
Abstract
Host cdc2-like kinase 1 (CLK1) is responsible for the alternative splicing of the influenza virus M2 gene during influenza virus infection and replication that has been recognized as a potential anti-influenza virus target. In this study, we showed that gallocatechin-7-gallate (J10688), a novel CLK1 inhibitor isolated from Pithecellobium clypearia Benth, exerted potent anti-influenza virus activity in vivo and in vitro. ICR mice were intranasally infected with a lethal dose of H1N1. Administration of J10688 (30 mg·kg−1·d−1, iv, for 5 days) significantly increased the survival rate of the H1N1-infected mice to 91.67% and prolong their mean survival time from 5.83 ± 1.74 days to 13.66 ± 1.15 days. J10688 administration also slowed down body weight loss, significantly alleviated influenza-induced acute lung injury, reduced lung virus titer, elevated the spleen and thymus indexes, and enhanced the immunological function. We further explored its anti-influenza mechanisms in the H1N1-infected A549 cells: as a novel CLK1 inhibitor, J10688 (3, 10, 30 μmol/L) dose-dependently impaired synthesis of the viral proteins NP and M2, and significantly downregulated the phosphorylation of splicing factors SF2/ASF and SC35, which regulate virus M2 gene alternative splicing. As a novel CLK1 inhibitor with potent anti-influenza activity in vitro and in vivo, J10688 could be a promising antiviral drug for the therapy of influenza A virus infection.
Similar content being viewed by others


Introduction
Influenza A virus (IAV) infection has become a global public health threat associated with significant morbidity and mortality. Currently, the limited therapeutic options against the influenza virus cannot cope with the rapid emergence of drug-resistant viruses [1, 2]. In addition, the emergence of highly pathogenic strains, such as H5N1 IAV, which emerged in 1996 in China [3], presents a tremendous challenge to currently available antiviral therapeutics. A new generation of antiviral agents, especially those targeting resistant strains, is urgently needed. Since it was reported that host factors play crucial roles in the viral life cycle and host immune response, an antiviral therapy aimed at these host factors was believed to be a more sophisticated strategy to minimize the chance of developing drug resistance [4].
Host cell cdc2-like kinase 1 (CLK1) is a critical element of several important regulatory pathways involved in pre-mRNA alternative splicing. Alternative splicing enriches the structural and functional variability of multiple protein isoforms involved in the genomic and proteomic regulatory network. CLK1 acts by phosphorylating the serine- and arginine-rich (SR) factors to direct the early events of spliceosome assembly, which are necessary for proper mRNA maturation [5, 6]. The family of SR factors includes SC35, 9G8, SRp20, and SF2/ASF. All these factors are not only essential for constitutive splicing but also are involved in regulating splicing in a concentration-dependent manner by influencing the selection of alternative splice sites [7]. The influenza virus takes advantage of the host RNA-processing machinery to initiate the alternative splicing that results in the expression of proteomic diversity. It was demonstrated that an SR protein, ASF/SF2, bound to CLK1 at the RNA-recognition motifs of ASF/SF2, which regulated the synthesis of M2 RNA, a splice variant of the IAV M gene [8]. It has also been reported that a small molecule inhibitor of CLK1, TG003, could reduce influenza virus replication by more than two orders of magnitude, an effect related to impaired splicing of the viral M2 mRNA [9]. Thus, CLK1 was assumed to be a potential host cell target for influenza virus treatment. Finding more CLK1 inhibitors could be an effective avenue of research for new anti-influenza drugs.
In a previous study, we established a reliable screening assay for CLK1 inhibitors, resulting in the discovery of several compounds with strong inhibitory activity against CLK1 [10]. Most of these compounds were validated to have anti-influenza virus effects. The most potent of these compounds, gallocatechin-7-gallate (J10688, Fig. 1a), was chosen as a candidate for further research. J10688 was one of the active components isolated from Pithecellobium clypearia Benth, a kind of traditional Chinese herbal medicine distributed in the south of China. We have also found that several chemical constituents from P. clypearia, including J10688, exhibited influenza virus neuraminidase-inhibitory activity and anti-inflammatory activity in vitro [11]. The pharmacokinetic properties of J10688 in Sprague–Dawley (SD) rats have also been studied to obtain a better understanding of its pharmacological function and mechanism [12]. In this study, the activity of J10688 was further investigated, including the anti-influenza H1N1 virus effect in vivo and the efficacy against H1N1, H3N2, and subtype B in vitro. Mechanistic studies were then conducted to investigate its target and mode of action. Our findings demonstrated that the CLK1 inhibitor J10688 is a potential antiviral agent that acts by influencing viral M2 mRNA splicing and through other complicated antiviral effects.

a The chemical structure of J10688. b Basic experimental protocol used to test J10688 in ICR mice infected with a mouse-adapted A/PR/8/4 H1N1 influenza virus. A lethal dose of H1N1 (10 LD50) was used to intranasally infect mice on day 0. After 24 h, all mice received J10688 (30, 10, and 3 mg/kg/day i.v.), oseltamivir (20 mg/kg/day, p.o.), TG003 (50 mg/kg/day, p.o.), or no treatment in the model groups (control, normal saline, p.o.) for five successive days. Survival rate (each group n = 12; c) and body weight (each group n = 12; d) of J10688-, oseltamivir- and TG003-infected mice.
Materials and methods
Viruses and cells
The human influenza virus strains A/PR/8/34 (H1N1), A/Sydney/5/97 (H3N2), and B/Jiangsu/10/2003 were kindly donated by the Institute for Viral Diseases Control and Prevention, Chinese Centers for Disease Control and Prevention. Viral stocks of the laboratory-adapted strains were propagated in 9-day-old embryonated chicken eggs for 48 h and stored at −80 °C. All viruses involved in the experiments of this paper were handled in a biological safety protection second-level laboratory.
Madin–Darby canine kidney (MDCK) cells and the human lung cancer cell line A549 were cultured in Dulbecco’s modified Eagle's medium (DMEM) and RPMI-1640 medium, respectively. Media were supplemented with 10% fetal bovine serum (FBS) and incubated in a 5% CO2 incubator.
Samples
J10688 was extracted and isolated from the leaves and twigs of P. clypearia, which were collected from Shankou (Beihai, China) and identified at the Chinese Academy of Medical Sciences. The purity of the compound was over 98%, calculated through peak area normalization using high-performance liquid chromatography [11]. All experimental reagents were purchased from Sigma-Aldrich, China, unless otherwise specified.
Mice
Male ICR mice, 6–8 weeks old, were purchased from Vital River Laboratory Animal Technology Co Ltd (Beijing, China), housed in independent ventilated cages and received pathogen-free food and water. All animals received care according to The Guide for the Care and Use of Laboratory Animals.
Therapeutic efficacy study in mice
Mouse-adapted influenza A/PR/8/34 (H1N1) virus stocks were diluted in PBS to a 10-fold LD50 concentration. Mice were anesthetized with diethyl ether and inoculated intranasally with 50 μL of virus or PBS. After 24 h, all the mice were intravenously administered J10688 or normal saline (model groups) and intragastrically administered the reference drugs oseltamivir, TG003, and normal saline (normal group) [13]. The mice were partly killed on day 6 after infection, and the lung tissues were collected and homogenized in 1 mL of Trizol® reagent (Life Technology, Lot# 15596-026) to extract the viral RNA for RT-qPCR assay. The lung, spleen, and thymus were dissected, and organ indexes were calculated using the formula below[14]:
Histological staining
The mice were killed, and the lungs were obtained and fixed in buffered 4% paraformaldehyde. The lungs were stained with hematoxylin and eosin as described previously [15].
Cytotoxicity test
A monolayer of MDCK cells or A549 cells was treated with different concentrations of J10688 and incubated at 37 °C under a 5% CO2 environment for 48 h. Subsequently, cell viability was detected with the MTT method [16]. The CC50 value (i.e., the concentration of compound that reduces the cell viability by 50%) was determined using GraphPad Prism 5 Software by plotting the percent cell viability as a function of compound concentration [17].
CPE reduction assay
To evaluate the pharmacological properties of J10688, three different time points for drug administration and three kinds of influenza subtype (A/PR/8/34 (H1N1), A/Sydney/5/97 (H3N2), and B/Jiangsu/10/2003) were selected in our experiments. (1) Pre-incubation assay: influenza viruses (100TCID50) were pre-incubated with serial dilutions of J10688 for 2 h at 37 °C before being added to MDCK cells for cell viability determination. Two hours later, all the supernatant was removed, and cell culture media with different dilutions of drugs were added. (2) Simultaneous treatment assay: J10688 was added to the cells along with the influenza viruses (100TCID50). Two hours later, all the supernatant was removed, and cell culture media with different drug dilutions were added. (3) Post-treatment assay: J10688 was added to the cells 2 h after the adsorption of the influenza virus (100TCID50). The plates were incubated at 37 °C in a humidified 5% CO2 atmosphere for 48 h. The CPE was then assessed using the MTT method as described above. The resulting spectrophotometric data were used to calculate the IC50 [18]. All these experiments were repeated at least three times.
Quantitative real-time PCR assay
A549 cells infected with influenza A/PR/8/34 (H1N1) virus (100TCID50) for 2 h were treated with J10688 at various concentrations. At 6 h post-infection, the total RNA was isolated. The primer sequences for qPCR of the influenza virus HA gene were 5’-CCTGCTCGAAGACAGCCACAACG-3’ (sense) and 5’-TTCCCAAGAGCCATCCGGCGA-3’ (antisense). GAPDH primers were used as internal controls of cellular RNAs: 5’-AGGCGTCGGAGGGCCCCCTC-3’ (sense) and 5’-AGGGCAATGCCAGCCCCAGCG-3’ (antisense). The amplification conditions were as follows: 94 °C for 30 s (one cycle), followed by 94 °C for 5 s, 55 °C for 15 s and 72 °C for 10 s (40 cycles). RT-qPCR was conducted using the CFX96 Realtime PCR system (Bio-Rad). The data were analyzed with the Bio-Rad CFX manager using the mode for normalized expression (2−△△Ct).
The viral load of influenza-infected mouse lungs was measured by the RT-qPCR method in the same way. The lungs of mice killed on the 6th day were sampled and homogenized in Trizol® reagent to extract total RNA. The primer sequences for qPCR of the influenza virus NP gene were 5’-CTGTTTTGGATCAACCGTC-3’ (sense) and 5’-CTGTTTTGGATCAACCGTC-3’ (antisense). The mouse GAPDH gene was used as the internal control of cellular RNAs, using the primer sequences 5’-AGCAGTCCCGTACACTGGCAAAC-3’ (sense) and 5’-TCTGTGGTGATGTAAATGTCCTCT-3’ (antisense).
Lymphocyte proliferation assay
The experiment was performed as described by Yu et al. [19]. The spleen was minced in PBS and passed through a fine steel mesh under aseptic conditions to obtain the cell suspension. After separating the red blood cells in NH4Cl and washing the rest three times, the cells were cultured in RPMI-1640 with 10% FBS. Mitogen-induced stimulation was performed by incubating equal volumes (100 μL) of lymphocyte suspension with either ConA (5 μg/mL) or LPS (10 μg/mL) in each well. RPMI-1640 medium (100 μL) was added to the cell suspension in the control wells. All cells were incubated in a 5% CO2 at 37 °C for 48 h, and the MTT method was used to determine the cell viability. The stimulation index of lymphocytes was calculated based on the following formula:
Indirect immunofluorescence microscopy
A549 cells were treated with J10688 (3, 10, and 30 μM) after being infected with influenza A/PR/8/34 (H1N1) virus (100TCID50) for 2 h. Twenty-four hours later, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100. Mouse monoclonal antibodies against IAV nucleoprotein (NP; Santa Cruz, Catalog# sc-80481) and M2 (Santa Cruz, Catalog# sc-32238) were incubated with the cells overnight at 4 °C, followed by Alexa Fluor® 488 conjugate (Cell Signaling Technology, #4408). The nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI, 1 μg/mL) for 10 min in the dark. Cell imaging and fluorescence were carried out using the Thermo Scientific ArrayScan Infinity (Thermo Fisher Scientific, USA).
Cytokine secretion assay (ELISA)
The lungs of mice killed on the sixth day were homogenized in PBS. The expression levels of IFN-γ, IL-6, TNF-α, and IL-1β in the mouse lungs were detected by an ELISA kit (ExCell Bio, Shanghai, China) according to the manufacturer’s protocol.
Statistical analysis
Results were expressed as the mean ± SD (n ≥ 3). The statistical significance of the data was evaluated by one-way ANOVA or independent tests. Differences were considered statistically significant when the P value was less than 0.05.
Results
Therapeutic efficacy of J10688 against A/PR/8/4 (H1N1) influenza virus in vivo
To validate the pharmacological function of J10688, the therapeutic efficacy for influenza virus-induced pneumonia in mice was evaluated. The protocol for this experiment is illustrated in Fig. 1b.
As shown in Fig. 1c, 91.67% of mice that received intravenous treatment with 30, 10, and 3 mg/kg/day J10688 survived (P < 0.0001), whereas all the mice that were administered normal saline died within 9 days. The reference drugs oseltamivir and TG003 also showed a strong protective effect. All mice survived after oral administration of 20 mg/kg/day oseltamivir, while the 60 mg/kg/day TG003-treated group showed a 91.67% survival rate. In addition, J10688 could significantly suppress body weight loss (Fig. 1d). The mean survival time is shown in Table 1. In comparison with the model group (mean survival time of 5.83 days), the mean survival time in the J10688 group (30 mg/kg/day) was statistically prolonged (13.58 days, P < 0.0001). Oseltamivir and TG003 prolonged the mouse survival times to 14.0 and 13.58 days, respectively.
J10688 inhibited influenza virus replication and protected the mouse lungs
The RT-qPCR method was used in this study to examine the viral NP mRNA expression level in H1N1-infected mouse lungs. The 2−ΔΔCt (Livak) algorithm was used to analyze the Ct values based on the approximate efficiencies of the target and reference genes. As shown in Fig. 2a, the expression level of the NP gene in all drug-treated groups declined, which means that all the drugs can effectively suppress A/PR/8/34 (H1N1) virus replication during viral pneumonia in the model mice. Compared with the results from the model group, J10688 administered at 30 mg/kg/day (P < 0.05) and 10 mg/kg/day (P < 0.05) could significantly downregulate the expression of NP mRNA.

Drug-protective effects on the lung tissues of mice infected by influenza virus H1N1 6 days prior. a Influenza viral NP mRNA levels in mouse lungs measured by RT-qPCR and normalized to GAPDH. J10688 treatment can dose-dependently decrease the expression of NP mRNA (**P < 0.01, *P < 0.05 vs. model). b Lung index. *P < 0.05 vs. model. c H&E staining of sectioned lungs, magnification 200 times. The pathological changes were evaluated based on hyperemia, bronchiole epithelium cell necrosis, lung exudates, alveolar interstitial pneumonia, and lung abscesses. c1 Normal group: the bronchial wall was not thickened in the lung, the lumen was clean, there was no inflammatory cell infiltration, and there was no alveolar wall thinning. c2 Model group: there was a large number of mononuclear cell infiltrates surrounding the alveoli and bronchus, the presence of pulmonary microvascular congestion, exhibition of bronchial cavity effusion and falling debris, and thickening of the alveolar interstitial edema. c3 Oseltamivir group; c4 TG003 group; c5 J10688, 30 mg/kg/day; c6 J10688, 10 mg/kg/day; c7 J10688, 3 mg/kg/day
The lung index data show that J10688 administered at 30 mg/kg/day provided protective effects against viral pneumonia at 6 days after infection (P < 0.05). Oseltamivir tended to inhibit an increase in the lung index due to influenza, while TG003 did not have this effect (Fig. 2b). Histopathologic examination (H&E staining) further showed that the alveolar damage and interstitial inflammatory infiltration in the lung tissues of the mice treated by J10688 were greatly ameliorated compared to those treated by normal saline (Fig. 2c).
J10688 inhibited the expression levels of inflammatory markers in mouse lungs
The pro-inflammatory cytokine interferon-gamma (IFN-γ) is well known for its important role in innate and adaptive immunity against intracellular infections. In this paper, IFN-γ was detected as an inflammatory marker, and its expression level in mouse lungs increased after infection by influenza virus. All drug treatment groups showed improved expression levels of IFN-γ to different degrees. Three other inflammatory cytokines were detected, IL-6, TNF-α, and IL-1β, and their expression levels in the group treated with J10688 at a dose of 30 mg/kg/day were significantly decreased (P < 0.05; Fig. 3). These data suggest that J10688 could modulate the inflammatory response in the lung.

The expression level of IFN-γ, IL-6, TNF-α, and IL-1β in infected mouse lungs. Lung tissue homogenates were prepared for detection of inflammatory cytokine by ELISA. Each group n = 6, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the model group
J10688 improved the immune function of the infected mice
On the sixth day after infection, some of the mice were killed to collect spleen and thymus samples. As depicted in Fig. 4a, a great change in the appearance of the spleen was observed in the model group after being infected with influenza, while J10688 improved the appearance of the spleen to the same extent as did the reference drugs. The spleen and thymus indexes of the model group decreased significantly in comparison with those of the normal group (##P < 0.01). In contrast, the J10688 treatment (30 mg/kg/day) increased the indexes (*P < 0.05, **P < 0.01), which means that J10688 can effectively inhibit lesions of the immune organs (Fig. 4b, c).

a Spleen picture from each group. b Spleen index of infected mice after 6 days. c Thymus index. d Stimulation index of ConA, which stimulates T-lymphocyte mitosis. e Stimulation index of LPS, which stimulates B-lymphocyte mitosis. ##P < 0.01, ###P < 0.001 compared with the model group. Each group n = 6, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the model group
ConA and LPS can stimulate mitosis of T and B lymphocytes, respectively, and the stimulation index is representative of the body’s immune status. As shown in Fig. 4d, e, the stimulation indexes decreased significantly (###P < 0.001) in the model groups, which meant that cellular and humoral immunity were suppressed. Treatment with J10688 could dose-dependently increase the stimulation index, effectively stimulating lymphocyte proliferation and improving the immune function.
Antiviral activity of J10688 on influenza viruses in vitro
The cytotoxicity of J10688 on MDCK and A549 cells was evaluated by the MTT assay. The results showed that J10688 had no significant cytotoxicity on MDCK and A549 cells (Fig. 5a, b), with both of the CC50 values above 100 μM. To verify the anti-influenza virus activity of J10688 in vitro, the reduction of influenza virus-induced cytopathic effects (CPEs) was evaluated in MDCK cells. Time-of-addition experiments were performed to determine the stage(s) at which J10688 exerted its antiviral effects. J10688 was added to MDCK cells at three distinct time points: after incubation with the virus for 2 h (pre-incubation treatment), at the same time as the viral infection (simultaneous treatment), or at 2 h after entry (post treatment). As shown in Fig. 5c-e, J10688 exhibited antiviral effects in the three different types of influenza viruses. There was a positive correlation between the concentration of J10688 and its corresponding inhibitory effects. When the virus was pre-incubated with J10688, the EC50s (i.e., half maximal effective concentration) for A/PR/8/34 (H1N1), A/Sydney/5/97 (H3N2), and B/Jiangsu/10/2003 were 1.69, 2.28, and 23.18 μM, respectively, while in the simultaneous treatment assay, the EC50s for H1N1, H3N2, and type B were 2.85, 12.47, and 57.61 μM, respectively. The EC50 values of J10688 ranged from 46.27 to 55.84 μM in the post-treatment assay of H1N1 and H3N2. J10688 only presented weak suppressive activity for type B, with an EC50 over 100 μM.

Antiviral activity of J10688 on influenza viruses in vitro. a Cell viability following J10688 treatment in MDCK cells or A549 cells (b) for 72 h, determined by MTT assay. c–e Inhibition rates of J10688 (from 0.04 to 100 μM) against A/PR/8/34 (H1N1), A/Sydney/5/97 (H3N2), and B/Jiangsu/10/2003. b Influenza viruses with different infection protocols in MDCK cells. c Pre-incubation treatment, d simultaneous treatment, e post treatment. Each concentration took up three wells for each assay, and three independent determinations were carried out
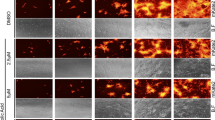
To investigate whether J10688 impaired the synthesis of viral proteins in host cells, A549 cells were infected with H1N1 virus with or without J10688 treatment. The concentrations of J10688 in the treated groups were 3, 10, and 30 μM. At 12 h post infection, the expression of the viral proteins was monitored by HSC using monoclonal antibodies against NP and M2. As depicted in Fig. 6a-d, a strong green fluorescence signal was observed in the model group cells without J10688 treatment. As demonstrated in the immunofluorescence staining test, the NP and M2 expression levels were remarkably decreased in a dose-dependent manner. Compared with the normal group, the protein expression levels of NP and M2 were higher in the normal group (P < 0.001). The expression levels of NP and M2 at doses of 10 and 30 μM J10688 were significantly lower (P < 0.01) compared with those of the model group.

J10688 reduced the expression levels of the viral nucleoprotein (NP), M2-iron channel protein (M2), and HA mRNA in H1N1-infected A549 cells. J10688, with a series of concentrations of 3, 10, and 30 μM, was added with influenza virus A/PR/8/34 (H1N1)-infected cells at 100TCID50. At 7 h post infection, NP and M2 localization was visualized using specific antibodies with an immunofluorescence microscope. The green fluorescence indicates the presence of the viral NP or M2 protein, while the blue fluorescence of DAPI indicates the nucleus of the cell. Photographs of NP (a) and M2 (b) were taken randomly from one representative experiment. In addition, the average fluorescence intensity of NP (c) and M2 (d) in each cell was recorded. e Influenza viral HA mRNA levels in each group were detected by RT-qPCR. ###P < 0.001, compared with the model; **P < 0.01, ***P < 0.001, compared with the model. In total, three independent experiments were performed
We then tested the effect of J10688 on the transcription of viral genes in infected cells using quantitative RT-qPCR of the influenza virus HA gene. A549 cells were infected with A/PR/8/34 (H1N1) virus and were incubated for 6 h in the presence of various drug concentrations. Total RNAs were isolated from infected cells, and RT-qPCR analysis was performed using primers specific for viral HA RNA. The qPCR results showed that treatment with J10688 drastically reduced the copy number of viral RNA synthesis at 6 h post infection in a dose-dependent manner (Fig. 6e). These results indicated that J10688 effectively inhibited the growth of H1N1 virus in A549 cells.
Effects of J10688 on the CLK1 pathway
Influenza makes one of the host splice factors, SF2/ASF, engage in its own M1 mRNA splicing to produce M2 pre-mRNA and, subsequently, M2 ion channel protein expression [9]. In a previous study, we found that J10688 is a potent inhibitor of CLK1 [10]. To investigate the anti-influenza virus mechanism of J10688, we carried out an in vitro experiment in which A549 cells were infected with A/PR/8/34 (H1N1) for 2 h, washed free of the virus, and then treated with J10688 (3, 10, and 30 μM). The total cellular protein was collected 6 h later and analyzed by western blot analysis. The results presented in Fig. 7a show that J10688 could significantly reduce the expression level of the influenza virus M2 protein in a dose-dependent manner, and there was nearly no M2 protein expression in the TG003 group. In contrast, the influenza M1 protein expression in each group had no significant changes. The intensity of M2/M1 increased by 3.5 times in the model group compared with that in the normal group. The J10688 treatment groups and the TG003 group tended to show decreased levels of M2/M1, thus evidencing suppression of influenza virus alternative splicing from M1 to M2.

In-depth analysis of the impact of the CLK1 inhibitor J10688 on influenza A virus infection. a A549 cells were treated with J10688 or TG003 (50 μM) and subsequently infected with influenza A/PR/8/6 H1N1 virus (100 TCID50) for 6 h. Protein levels of spliced M2 and M1 after inhibition of CLK1 by J10688 or TG003. b The impact of J10688 or TG003 on the phosphorylation level of host SF2/ASF after infection with influenza virus. c The impact of J10688 or TG003 on the phosphorylation level of host SC35 after infection with influenza virus. ##P < 0.01, ###P < 0.001 compared with the normal, *P < 0.05, **P < 0.01, ***P < 0.001 compared with the model.
CLK1 regulates alternative splicing of the SR protein by phosphorylation. SR proteins are essential splicing factors that are regulated through multisite phosphorylation of their RS (arginine/serine-rich) domains [5, 20]. SC35 and SF2/ASF are splicing enhancer elements that interact with M2 mRNA alternative splicing and become over-phosphorylated after virus infection. J10688 and TG003 treatment significantly inhibited excessive phosphorylation of SC35 and SF2/ASF (Fig. 7b, c).
Discussion
Influenza is a highly contagious disease that can cause high morbidity and mortality in an epidemic. The clinical utility of anti-influenza viral drugs has been limited due to the drugs’ severe side effects and the occurrence of resistant strains. Hence, the discovery of novel anti-influenza virus drugs is in urgent demand. Traditional Chinese medicinal herbs may be a potential alternative medicine for treatment of this disease. Recently, a number of clinical trials of traditional Chinese medicines for influenza have been conducted [21]. J10688 is an effective compound isolated from the traditional Chinese medicinal plant P. clypearia Benth. It is widely used as a folk medicine for the treatment of upper respiratory tract infections, pharyngitis, laryngitis, and acute tonsillitis [22]. In China, P. clypearia has been used as an anti-inflammatory drug in clinics for many years. Here, we investigated its antiviral effects in vivo and in vitro. Its antiviral mechanism was also further explained/discussed.
The inhibitory activity of J10688 against H1N1 in vivo was tested. The intravenous administration of J10688 showed positive effects in the H1N1-infected mice, including increasing the survival rate, prolonging the mean survival time, and decreasing the lung index and lung consolidation. It also had some effects on inflammatory cytokine secretion. In contrast to the model group, which experienced remarkable body weight loss and a 100% mortality rate on day 14, J10688 treatment brought about beneficial effects with respect to these indicators. Notably, the mortality rate decreased by 91.67% after J10688 treatment. Our previous study on the pharmacokinetics and tissue distribution of J10688 proved that it has a limited oral bioavailability but a wide distribution across all organs and tissues after intravenous administration, accumulating especially in lung tissues, where the influenza virus mainly occurs [12]. This drug distribution to the target organ provides the basis of the pharmacological activity of J10688 in vivo.
It has been reported that the pathological injury caused by H1N1 infection is mostly due to host inflammatory responses to the virus, rather than to direct viral destruction of respiratory epithelia [23]. The morphology is closely associated with intracellular biochemical changes. In this study, H&E staining revealed severe pneumonia in the model group, including hyperemia, inflammatory cellular infiltration, bronchiole epithelium cell necrosis, lung exudates, alveolus interstitial pneumonia, and lung abscess. J10688 treatment improved the symptoms in some ways, which suggests that J10688 exerted anti-influenza virus effects in vivo.
Investigation of the antiviral effects of J10688 in vitro included time-of-addition assays to identify the stage(s) in which J10688 acted. The relative inhibitory rates against the CPE in the three administrations were as follows: pre-incubation treatment > simultaneous treatment >post treatment. This tendency was found for three kinds of viruses. There was a marked difference in the antiviral effects when the compound was added before infection compared to post-infection treatment (1.7 vs. 46 µM). This clearly indicated that part of the J10688 mechanism of action is related to viral entry. In addition, the difference between pre-incubation treatment and simultaneous treatment suggested that J10688 may directly inactivate the virus. It was also shown that J10688 treatment could decrease the expression level of virus HA mRNA, NP, and M2 protein in a dose-dependent manner. All these findings suggest that J10688’s inhibition of influenza virus replication may have both preventive and therapeutic effects on influenza virus infection.
Alternative splicing plays an important role during the processing of influenza virus M2 mRNA. The influenza virus M1 mRNA has two alternative 5’ splice sites. One is the distal 5’ splice site, producing mRNA that closely fits the consensus 5’ splice sequence in uninfected cells. The other one is the proximal 5’ splice site used in influenza virus-infected cells, which generates the M2 mRNA encoding the M2 ion-channel protein [24]. The SR proteins, as the constitutive splicing factors, can favor the use of the proximal 5’ splicing site [25]. Phosphorylation of SR affects alternative splicing, which is regulated by a family of kinases termed CLK1 [26]. SR proteins are evolutionarily conserved phosphoproteins that bind to pre-mRNA molecules to promote both constitutive and alternative splicing [27]. Thus, CLK1 controls M2 protein expression in an indirect way. CLK1 has been recognized as an important host protein for influenza virus treatment, and the antiviral effects of its inhibitor, TG003, have been proven [9]. In a previous study, we found that J10688 exerted a CLK1-inhibitory effect with an IC50 of 2.7 μM [10]. Here, we found that the influenza viral M2/M1 ratio significantly decreased in the J10688-treatment groups, and the phosphorylation level of the splicing factors SF2/ASF and SC35 declined, which indicates that J10688 executed its antiviral effects partially through the CLK1 pathway.
In this paper, a promising anti-influenza virus candidate, J10688, has shown potential for influenza infection treatment. The anti-influenza virus efficiency and the molecular mechanism of J10688 were initially discussed. The host CLK1 inhibitory effect of J10688 has been proposed to be the main mechanism of its anti-influenza virus activity. Furthermore, J10688 could also elevate the immune organ indexes, enhance the immunological function, and downregulate some main inflammatory factors. All these results implied multiple complex antiviral mechanisms of J10688, which will be further investigated in our future work.
References
Gillman A. Risk of resistant avian influenza A virus in wild waterfowl as a result of environmental release of oseltamivir. Infect Ecol Epidemiol. 2016;6:32870.
Tamura D, DeBiasi RL, Okomo-Adhiambo M, Mishin VP, Campbell AP, Loechelt B, et al. Emergence of multidrug-resistant influenza A(H1N1)pdm09 virus variants in an immunocompromised child treated with oseltamivir and zanamivir. J Infect Dis. 2015;212:1209–13.
Bui C, Bethmont A, Chughtai AA, Gardner L, Sarkar S, Hassan S, et al. A systematic review of the comparative epidemiology of avian and human influenza A H5N1 and H7N9-lessons and unanswered questions. Transbound Emerg Dis. 2016;63:602–20.
Watanabe T, Kawaoka Y. Influenza virus-host interactomes as a basis for antiviral drug development. Curr Opin Virol. 2015;14:71–8.
Aubol BE, Plocinik RM, Keshwani MM, McGlone ML, Hagopian JC, Ghosh G, et al. N-terminus of the protein kinase CLK1 induces SR protein hyperphosphorylation. Biochem J. 2014;462:143–52.
Ninomiya K, Kataoka N, Hagiwara M. Stress-responsive maturation of Clk1/4 pre-mRNAs promotes phosphorylation of SR splicing factor. J Cell Biol. 2011;195:27–40.
Sahebi M, Hanafi MM, van Wijnen AJ, Azizi P, Abiri R, Ashkani S, et al. Towards understanding pre-mRNA splicing mechanisms and the role of SR proteins. Gene. 2016;587:107–19.
An J, Nakajima T, Shibata H, Arimura T, Yasunami M, Kimura A. A novel link of HLA locus to the regulation of immunity and infection: NFKBIL1 regulates alternative splicing of human immune-related genes and influenza virus M gene. J Autoimmun. 2013;47:25–33.
Karlas A, Machuy N, Shin Y, Pleissner KP, Artarini A, Heuer D, et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–22.
Zu M, Li C, Fang JS, Lian WW, Liu AL, Zheng LS, et al. Drug discovery of host CLK1 inhibitors for influenza treatment. Molecules. 2015;20:19735–47.
Kang J, Liu C, Wang H, Li B, Li C, Chen R, et al. Studies on the bioactive flavonoids isolated from Pithecellobium clypearia Benth. Molecules. 2014;19:4479–90.
Li C, Song X, Song J, Pang X, Wang Z, Zhao Y, et al. Pharmacokinetic study of gallocatechin-7-gallate from Pithecellobium clypearia Benth. in rats. Acta Pharm Sin B. 2016;6:64–70.
Liu Z, Yang ZQ, Xiao H. Antiviral activity of the effective monomers from Folium Isatidis against influenza virus in vivo. Virol Sin. 2010;25:445–51.
Hayashi K, Imanishi N, Kashiwayama Y, Kawano A, Terasawa K, Shimada Y, et al. Inhibitory effect of cinnamaldehyde, derived from Cinnamomi cortex, on the growth of influenza A/PR/8 virus in vitro and in vivo. Antivir Res. 2007;74:1–8.
Fischer AH, Jacobson KA, Rose J, Zeller R. Hematoxylin and eosin staining of tissue and cell sections. Cold Spring Harbor Protoc. 2008;2008: pdb.prot4986. https://doi.org/10.1101/pdb.prot4986.
Xu J, Wang J, Deng F, Hu Z, Wang H. Green tea extract and its major component epigallocatechin gallate inhibits hepatitis B virus in vitro. Antivir Res. 2008;78:242–9.
Zu M, Yang F, Zhou W, Liu A, Du G, Zheng L. In vitro anti-influenza virus and anti-inflammatory activities of theaflavin derivatives. Antivir Res. 2012;94:217–24.
Smee DF, Julander JG, Tarbet EB, Gross M, Nguyen J. Treatment of oseltamivir-resistant influenza A (H1N1) virus infections in mice with antiviral agents. Antivir Res. 2012;96:13–20.
Yu J, Shi FS, Hu S. Improved immune responses to a bivalent vaccine of Newcastle disease and avian influenza in chickens by ginseng stem-leaf saponins. Vet Immunol Immunopathol. 2015;167:147–55.
Duncan PI, Stojdl DF, Marius RM, Bell JC. In vivo regulation of alternative pre-mRNA splicing by the Clk1 protein kinase. Mol Cell Biol. 1997;17:5996–6001.
Wang C, Cao B, Liu Q-Q, Zou Z-Q, Liang Z-A, Gu L, et al. Oseltamivir compared with the Chinese traditional therapy Maxingshigan–Yinqiaosan in the treatment of H1N1 influenza: a randomized trial. Ann Intern Med. 2011;155:217–25.
Li Y, Leung K-T, Yao F, Ooi LS, Ooi VE. Antiviral flavans from the leaves of Pithecellobium c lypearia. J Nat Prod. 2006;69:833–5.
Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, et al. Requirement of MIP-1 alpha for an inflammatory response to viral infection. Science. 1995;269:1583–5.
Shih SR, Nemeroff ME, Krug RM. The choice of alternative 5’ splice sites in influenza virus M1 mRNA is regulated by the viral polymerase complex. Proc Natl Acad Sci USA. 1995;92:6324–8.
Shih SR, Krug RM. Novel exploitation of a nuclear function by influenza virus: the cellular SF2/ASF splicing factor controls the amount of the essential viral M2 ion channel protein in infected cells. EMBO J. 1996;15:5415–27.
Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem. 2004;279:24246–54.
Jakubauskiene E, Vilys L, Makino Y, Poellinger L, Kanopka A. Increased serine-arginine (SR) protein phosphorylation changes pre-mRNA splicing in hypoxia. J Biol Chem. 2015;290:18079–89.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81673480), the Beijing Natural Science Foundation (7152103), the CAMS Initiative for Innovative Medicine (CAMS-I2M; 2016-I2M-3-007), the National Great Science and Technology Projects (2012ZX09301002-2013HXW-11, 2014ZX09507003-002), and the International Collaboration Project (2011DFR31240).
Author contributions
The contributions of the respective authors are as follows: A-lL and G-hD designed the research; CL, W-wL, L-jX, X-cP, and HJ performed the research; and CL wrote the paper. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Li, C., Xu, Lj., Lian, Ww. et al. Anti-influenza effect and action mechanisms of the chemical constituent gallocatechin-7-gallate from Pithecellobium clypearia Benth. Acta Pharmacol Sin 39, 1913–1922 (2018). https://doi.org/10.1038/s41401-018-0030-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0030-x
Keywords
This article is cited by
-
Cdc2-like kinases: structure, biological function, and therapeutic targets for diseases
Signal Transduction and Targeted Therapy (2023)
-
Substantial effect of phytochemical constituents against the pandemic disease influenza—a review
Future Journal of Pharmaceutical Sciences (2021)
