xenografts in nude mice
Female BALB/c nude mice (5–6 weeks old) purchased from Shanghai Slac Laboratory Animal Co. Ltd. were used to establish xenograft models with the human MM cell lines LP1 and RPMI-8226. When tumors were palpable, mice were randomly divided into three groups. One group received vehicle as the control, and the other two groups were orally administered mebendazole at a dosage of 50 or 100 mg/kg body weight once daily for 20 days. Body weight and tumor volumes were measured every other day (tumor volume = tumor length × width2/2). At the end of this experiment, tumors and blood samples were collected for further studies. All experiments involving mice were reviewed and approved by the Review Board of Animal Care and Use of Soochow University.
Statistical analyses
Statistical differences between the control and the experimental groups were analyzed by Student’s t-test.
Results
Identification of c-Maf/USP5 inhibitors by a mid-throughput screen
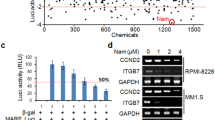
c-Maf is sufficient to induce myelomagenesis [12] and confers chemoresistance to proteasomal inhibitors such as bortezomib [6], the mainstay anti-MM agent. Our previous studies showed that targeting USP5 leads to c-Maf degradation and MM cell death, suggesting that the USP5/c-Maf axis is a potential therapeutic target in MM [8]. To identify anti-MM inhibitors of the USP5/c-Maf axis, we established a screening system in HEK293T cells based on MARE-driven luciferase activity in the presence of c-Maf and USP5 (Fig. 1a). These cells were treated for 24 h with each drug from an FDA-approved drug library before luciferase activity was measured in cell lysates. The results showed that 30 out of the 1700 drugs decreased luciferase activity by >50% in the presence of USP5 and the transcription factor c-Maf (Fig. 1b). These drugs included the anti-MM agent daunorubicin (DNR), the antileukemia agent vincristine (VCR), and 12 other cytotoxic FDA-approved anticancer drugs. Several drugs that have not yet been approved for cancer treatment were also identified. These drugs included artesunate (ART), an antimalarial agent; mebendazole (MBZ), an antiparasitic agent; nicergoline (NCG), for the treatment of cognitive, affective, and behavioral disorders of older people; and tolazamide (TLZ), for the treatment of type 2 diabetes. In the second screen, the MM cell line LP1, which expresses a high level of c-Maf [5, 8], was treated with all of the above drugs for 24 h, followed by IB assays to evaluate PARP cleavage, a hallmark of apoptosis, and c-Maf degradation. The results showed that the classic cytotoxic anticancer drugs daunorubicin and vincristine induced MM cell apoptosis at nanomolar concentrations, in association with c-Maf degradation (Fig. 1c). However, further studies showed that these two drugs downregulated the transcription of c-Maf at the mRNA level (Fig. 1d). Nicergoline and tolazamide did not show marked PARP cleavage activity. Artesunate and mebendazole induced MM cell apoptosis as evidenced by PARP cleavage and c-Maf degradation. Because artesunate has been well studied in MM cell apoptosis [13, 14] and it downregulated c-Maf mRNA expression (Fig. 1d), mebendazole was selected for further studies. Mebendazole decreased c-Maf protein expression but had no effects on its mRNA level (Fig. 1d), and its potential application in myeloma treatment has not yet been reported.

Identification of mebendazole as a promoter of c-Maf protein degradation. a The system for screening c-Maf/USP5 inhibitors. b HEK293T cells were plated in 96-well plates (10 000 cells per well) 24 h before being cotransfected with plasmids, including the c-Maf, USP5 and pMARE.Luci plasmids. On the third day, cells were treated with each compound from the drug library for 24 h before being lysed for luciferase assays. The activities of the compounds were expressed as log2 (sample luciferase RLU/control luciferase RLU). Drugs associated with a value of log2 < -1 were considered potential inhibitors. c, d LP1 cells were treated with candidate drugs at the indicated concentrations for 24 h, followed by IB assays and RT-PCR for the indicated proteins/genes
Mebendazole inhibits the USP5/c-Maf axis and induces c-Maf degradation via the ubiquitin-proteasome pathway
Previous studies have demonstrated that the deubiquitinase USP5 stabilizes c-Maf by binding to c-Maf and suppressing its polyubiquitination [8]. The above results showed that mebendazole downregulated c-Maf protein expression but had no effect on its mRNA expression (Fig. 1); thus, we sought to determine whether mebendazole could suppress USP5 activity via c-Maf ubiquitination. To this end, we first measured the c-Maf and USP5 levels in mebendazole-treated MM cells. The results showed that mebendazole downregulated the protein expression of both USP5 and c-Maf in both LP1 and RPMI-8226 cells (Fig. 2a, b). As expected, mebendazole failed to downregulate c-Maf mRNA expression, as shown by the RT-PCR assay results (Fig. 2a, b). Notably, we found that USP5 mRNA expression was also decreased by mebendazole, suggesting that mebendazole might suppress USP5 transcription.

Mebendazole inhibits USP5 expression and induces c-Maf degradation via the ubiquitin-proteasome pathway. a LP1 and RPMI-8226 cells were treated with MBZ at the indicated concentrations for 24 h, followed by IB assays for c-Maf and USP5. Total RNA was extracted for RT-PCR to assess c-Maf and USP5 expression. GAPDH was used as the internal control. b The c-Maf and USP5 expression levels from a were analyzed by densitometry. c LP1 and RPMI-8226 cells were treated with MBZ or MG132 at the indicated concentrations for 24 h, followed by immunoblotting for c-Maf and GAPDH. d HEK293T cells were cotransfected with the HA-c-Maf and Myc-USP5 plasmids for 48 h, followed by MBZ treatment for 24 h and subsequent IP/IB assays. e LP1 and RPMI-8226 cells were treated with MBZ at the indicated concentrations for 24 h, followed by cell lysate preparation and IP/IB assays. f HEK293T cells were cotransfected with the HA-c-Maf, Myc-USP5 and Flag-Ub plasmids for 48 h. Cells were then treated with MBZ for 24 h before being lysed and subjected to IP/IB assays as indicated. g LP1 and RPMI-8226 cells were treated with MBZ at the indicated concentrations for 24 h, followed by IP/IB assays
Because c-Maf turnover is processed via the proteasome, we sought to determine whether mebendazole-induced c-Maf degradation could be reversed by proteasomal inhibitors. To this end, LP1 and RPMI-8226 cells were treated with mebendazole followed by treatment with MG132. The IB assay results showed that mebendazole downregulated c-Maf but that this downregulation was abolished by MG132, a typical proteasomal inhibitor (Fig. 2c), suggesting that mebendazole induces c-Maf degradation via the ubiquitin-proteasome pathway. Because USP5, as a deubiquitinase of c-Maf, binds to and suppresses c-Maf polyubiquitination, we sought to determine whether mebendazole could interfere with the association of c-Maf and USP5. Therefore, we first evaluated the interaction between USP5 and c-Maf in the presence of mebendazole. The results showed that mebendazole decreased the USP5 content in the c-Maf immunoprecipitates in both exogenous and endogenous contexts, as shown in HEK293T and MM cells, respectively (Fig. 2d, e), suggesting that the interaction between USP5 and c-Maf was disrupted by mebendazole. Next, we evaluated the polyubiquitination levels of c-Maf in HEK293T, LP1 and RPMI-8226 cells in the presence of mebendazole. The IP/IB assay results showed that mebendazole increased the polyubiquitination levels of c-Maf in all three cell lines (Fig. 2f, g). These findings thus confirmed the hypothesis that mebendazole prevented USP5-mediated c-Maf deubiquitination and led to c-Maf degradation.
Mebendazole suppresses the transcriptional activity of c-Maf
c-Maf is involved in myelomagenesis as a transcription factor by promoting the transcription of its target genes, including CCND2, ITGB7, and ARK5 [4]. The above results demonstrated that mebendazole suppressed the USP5/c-Maf axis and promoted c-Maf degradation; therefore, mebendazole might downregulate the transcriptional activity of c-Maf. To confirm this hypothesis, we used firefly luciferase as the reporter under the regulation of a c-Maf recognition element (MARE). HEK293T cells cotransfected with MARE.luci, c-Maf, and USP5 were treated with mebendazole for 24 h, followed by a luciferase assay. Ectopic expression of USP5 significantly increased the transcriptional activity of c-Maf in terms of the measured luciferase activity (Fig. 3a). Notably, this activity was markedly inhibited by mebendazole in a concentration-dependent manner. In contrast, another deubiquitinase, USP7, also increased the transcriptional activity of c-Maf, but luciferase activity driven by USP7 was much less affected by mebendazole than luciferase activity driven by USP5 (Fig. 3a). To confirm this finding, c-Maf-expressing LP1 and RPMI-8226 and c-Maf-deficient U266 cell lines were treated with mebendazole for 24 h at increasing concentrations, followed by RT-PCR to measure the gene transcription levels. The results showed that mebendazole decreased the mRNA levels of all three genes—CCND2, ARK5, and ITGB7—that were under the control of c-Maf in LP1 and RPMI-8226 cells that express c-Maf but not in U266 cells that lack c-Maf (Fig. 3b). To confirm these findings, we next performed quantitative real-time PCR to measure the expression of all genes. Consistent with the results of the above assays, mebendazole downregulated the transcription of USP5 (Fig. 3c), CCND2 (Fig. 3e), ITGB7 (Fig. 3f), and ARK5 (Fig. 3g) but had no effects on c-Maf transcription (Fig. 3d). Therefore, mebendazole probably affects c-Maf transcriptional activity by inducing c-Maf degradation via the inhibition of USP5.

Mebendazole inhibits the transcriptional activity of c-Maf. a HEK293T cells were cotransfected with the HA-c-Maf, pMARE.Luci and Myc-USP5 or Flag-USP7 plasmids. After 48 h, cells were treated with MBZ in a dose-dependent manner, as indicated, for another 24 h. Cells were harvested for luciferase activity measurements and IB assays. b RPMI-8226, LP1, and U266 cells were treated with MBZ in a dose-dependent manner for 24 h, followed by total RNA extraction and RT-PCR analysis. c–g LP1 and RPMI-8226 cells were treated with MBZ at the indicated concentrations for 24 h, followed by quantitative real-time PCR to measure the expression of genes, including USP5 (c), c-Maf (d), CCND2 (e), ITGB7 (f), and ARK5 (g). *P < 0.05, **P < 0.01, ***P < 0.001
Mebendazole preferentially induces the apoptosis of MM cells that express c-Maf
Because the USP5/c-Maf axis plays a critical role in promoting MM cell proliferation and survival, and interference with USP5 and c-Maf leads to MM cell apoptosis [8], we next evaluated the effects of mebendazole on MM cell survival and apoptosis. To this end, the MM cell lines LP1, RPMI-8226, and U266 were exposed to mebendazole for 24 h at increasing concentrations, followed by IB assays. As shown in Fig. 4a, the cleavage and activation of the apoptotic markers PARP and caspase-3 were significantly increased in both LP1 and RPMI-8226 cells that express c-Maf. In contrast, a lower level of PARP cleavage fragments was detected in U266 cells that lack c-Maf (Fig. 4a, b). Caspase-3, one of the major apoptotic executor enzymes, was not activated in U266 cells (Fig. 4a-c). These findings suggest that mebendazole probably preferentially induces the apoptosis of c-Maf-expressing MM cells. To further confirm this hypothesis, all three MM cell lines were exposed to mebendazole at a very low concentration (0.5 μM) followed by Annexin V/PI staining and flow cytometric analyses. As shown in Fig. 4d, 0.5 μM mebendazole induced marked apoptosis in LP1 and RPMI-8226 cells but not in U266 cells, suggesting that U266 cells are less sensitive to mebendazole due to their deficiency in c-Maf expression. Therefore, mebendazole, especially at low concentrations, preferentially induces MM cell apoptosis in a manner dependent on the expression of c-Maf.

Mebendazole preferentially induces the apoptosis of c-Maf-expressing MM cells. a LP1, RPMI-8226, and U266 cells were treated with MBZ at the indicated concentrations for 24 h, followed by IB assays to evaluate the expression of apoptotic proteins, as indicated. b The ratio of cleaved PARP to pro-PARP and c the ratio of cleaved Caspase-3 to pro-Caspase-3. d MM cells were treated with MBZ (0.5 µM) for 12 h, followed by Annexin V-FITC and PI staining and flow cytometric analyses. ***P < 0.001
Mebendazole exerts synergistic antimyeloma effects with a USP5 inhibitor
Previous studies identified WP1130 as a small molecule compound that inhibits the deubiquitinase activity of USP5 [15]. In the above studies, mebendazole was demonstrated to downregulate USP5 expression and disrupt the interaction between USP5 and c-Maf. We sought to determine whether these two agents could synergize to induce MM cell apoptosis. To this end, LP1 and RPMI-8226 cells were treated with WP1130 and mebendazole alone or in combination (at a very low concentration; 1 μM for WP1130 and 0.1 μM for mebendazole), followed by IB assays to investigate PARP cleavage, a hallmark of cell apoptosis. The results showed that PARP was not markedly cleaved by WP1130 or mebendazole due to the very low concentrations used; however, it was significantly cleaved by cotreatment with these two agents. Notably, c-Maf expression was also decreased by cotreatment (Fig. 5a, b). Therefore, combined treatment with mebendazole and WP1130 demonstrated synergy against MM. We next examined the effects of mebendazole in combination with daunorubicin, a typical anti-MM drug. These two drugs alone induced limited PARP cleavage, but combined treatment significantly increased PARP cleavage (Fig. 5c, d). Collectively, these results thus showed marked synergy between mebendazole and USP5 inhibitors or anti-MM drugs such as daunorubicin.

Mebendazole enhances MM cell apoptosis induced by WP1130 and daunorubicin (DNR). a LP1 and RPMI-8226 cells were treated with MBZ (0.1 µM) and WP1130 (1 µM) alone or in combination for 24 h, followed by IB assays for c-Maf, PARP and GAPDH. b The ratio of cleaved PARP to pro-PARP from a. c LP1 and RPMI-8226 cells were treated with MBZ (0.1 µM) and DNR (100 nM) alone or in combination for 24 h, followed by IB assays for c-Maf, PARP, and GAPDH. d The ratio of cleaved PARP to pro-PARP from c. **P < 0.01, ***P < 0.001
Mebendazole delays the growth of human multiple myeloma xenografts in a nude mouse model
The above studies provided strong evidence that mebendazole inhibits the USP5/c-Maf axis and induces MM cell apoptosis. To further investigate the effects of mebendazole against human MM in vivo, two myeloma xenograft models were established by subcutaneous injection of LP1 or RPMI-8226 cells in the right flanks of nude mice. When the tumors were ~5 mm in diameter, mice in each model were randomly divided into three groups. Each group was orally administered vehicle or mebendazole at a dose of 50 or 100 mg/kg for a continuous 20 days. The tumor sizes and mouse body weights were monitored every other day. Tumor growth was significantly decreased by mebendazole, and this effect was dose-dependent (Fig. 6a, b). At the dose of 100 mg/kg, mebendazole almost completely suppressed the growth of myeloma xenografts in both models (Fig. 6a, b). However, mebendazole did not show toxicity to mice, as shown via body weight measurements (Fig. 6c, d) and biochemical analyses of blood. There were no marked alterations in the levels of alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), urea, creatinine (CREA), or albumin in mice treated with mebendazole compared with those levels in vehicle-treated mice (Tables 1 and 2). Collectively, these results suggested that mebendazole was well-tolerated. Subsequently, we analyzed the effects of mebendazole on MM xenografts. In the IB assays on tumor tissues from treated mice, USP5 and c-Maf were decreased in tumor tissues from mebendazole-treated mice but not in tumor tissues from untreated mice (Fig. 6e, f). Moreover, caspase-3 and PARP were also cleaved in tissues from mebendazole-treated mice (Fig. 6e, f), suggesting that mebendazole also induces MM cell apoptosis in vivo. Taken together, these results indicated that mebendazole displays significant anti-MM activity in vivo by inhibiting the USP5/c-Maf axis.

Mebendazole delays myeloma tumor growth in nude mice without toxicity. Human multiple myeloma cells (LP1 and RPMI-8226) were injected subcutaneously into nude mice at a density of 30 million cells/site. When tumors were palpable, mice were orally administered vehicle or MBZ (50 mg/kg or 100 mg/kg body weight) in PBS containing 50% sesame oil daily for a continuous 20 days. Tumor volumes (a, b) and body weights (c, d) were monitored every other day. **P < 0.01; ***P < 0.001. e, f Tumor samples from each model were subjected to IB analyses for USP5, c-Maf, caspase-3, and PARP with specific antibodies
Discussion
The above studies identified mebendazole as an inhibitor of the USP5/c-Maf axis and indicated that it displays potent antimyeloma activity in vitro and in vivo.
Mebendazole, a benzimidazole, is a well-tolerated, highly effective broad-spectrum anthelmintic used for the treatment of various intestinal infections with pinworms, hookworms, or roundworms. Recent studies found that mebendazole also displays anticancer activity against several types of cancer, including colon cancer [16, 17], brain tumor [18], glioblastoma multiforme [19], non-small cell lung cancer [20], and melanoma [21]. The initial anticancer mechanistic studies focused on its antimicrotubule activity [22], but mebendazole probably operates through diverse mechanisms dependent on the cancer type. It was found that mebendazole can stabilize p53, p21, and MDM2 in lung cancers [23, 24] but downregulates the prosurvival protein Bcl-2 through phosphorylation in chemoresistant melanoma cells [21]. Mebendazole blocks the hedgehog/SMO pathway in basal cell carcinoma [25] but inhibits the TRAF2- and NCK-interacting kinase in colorectal cancer [26]. All of the above studies suggest that mebendazole elicits diverse anticancer effects. In the present study, we found that mebendazole displays potent antimyeloma activity at low concentrations by inhibiting the USP5/c-Maf axis.
c-Maf is an oncogenic transcription factor that is frequently dysregulated in MM and contributes to myelomagenesis by promoting the expression of several important genes involved in cancer cell proliferation, survival and metastasis, including CCND2, ITGB7, CCR1, and ARK5 [27]. Inhibiting c-Maf thus leads to regression of MM tumors and inhibition of MM cell proliferation [4, 8]. Our recent studies demonstrated that the c-Maf protein is processed via the ubiquitin-proteasome pathway under the control of the ubiquitin-conjugating enzyme UBE2O [11], the ubiquitin ligase HERC4 [10] or TMEPAI/NEDD4 [28]. c-Maf ubiquitination can be reversed by the deubiquitinase USP5 because USP5 prevents c-Maf from polyubiquitination and degradation [8]. Moreover, inhibiting USP5 by genetic shRNA or small molecule inhibitors such as WP1130 leads to MM cell apoptosis [8]. Collectively, these findings thus establish a rationale for treating MM by targeting the USP5/c-Maf axis. As a principle of concept, the present study identified mebendazole as an agent with potent antimyeloma activity from the FDA-approved drug library. The mechanistic investigation showed that mebendazole not only prevents the interaction between USP5 and c-Maf but also suppresses USP5 transcription, thus inducing c-Maf proteasomal degradation.
Notably, mebendazole is cytotoxic to various cancer cells, including MM and leukemia cells, at higher concentrations (data not shown); however, it elicits selective anti-MM activity at low concentrations. As shown in the present study, at low molar concentrations, such as 0.5 µM, mebendazole induced the apoptosis of LP1 and RPMI-8226 cells that express c-Maf (Fig. 4) but was less effective in U266 cells that lack c-Maf, suggesting that c-Maf is a key factor in MM cell death induced by low concentrations of mebendazole. We also found that 0.1 µM mebendazole can markedly enhance the anti-MM activity of other agents. Mebendazole synergizes with WP1130, an inhibitor of USP5, because mebendazole downregulates USP5 expression and WP1130 inhibits USP5 deubiquitinase activity. Mebendazole also enhances antimyeloma activity in combination with daunorubicin, a major antimyeloma drug. These results indicate that mebendazole exerts potent antimyeloma activity alone or in combination with other anticancer agents. However, mebendazole does not show high toxicity, as seen in the tumor-bearing nude mice. Actually, as an oral antiparasitic agent, mebendazole has historically been well demonstrated to be safe in humans.
In summary, the present study identified mebendazole as a potent and promising antimyeloma agent via its inhibition of the USP5/c-Maf axis. This report is the first on treating myeloma by targeting c-Maf ubiquitination and proteasomal degradation. Given its high safety and potent anti-MM activity alone or in combination with other agents, mebendazole is guaranteed to be repurposed for personalized MM therapy.
References
Raab MS, Podar K, Breitkreutz I, Richardson PG, Anderson KC. Multiple myeloma. Lancet. 2009;374:324–339.
Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14:100–113.
Perrot A, Corre J, Avet-Loiseau H. Risk stratification and targets in multiple myeloma: from genomics to the bedside. Am Soc Clin Oncol Educ Book. 2018;38:675–680.
Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, et al. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell. 2004;5:191–199.
Neri P, Ren L, Azab AK, Brentnall M, Gratton K, Klimowicz AC, et al. Integrin beta7-mediated regulation of multiple myeloma cell adhesion, migration, and invasion. Blood. 2011;117:6202–6213.
Qiang YW, Ye S, Chen Y, Buros AF, Edmonson R, van Rhee F, et al. MAF protein mediates innate resistance to proteasome inhibition therapy in multiple myeloma. Blood. 2016;128:2919–2930.
Chen G, Xu X, Tong J, Han K, Zhang Z, Tang J, et al. Ubiquitination of the transcription factor c-MAF is mediated by multiple lysine residues. Int J Biochem Cell Biol. 2014;57:157–166.
Wang S, Juan J, Zhang Z, Du Y, Xu Y, Tong J, et al. Inhibition of the deubiquitinase USP5 leads to c-Maf protein degradation and myeloma cell apoptosis. Cell Death Dis. 2017;8:e3058.
Lacey E. The role of the cytoskeletal protein, tubulin, in the mode of action and mechanism of drug resistance to benzimidazoles. Int J Parasitol. 1988;18:885–936.
Zhang Z, Tong J, Tang X, Juan J, Cao B, Hurren R, et al. The ubiquitin ligase HERC4 mediates c-Maf ubiquitination and delays the growth of multiple myeloma xenografts in nude mice. Blood. 2016;127:1676–1686.
Xu Y, Zhang Z, Li J, Tong J, Cao B, Taylor P, et al. The ubiquitin-conjugating enzyme UBE2O modulates c-Maf stability and induces myeloma cell apoptosis. J Hematol Oncol. 2017;10:132.
Morito N, Yoh K, Maeda A, Nakano T, Fujita A, Kusakabe M, et al. A novel transgenic mouse model of the human multiple myeloma chromosomal translocation t(14;16)(q32; q23). Cancer Res. 2011;71:339–348.
Holien T, Olsen OE, Misund K, Hella H, Waage A, Rø TB, et al. Lymphoma and myeloma cells are highly sensitive to growth arrest and apoptosis induced by artesunate. Eur J Haematol. 2013;91:339–346.
Papanikolaou X, Johnson S, Garg T, Tian E, Tytarenko R, Zhang Q, et al. Artesunate overcomes drug resistance in multiple myeloma by inducing mitochondrial stress and non-caspase apoptosis. Oncotarget. 2014;5:4118–4128.
Kapuria V, Peterson LF, Fang D, Bornmann WG, Talpaz M, Donato NJ, et al. Deubiquitinase inhibition by small-molecule WP1130 triggers aggresome formation and tumor cell apoptosis. Cancer Res. 2010;70:9265–9276.
Nygren P, Fryknas M, Agerup B, Larsson R. Repositioning of the anthelmintic drug mebendazole for the treatment for colon cancer. J Cancer Res Clin Oncol. 2013;139:2133–2140.
Williamson TBR, Staedtke V, Huso D, Riggins GJ. Mebendazole and a non-steroidal anti-inflammatory combine to reduce tumor initiation in a colon cancer preclinical model. Oncotarget. 2016;7:68571–68584.
Bai RY, Staedtke V, Wanjiku T, Rudek MA, Joshi A, Gallia GL, et al. Brain penetration and efficacy of different mebendazole polymorphs in a mouse brain tumor model. Clin Cancer Res. 2016;21:3462–3470.
Bai RY, Staedtke V, Aprhys CM, Gallia GL, Riggins GJ. Antiparasitic mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro Oncol. 2011;13:974.
Sasaki J, Ramesh R, Chada S, Gomyo Y, Roth JA, Mukhopadhyay T, et al. The anthelmintic drug mebendazole induces mitotic arrest and apoptosis by depolymerizing tubulin in non-small cell lung cancer cells. Mol Cancer Res. 2002;1:1201–1209.
Doudican N, Rodriguez A, Osman I, Orlow SJ. Mebendazole induces apoptosis via Bcl-2 inactivation in chemoresistant melanoma cells. Mol Cancer Res. 2008;6:1308.
Spagnuolo PA, Hu J, Hurren R, Wang X, Gronda M, Sukhai MA, et al. The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood. 2010;115:4824–4833.
Mukhopadhyay T, Sasaki J, Ramesh R, Roth JA. Mebendazole elicits a potent antitumor effect on human cancer cell lines both in vitro and in vivo. Clin Cancer Res. 2002;8:2963–2969.
Dakshanamurthy S, Issa NT, Assefnia S, Seshasayee A, Peters OJ, Madhavan S, et al. Predicting new indications for approved drugs using a proteochemometric method. J Med Chem. 2012;55:6832–6848.
Larsen AR, Bai RY, Chung JH, Borodovsky A, Rudin CM, Riggins GJ, et al. Repurposing the antihelmintic mebendazole as a hedgehog inhibitor. Mol Cancer Ther. 2015;14:3–13.
Tan Z, Chen L, Zhang S. Comprehensive modeling and discovery of mebendazole as a novel TRAF2- and NCK-interacting kinase inhibitor. Sci Rep. 2016;6:33534.
Rasmussen T, Knudsen LM, Dahl IMS, Johnsen HE. C-MAF oncogene dysregulation in multiple myeloma: frequency and biological relevance. Leuk Lymphoma. 2003;44:1761–1766.
Du Y, Liu Y, Xu Y, Juan J, Zhang Z, Xu Z, et al. The transmembrane protein TMEPAI induces myeloma cell apoptosis by promoting degradation of the c-Maf transcription factor. J Biol Chem. 2018;293:5847–5859.
Acknowledgements
This work was partly supported by the National Natural Science Foundation of China (81320108023 to XM, 81600171 to ZZ, and 81770215 to BC), the Natural Science Foundation of Jiangsu Higher Education Institutes of China (17KJA180010 to XM), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD to XM), and the Jiangsu Key Laboratory for Neuro-Psycho-Diseases (BK2013003 to XM). The study was also partly supported by the Suzhou Key Laboratory for Pediatric Leukemia (SZS201615 to XM) and the Suzhou Key Medical Center (Szzx201506 to YZ).
Author information
Authors and Affiliations
Contributions
XM, ZZ, and TZ designed the study; XC, YX, XW, ZZ, PL, and BC conducted the experiments; XM, XC, YZ, QW, and TZ analyzed the data; and XM and XC wrote the manuscript.
Corresponding authors
Rights and permissions
About this article

Cite this article
Chen, Xh., Xu, Yj., Wang, Xg. et al. Mebendazole elicits potent antimyeloma activity by inhibiting the USP5/c-Maf axis. Acta Pharmacol Sin 40, 1568–1577 (2019). https://doi.org/10.1038/s41401-019-0249-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-019-0249-1
Keywords
This article is cited by
-
The role and regulation of Maf proteins in cancer
Biomarker Research (2023)
-
A pan-cancer analysis of the role of USP5 in human cancers
Scientific Reports (2023)
-
Mebendazole, an anti-helminth drug, suppresses inflammation, oxidative stress and injury in a mouse model of ulcerative colitis
Scientific Reports (2022)
-
Deubiquitinases in hematological malignancies
Biomarker Research (2021)
-
Suppression of USP7 induces BCR-ABL degradation and chronic myelogenous leukemia cell apoptosis
Cell Death & Disease (2021)
