
Abstract
Background
Infants born extremely premature are at increased risk for health complications later in life for which neonatal inflammation may be a contributing biological driver. Placental CpG methylation provides mechanistic information regarding the relationship between prenatal epigenetic programming, prematurity, neonatal inflammation, and later-in-life health.
Methods
We contrasted CpG methylation in the placenta and neonatal blood spots in relation to neonatal inflammation in the Extremely Low Gestational Age Newborn (ELGAN) cohort. Neonatal inflammation status was based on the expression of six inflammation-related proteins, assessed as (1) day-one inflammation (DOI) or (2) intermittent or sustained systemic inflammation (ISSI, inflammation on ≥2 days in the first 2 postnatal weeks). Epigenome-wide CpG methylation was assessed in 354 placental samples and 318 neonatal blood samples.
Results
Placental CpG methylation displayed the strongest association with ISSI (48 CpG sites) but was not associated with DOI. This was in contrast to CpG methylation in blood spots, which was associated with DOI (111 CpG sites) and not with ISSI (one CpG site).
Conclusions
Placental CpG methylation was strongly associated with ISSI, a measure of inflammation previously linked to later-in-life cognitive impairment, while day-one neonatal blood methylation was associated with DOI.
Impact
-
Neonatal inflammation increases the risk of adverse later-life outcomes, especially in infants born extremely preterm.
-
CpG methylation in the placenta and neonatal blood spots were evaluated in relation to neonatal inflammation assessed via circulating proteins as either (i) day-one inflammation (DOI) or (ii) intermittent or sustained systemic inflammation (ISSI, inflammation on ≥2 days in the first 2 weeks).
-
Tissue specificity was observed in epigenetic–inflammatory relationships: placental CpG methylation was associated with ISSI, neonatal blood CpG methylation was associated with DOI.
-
Supporting the placental origins of disease framework, placental epigenetic patterns are associated with a propensity for ISSI in neonates.
Similar content being viewed by others


Introduction
Infants born extremely preterm have an increased risk for neonatal complications, including respiratory distress, necrotizing enterocolitis (NEC), sepsis, chronic lung disease, and perinatal brain injury.1,2,3 Later in life, they also have higher rates of neurodevelopmental disorders including cerebral palsy, cognitive impairment, epilepsy, and a variety of psychiatric disorders compared to those born at term.1,4,5 Many of these later-in-life health outcomes have been associated with neonatal inflammation, providing mechanistic insight into the connections between extreme prematurity, neonatal inflammatory processes, and later-life health outcomes.2,6,7,8,9,10,11 In the Extremely Low Gestational Age Newborn (ELGAN) Study, increased levels of neonatal inflammation-related proteins have been tied to attention problems and cognitive impairment later-in-life.6,7,8,11 Furthermore, these associations were found to be significantly stronger for neonates with intermittent or sustained inflammation (ISSI) over the first 2 weeks of life compared to those with inflammation observed on a single day, for example, “day one inflammation” (DOI).6,7,9,11 These findings highlight the importance of distinguishing between ISSI, which reflects either persisting or recurrent inflammation, versus DOI, which likely reflects inflammation initiation prior or very soon after birth, when evaluating the role of epigenetic signatures in prematurity-related developmental outcomes.
Among potential antecedents of neonatal inflammation, placental functioning warrants consideration.12 The placenta is a temporary organ that is critical for fetal growth and survival as it is responsible for providing nutrients, removing waste, and serving as a conduit for neuroendocrine and immunologic signaling from the mother.1,13 The placenta is considered the master regulator of the intrauterine environment, and as a result, it sets the foundation for early and later-in-life health trajectories.1,13 Mounting evidence suggests that placental characteristics are associated with developmental health outcomes, such as childhood body mass index (BMI) and neurodevelopmental impairment.1,14,15,16,17,18,19 It has been suggested that the placenta may be linked to later-in-life outcomes by playing a role in a “first hit” of inflammation, sensitizing the fetus for neonatal inflammation and adverse outcomes.20 The concept of in utero factors influencing disease risk in childhood and adulthood constitutes the developmental origins of health and disease (DOHaD) framework.21 We, and others, hypothesize that the placental epigenome may orchestrate prenatal programming, as posited by DOHaD, either by directly influencing fetal physiology or by altering placental functioning.1,17,21
One form of epigenetic modification is CpG methylation, which can influence the signaling of key biological pathways. CpG methylation suppresses or activates gene expression depending on its location within the genome.22 Placental CpG methylation is posited as a major mechanism through which the placenta dynamically responds to prenatal exposures and transduces their effects onto the growth and development of the fetus.23 For example, placental CpG methylation has been tied to environmental chemical exposures, such as toxic metals and air pollution,24,25,26,27 maternal distress during pregnancy,28 and maternal drug exposure.29 In addition, ELGAN researchers have shown that CpG methylation patterns in the placenta are associated with childhood cognitive impairment, underscoring the long-lasting programming effect of the placenta.14,15,16 While ISSI in the neonate and placental epigenetics have been individually tied to later-in-life cognitive impairment within the ELGAN study, relationships between these two early life processes have not been studied.
In many epigenetic association studies, neonatal blood is evaluated as a measure of early-life CpG methylation.30 For example, neonatal blood methylation, assessed through the collection of dried blood spots, has been linked to childhood BMI and autism.31,32,33,34 However, methylation patterns are known to vary across tissue and time, and thus CpG methylation signatures are likely distinct between blood and placenta.30,35,36,37 Analyzing both placental CpG methylation and neonatal blood spot CpG methylation, each of which represent likely different epigenetic profiles, will provide additional insight into the tissue specificity of CpG methylation signatures as they relate to neonatal inflammation.
In the present study, we contrasted the epigenome-wide CpG methylation patterns in placenta and neonatal blood samples as they related to DOI, a single-day measure of inflammation, and ISSI, a multi-day measure of inflammation over the first 2 postnatal weeks. Inflammation during postnatal days 1–14 was assessed in neonatal blood by measuring six targeted pro-inflammatory proteins previously found within the ELGAN cohort to be associated with later-life cognitive impairment.6,7,8 Based on existing literature, we hypothesized that we would observe tissue-specific associations of placental and neonatal blood CpG methylation with neonatal inflammation and that these associations would vary in relation to DOI versus ISSI.
Methods
The ELGAN cohort
The ELGAN study is a prospective cohort study designed to examine the risk of structural and functional brain disorders in extremely preterm children (birth at <28 weeks of gestation).38 Between 2002 and 2004, women delivering before 28 weeks of gestation were asked to enroll in the study from 14 participating institutions across 5 states (North Carolina, Massachusetts, Michigan, Illinois, Connecticut). Gestational age was estimated using fetal ultrasound before the 14th gestational week for most participants (62%), followed by later fetal ultrasound, self-reported last menstrual period, and neonatal intensive care unit records. At delivery, demographic and clinical information was gathered using structured data collection forms. The study was approved by the Institutional Review Board at each participating ELGAN site.
A summary of the study population selection is provided in Fig. S1. Overall, 1249 mothers of 1506 infants were enrolled. Of these infants, 996 children had neonatal blood samples for measurement of inflammation-related proteins and survived to age 10 years. At age 10 years, out of these 996 children, 889 (92%) of them were reached for follow up. Consequently, the relationship between neonatal inflammatory proteins and later-life neurological impairment has been studied in this sample population.8,39 Given the established relationships in this population, these 889 participants were the starting cohort for our analysis.
Of the 889, 415 participants had placenta biospecimens analyzed for CpG methylation (detailed further below). All 415 participants with placental CpG methylation characterized had available data for circulating inflammation-related proteins in neonatal blood. Of the 415 subjects with placental specimens available, 390 also had neonatal blood available for CpG methylation analysis. Of the 415 participants in the placental methylation group, 411 passed quality assessment and quality control (QA/QC) steps for the methylation data (described in detail below). Of the 390 participants in the neonatal blood methylation group, 368 passed QA/QC steps.
The ELGAN cohort has a large proportion of multiple pregnancies, given the higher risk of preterm birth in multiple pregnancies. Since the presence of multiple infants from the same mother within the cohort violates the independence of observations, an assumption required for later statistical modeling, all singletons were selected for inclusion and, among multiples, one infant was randomly selected to be included. This resulted in the final sample for the placental methylation group being 354 and the final sample for neonatal blood methylation group being 318.
Neonatal blood collection
Peripheral blood spots were collected at multiple time points in the first 2 weeks of participating infants’ lives: day-1 (range, 1–3 days), day-7 (range, 5–8 days), and day-14 (range, 12–15 days). Blood spots were collected on filter paper (Schleicher & Schuell 903, GE Healthcare, Chicago, IL) and stored at −70 °C in sealed bags with desiccant until processing.
Day-1, day-7, and day-14 blood samples were assessed for circulating inflammation-related proteins (described in further detail below). The proteins were measured in the Laboratory of Genital Tract Biology, Brigham and Women’s Hospital. Because the volumes of blood on the filter paper varied, each fixed spot area was eluted in the same volume of elution buffer and the concentration of each protein was normalized to total protein concentration. All samples were quantified twice and the mean was used for subsequent analyses. Detailed description of protocols for protein elution, procedures for measuring concentrations, as well as absolute values and ranges of proteins quantified have been described elsewhere.8,39,40,41,42
Day-1 neonatal blood spots were further assessed for CpG methylation. To extract DNA from the dried blood spots, 3 mm diameter spots were punched from the filter paper, lysed in Proteinase K solution from the EZ1 DNA Investigator kit (Qiagen, Germantown, MD), and shaken in a thermomixer. The resulting supernatant was processed using the Qiagen EZ1 Advanced instrument according to the manufacturer’s protocol. The quantity of DNA was assessed using a DropSense 96 Spectrophotometer (Trinean, Pleasanton, CA), with a minimum of 20 ng DNA considered acceptable for methylation analysis.
Placental tissue collection
Placental tissue collection within the ELGAN cohort has been described in detail elsewhere.29,43,44 In summary, at delivery, placentas were collected, placed in a sterile basin, and biopsied. During biopsy, the amnion was pulled back to expose underlying chorion and a sample (<1 g) of tissue was extracted from the base of the chorion by applying traction to the chorion and underlying trophoblast tissue. Samples were placed into a sterile 2 mL cryovial, submerged in liquid nitrogen, and stored in a −80 °C freezer. To process the placenta samples for DNA extraction, banked samples were kept on dry ice and sectioned into approximately 0.025 g segments with a sterile disposable dermal curette, washed in sterile 1× phosphate-buffered saline (PBS) (Fisher Scientific, Waltham, MA) to reduce any potential blood contamination, and immediately refrozen in homogenization tubes. Tissue segments were homogenized with a sterile stainless-steel bead (Qiagen) in RLT Plus lysis buffer (Qiagen) using the TissueLyserII instrument (Qiagen). Homogenized samples were centrifuged to pellet cellular debris and the bead and clarified lysates were transferred to a new microfuge tube and stored at −80 °C until nucleic acid extraction. The ALLPrep DNA/RNA/miRNA Universal Kit (Qiagen) was used to isolate DNA. A total of 250–500 ng of input DNA was bisulfite treated using the Zymo EZ DNA Methylation Kit (Zymo Research, Irvine, CA).
Placental and neonatal blood methylation quantification
For both neonatal blood and placenta, genome-wide DNA methylation was profiled using Infinium MethylationEPIC BeadChips (Illumina, San Diego, CA). This platform simultaneously measures DNA methylation at over 850,000 sites at single-nucleotide resolution. Placental and neonatal blood DNA samples were analyzed separately as both tissues were not available at the same time. Within tissue type, samples were randomly allocated to different plates and chips to minimize confounding. Raw methylation image files were processed, including annotation to genes and genomic location, using the minfi (v1.26.0) package in R.45
Methylation data processing and QA/QC
For both the placental and neonatal blood methylation data, β values (the ratio of methylated to unmethylated signal intensities) were calculated for each locus. To generate final β values, sex mismatches, failed probes, and outliers were removed, the data were normalized, and batch effects were assessed. Sex mismatches were identified as samples in which the male/female sex empirically estimated from the methylation data differed from that recorded in the demographic data. Estimation of sex was based on the median values of measurements on the X and Y chromosomes, respectively. Failed probes were identified using detection p values comparing the total DNA signal (methylated + unmethylated) for each position to the background signal level, estimated using negative control positions. A probe was considered a failed probe if the detection p value was >0.01. Two threshold filters were employed based on failed probes. First, a probe-based filter was applied in which probes that failed for >10% of samples were removed. Second, a sample-based filter was employed in which samples with >5% failed probes were removed from the placenta data and samples with >15% failed probes were removed from the dried blood spot data. Different stringencies were applied based on balancing maximal data quality of the included data with maximal data point inclusion. Sex mismatches and failed probes were identified using the minfi (v1.26.0) package in R.45 For the placental methylation data, these steps removed 4 samples for sex mismatches and removed zero samples based on the sample-based filter, resulting in a total 411 samples. For the dried blood spot methylation data, these steps removed 18 samples for sex mismatches and 4 samples based on the sample filter, resulting in a total 368 samples. Additionally, 1597 (of 850,000) probes from the placental methylation data and 79,458 (of 850,000) probes from the dried blood spot methylation data were removed through the probe-based filter. The ShinyMethyl (v.1.26.0) package in R was used simultaneously to the above steps to visually verify and check for outliers.46
The data were then normalized utilizing normal-exponential out-of-band (noob) correction method and functional normalization within the minfi package. This normalization procedure is a between-array normalization method, initially designed for the Illumina Infinium HumanMethylation450 platform, although validated to work for the HumanMethylationEPIC platform.47 Batch effects were identified using principal components analysis in combination with visualizing data using plate position, chip, array, and date covariates. Plate position was determined to potentially be inducing batch effects in the placenta methylation data and plate number was determined to potentially be inducing batch effects in the dried blood spot methylation data. Potential batch effects were removed utilizing the ComBat function within the sva (v3.38.0) R package.48,49 Following batch effect correction, final β values were generated and then converted to M values ([log2(β/(1 − β)]), as these are more statistically valid for subsequent differential expression analysis.50
Cell-type estimation
As different cell types have variation in DNA methylation levels, this may confound associations. To control for this effect, cell-type proportions were empirically estimated in both the placental and neonatal blood methylation data. Cell-type estimation for neonatal blood samples was conducted utilizing the flowsorted.blood.epic R package (v1.8.0) for neonatal blood data utilizing the cord blood-derived reference library.51,52 Cell-type estimation for the placental data was conducted utilizing the planet R package (v0.99.4),53 calculating an average of the estimation from the first and third trimester placenta reference library. We averaged between first and third trimester libraries in order to best capture the cell-type variation present in the second trimester placenta samples used in this study (samples ranged from 161 to 191 gestational days or 23–28 weeks).
Definition of DOI and ISSI
In the ELGAN study, 27 inflammatory and neurotrophic proteins were previously selected for measurement based on their roles as cytokines, cytokine receptors, chemokines, adhesion molecules, metalloproteinases, other markers of inflammation, or as potential endogenous protectors against inflammation-related injury.54,55 Of these 27, 6 inflammation-related proteins have been associated most consistently with neurologic outcomes in previous ELGAN studies and were thus the focus of this study.6,8,39,56,57 These six proteins are: interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), intercellular adhesion molecule-1 (ICAM-1), interleukin-8 (IL-8), serum amyloid A (SAA), and C-reactive protein (CRP). As protein levels varied with gestational age and postnatal day of collection, protein levels were normalized within groupings of gestational age category (23–24, 25–26, and 27 weeks) and day of blood collection (1, 7, and 14).42
To generate variables that captured the data on protein levels well but reduced the dimensionality of the multiple days of measurements for six different inflammation-related proteins, we followed definitions used in previous studies by ELGAN investigators. Specifically, we used: (1) DOI and (2) ISSI.6,8,39,56,57 These were generated in the following steps. First, for each protein on each day, a dichotomous variable was generated whereby a subject was assigned as having a high level of a protein if the level was at or above the 75th percentile for a particular gestational age and day of sampling; otherwise, samples were considered to have a low-to-normal level. Participants were classified as having DOI if they had a high level of ≥3 of the 6 proteins (listed above) in the day-1 neonatal blood sample. Participants were classified as having ISSI if they had a high level of ≥3 of the 6 proteins in at least 2 of the blood samples collected during the first 2 postnatal weeks (on days 1, 7, and 14). Thus, ISSI can represent inflammation profiles in which inflammation is sustained or there are multiple intermittent episodes.9 The categories of DOI and ISSI were not mutually exclusive, whereby infants with DOI could go on to also have ISSI, or not.
Statistical analysis
For the assessment of both placental and neonatal blood CpG methylation, two epigenome-wide association studies (EWAS) models were fit: (1) evaluating the association between CpG methylation and ISSI, and (2) evaluating the association between CpG methylation and DOI. For each of these, separate robust linear regressions were fit to estimate the relationships between inflammation status and M values at each CpG loci. Based on a direct acyclic graph analysis, the following confounders were selected a priori: fetal sex (male/female), maternal smoking during pregnancy (yes/no), pre-pregnancy BMI (<18.5, 18.5–<30, 30+), and socioeconomic status (Fig. S2). Dagitty (v3.0) was used to define the minimally sufficient set of covariates.58 Socioeconomic status (SES) was operationalized as a summative count/"score" of: less than college education, single marital status, eligibility for the Supplemental Nutrition Assistance Program (SNAP), and public health insurance, as has been previously associated with placental DNA methylation by our team.59 For the selected covariates, 7 (2%) subjects were missing data on maternal smoking, 13 (4%) subjects were missing data for maternal pre-pregnancy BMI and 5 (1%) subjects were missing data on the cumulative SES score variable. When missing, these covariates were imputed utilizing random forest modeling within the missForest R package (v1.4).60 In addition to the above listed confounders, the first two eigenvectors based on principal components analysis of the cell-type proportions were included as covariates in each model in order to account for methylation variance due to different cell-type proportions. To control for multiple testing, p values were adjusted utilizing the false discovery rate and Benjamini–Hochberg (B-H)-adjusted p values/q-values <0.1 were considered statistically significant.61 This cut-off was chosen in order to balance Type I/Type II error trade-offs and to maximize the identification of all positive hits, given the novel nature of this research question. In all presentations of EWAS model results, we also detail if the CpG site is significant at q-value < 0.05, a stricter cut-off, or p value < 9 × 10−8, an even more stringent cut-off proposed by Mansell et al.62 Q–Q plots for all models are shown in Fig. S3. As a comparison, EWAS models utilizing the same covariates were fit to day-7 only inflammation as well.
Pathway enrichment analysis
To further understand the biological implications of the inflammation associated CpG sites, the loci were annotated to their respective genes using the MethylationEPIC beadchip annotation file provided by Illumina and accessible online.63 All genes that were annotated to an inflammation-associated CpG site (B-H-adjusted p < 0.1) were used as input data, with the results from each model being analyzed separately. Canonical pathway and network enrichment analyses were then carried out based on the annotated genes utilizing the Ingenuity Knowledge Database (Ingenuity Pathway Analysis, Qiagen, Redwood City). Over-represented canonical pathways were defined as those containing more differentially methylated genes than expected by random chance, as based on q-values calculated from a right-tailed Fisher’s Exact Test (significance defined at <0.05).61 Networks were constructed based on known protein–protein interactions and other molecular interactions and were ranked based on right-tailed Fisher’s Exact test p values, indicating the likelihood of observing a network containing at least the same number of proteins encoded by differentially methylated genes by chance, in comparison to random selections of other genes within the genome.
Overlap and correlation between inflammation-associated methylation in placental and neonatal blood samples
To evaluate persistence of inflammation-associated methylation signatures, we examined whether any CpG sites were associated with either ISSI or DOI in both tissues. We additionally calculated Pearson correlation coefficients and associated p values for methylation β values between placental and neonatal blood for ISSI- or DOI-associated CpGs in either tissue. Significant correlations were considered as p < 0.05.
Results
Study subject characteristics
General characteristics of the study participants are described in Table 1. A total of 354 subjects were included in the analysis of placental DNA methylation, and of these, 318 (87.9%) were also included in the analysis of neonatal blood methylation. Distributions of key maternal demographic characteristics and inflammation-related protein levels did not vary substantially between the placenta sample and the neonatal blood subset. Most mothers did not smoke during pregnancy (307, 88.5%) and most either self-identified their race as non-Hispanic white (209, 59.7%) or Black (105, 30%). Most mothers were between the ages of 21 and 35 years and had at least 12 years of education. Just over half of mothers were married and just over a third received public health insurance (i.e., Medicaid). Of the placenta samples, 181 (51.1%) were from male offspring, and of the neonatal blood samples, 172 (54.1%) were from male offspring. When comparing the analytical cohort used in this study (n = 354, or the n = 318 neonatal blood subset) to the ELGAN parent cohort (n = 889), there were no notable differences in distributions of key covariates or the inflammation measures, other than for marital status. The analytical cohort used in this study had a lower percentage of married women represented than the parent study, 55.1% versus 78.3%, respectively.
Placental CpG methylation is associated with ISSI
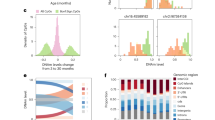
No CpG sites in the placenta were associated with DOI. However, 48 CpG sites were differentially methylated in relation to ISSI (Table 2, Manhattan plot: Fig. 1, and Volcano plot: Fig. S4). Of these 48 CpG sites, 32 were annotated to genes. The top three most significantly differentially methylated CpG sites were annotated to Derlin 1 (DERL1), Aspartyl Aminopeptidase (DNPEP), and ATP/GTP binding carboxypeptidase 1 (AGTPBP1), respectively. The associated CpG sites for these genes had varied genomic locations relative to the gene including being located in the 5′ untranslated region (UTR) between the transcription start site (TSS) and the ATG start site (5′UTR) (DERL1, DNPEP), first exon (LINC01107), between the stop codon and poly A signal (3′UTR) (LINC01107), 0–200 bases upstream of the TSS (TSS200) (DNPEP), and 200–1500 bases upstream of the TSS (TSS1500) (DNPEP). Table 2 details the top 25 most significant CpG sites, and all significant CpG sites are detailed in Table S1. Of these 48 CpG sites, the majority (40, 83.3%) demonstrated increased methylation in relation to higher ISSI (BetaDiff > 0). No CpG sites that were differentially methylated in relation to ISSI were located within genes encoding for the inflammation-related proteins measured.

a Placental CpG sites associated with day-one inflammation (DOI); b Placental CpG sites associated with intermittent or sustained inflammation (ISSI). The red horizontal line indicates q < 0.05 cut-off and the blue horizontal line indicates the q < 0.1 cut-off. There is no red line in plot (a) as no CpGs reached close to this significance level.
The top upstream regulators for these 32 annotated genes included A-Kinase Anchoring Protein 13 (AKAP13) (p = 1.51 × 10−03), MAGE family member 11 (MAGEA11) (p = 3.02 × 10−03), and Autocrine Motility Factor Receptor (AMFR) (p = 6.03 × 10−03). Furthermore, a significant biological network among these genes was identified with Tumor Protein 53 (TP53) as a central node, as well as Transforming Growth Factor-Beta (TGF-β) Receptor Type 2 (TGFBR2), and Epidermal Growth Factor Receptor (EGFR) as other nodes, and cellular development and proliferation as network-associated functions (Fig. 2 and Table S5). Notably, cancer and neurological disease were among top diseases and biological functions identified as enriched among these genes (p = 4.88 × 10−2 and p = 4.63 × 10−2, respectively). Top enriched canonical pathways included Calcium Transport I and Antigen Presentation Pathway. A summary of all pathway analysis findings can be found in Table S5.

Significantly enriched biological network among differentially methylated genes in the placenta in relation to ISSI.
Neonatal blood spot CpG methylation is associated with DOI rather than ISSI
A total of 111 CpG sites (annotated to 77 genes) were differentially methylated in neonatal blood in relation to DOI (Table 3, Manhattan plot: Fig. 3, and Volcano plot: Fig. S4). The most significantly differentially methylated CpG sites were annotated to the following genes: Transmembrane Protein 260 (TMEM260), Monofunctional C1-Tetrahydrofolate Synthase (MTHFD1L), and Myeloid Cell Nuclear Differential Antigen (MNDA). For each of these genes, the CpG site that was differentially methylated was located either 200–1500 bases upstream of the TSS (labeled as TSS1500) (MNDA) or within the 5′UTR (TMEM260, MTHFD1L). Table 3 details the top 25 most significant of these CpG sites, and all the significant CpG sites are detailed in Table S2. Of the 111 CpG sites, 39 (35.1%) demonstrated increased methylation levels in relation to higher DOI levels (BetaDiff >0). No inflammation-associated CpG sites were annotated to genes corresponding to the inflammation-related proteins used to define DOI. The 77 annotated genes were evaluated for enriched biological pathways and networks. Krueppel-like Factor 5 (KLF5) (p = 3.18 × 10−03), Immunoglobulin Heavy Constant Gamma 3—Immunoglobin Heavy Constant Gamma 1 (IGHG3–IGHG1) Readthrough (p = 3.37 × 10−03), and Signal Transducer and Activator of Transcription 1 and 3 (Stat 1/3) (p = 3.37 × 10−03) were identified as upstream regulators (Table S5). Top canonical pathways included Histidine Degradation III (2.87 × 10−04), Integrin Signaling (5.38 × 10−04), and Phagosome Formation (1.06 × 10−03) (Table S5). Only 1 CpG site was associated with ISSI in neonatal blood (Table 3 and Fig. 3) and this CpG site was not annotated to a gene.

a Day-1one neonatal blood spot CpG sites associated with day-one inflammation (DOI); b Day-one neonatal blood spot CpG sites associated with intermittent or sustained inflammation (ISSI). The red horizontal line indicates q < 0.05 cut-off and the blue horizontal line indicates the q < 0.1 cut-off.
Overlap in differentially methylated CpG sites across models
There was no overlap in the annotated genes between the DOI- and ISSI-associated CpG sites in either placenta or neonatal blood samples. Furthermore, the majority of DOI- or ISSI-associated CpG sites did not exhibit significant correlation between placental and neonatal blood β values (Table S6). Note that for the 48 CpG sites associated with ISSI in placental tissue, 23 were removed in the neonatal blood methylation QA/QC, therefore 25 were assessed for correlation between tissues. Of the 25, 4 (17.3%) were significantly correlated between placental and neonatal blood tissues, with 3 being positively correlated. The single CpG site associated with ISSI in neonatal blood was significantly positively correlated across both tissues. Of the 111 DOI-associated CpGs in neonatal blood, 24 (21.6%) were significantly correlated between tissues, 18 of which were positive correlations.
We also evaluated for an association between placenta and neonatal blood methylation with day-7 inflammation. A total of 4 CpG sites were differentially methylated in neonatal blood and 287 in placenta in relation to day-7 inflammation. Complete results of the EWAS models for day-7 inflammation can be found in Table S3 for neonatal blood and Table S4 for placenta. Interestingly, there was also minimal overlap in the day-7 findings with both the DOI and ISSI results. Specifically, only two genes, namely, Transcription Elongation Regulator 1 (TCERG1) and Synaptotagmin Like 3 (SYTL3), were differentially methylated in the placenta in relation to both ISSI and day-7 inflammation. For TCERG1, this corresponded to the same CpG site, cg23554212, whereas for SYTL3 this corresponded to two different CpG sites. Two other genes, Long Intergenic Non-Protein Coding RNA 299 (LINC00299) and Translocation Associated Membrane Protein 2 (TRAM2), contain CpG sites that were differentially methylated in neonatal blood in relation to DOI and in placenta in relation to day-7 inflammation. However, the specific CpG sites within the genes differentially methylated were not the same. Thus, striking tissue specificity was observed in the relationship between CpG methylation, DOI, and ISSI (Fig. 4).

A schematic demonstrating the tissue-specific associations among DNA methylation in the placenta, DNA methylation in neonatal blood spots, DOI, and ISSI.
Discussion
Neonatal inflammation, assessed through elevated levels of circulating inflammatory proteins early in life, has been associated with increased risk of neurodevelopmental disabilities later-in-life among those born extremely preterm.2,6,8,39,40,64,65,66 These later-in-life outcomes have also been tied to altered placental epigenetic modifications, suggesting a prenatal programming effect.1,14,15,16 However, the underlying mechanisms that connect extreme prematurity, placental epigenetic regulation, and neonatal inflammation remain understudied. In the present study, we sought to compare and contrast the associations between CpG methylation signatures in the placenta and neonatal blood as they relate to DOI and ISSI, based on circulatory pro-inflammatory proteins previously tied to cognitive impairment in ELGANs.6,7,8 Our results show striking tissue specificity in the relationship between CpG methylation and neonatal inflammation. Specifically, placental CpG methylation was primarily associated with ISSI while neonatal blood CpG methylation was primarily associated with DOI.
In the placenta, methylation levels of 48 CpG sites were associated with intermittent or sustained neonatal inflammation, or ISSI. These 48 CpG sites mapped to 32 genes that, when analyzed for enriched biological pathways, displayed a network with nodes including TP53, TGFBR2, and EGFR, each of which is known for their roles in cellular proliferation, including tumor development/suppression and signaling. The most significantly differentially methylated gene was DERL1, which contained a hypermethylated CpG site in the 5′UTR in relation to ISSI. Hypermethylation of sites in the 5′UTR has been previously shown to have a significant negative correlation with gene expression in cord blood, underscoring this methylation mark’s potential gene-silencing effect.67 Increased DERL1 expression is linked with cellular proliferation, thus an epigenetic downregulation in DERL1 may have implications for proper cellular proliferation.68,69,70 The finding that epigenetic regulation of placental genes involved in cellular proliferation is tied to neonatal inflammation is notable, as cellular proliferation is critical to proper placental development and formation.71 This reinforces the links between placental dysfunction and inflammation, the interplay of which has been linked to preterm birth pathogenesis, including within the ELGAN study.72,73 These findings further suggest that disordered placentation and/or placental dysfunction, tied to preterm birth, may also be involved in transducing a propensity for continued inflammation beyond birth to the neonate.
The finding that placental CpG methylation is associated with neonatal ISSI, which is known to increase risk for several neurodevelopmental disorders, gives further weight to the prenatal programming, or DOHaD, hypothesis.6,7,8,56,65 Genes in the ISSI-associated enriched biological network, specifically TP53 and the TGF-β family, have each been tied to regulation of neuronal differentiation, development, and function.74,75 In particular, the TGF-β family is involved in the early formation of the nervous system in embryogenesis and also plays a critical role in modulating inflammatory responses.74 Furthermore, calcium signaling was identified as an enriched canonical pathway among differentially methylated genes in relation to ISSI. Interestingly, dysregulated calcium signaling has been tied to altered neuronal signaling and subsequent neurodegeneration.76 Antigen presentation pathway—the vital process through which antigens are presented to immune cells to initiate adaptive immune system responses—was another enriched canonical pathway, suggesting potential epigenetic dysregulation of a critical function of immunity. In turn, prenatal immune activation has been shown to alter fetal brain development.77,78,79,80 Lastly, in addition to enriched pathways, specific differentially methylated genes have been linked to neurocognitive outcomes, including AGTPBP1, which has been found to be involved in neuropathology and behavioral deficit in animal models and human studies, and DNPEP, methylation of which has been linked to autism spectrum disorder.81,82 These pathways and genes provide potential biological links between placental epigenetic alterations associated with ISSI and neurodevelopment that warrants further research.
By showing that placental CpG methylation is linked to neonatal ISSI, the present study also lays the foundation for further investigation into the mediating role of ISSI in transducing prenatal insults to later-in-life neurodevelopmental outcomes. Our team has previously established that placental epigenetic variation is associated with later-in-life outcomes. Specifically, we have shown that methylation levels of critical hypothalamic–pituitary–adrenal (HPA) axis genes in the placenta are predictive of cognitive function at age 10 years.15 We also recently identified a placental multi-omics signature predictive of intellectual and social impairment at age 10 years.14 Interestingly, when we compare the 32 genes with differentially methylated sites in relation to ISSI in this study, we did not find any overlap with the genes reported in these previous two studies. This suggests that there are multiple pathways through which placental epigenetics influences neurodevelopment. For example, Meakin et al. focused on methylation of genes within a critical endocrine pathway (HPA axis), which may represent a pathway that can respond to initiators (e.g., psychological stress) other than inflammation. The current study provided initial evidence for a pathway connecting placental methylation and later-in-life cognitive outcomes via the mediating role of ISSI, a hypothesis that requires further validation. Taking previous studies and this study together highlights the multidimensional nature of the relationship between the placenta and the neurodevelopmental outcomes.
In contrast to the findings in placenta, neonatal blood spot methylation was not associated with ISSI, but rather with DOI. Specifically, methylation of 111 CpG sites (77 genes) in neonatal blood was associated with DOI. In contrast to ISSI, DOI in the ELGAN cohort has not been associated strongly with later-in-life cognitive outcomes.6 These data suggest that the CpG methylation patterns in neonatal blood may reflect a short-term inflammatory response to the extra-uterine environment rather than a longer-term programming regulating ISSI. Supporting this, enriched canonical pathways among DOI-associated genes included Phagosome Formation, a cellular response to infection, and Integrin Signaling, which is tightly linked to inflammation and infection as well as etiology of neonatal outcomes such as NEC and retinopathy of prematurity.83,84 It is worth noting that DOI and blood spot methylation were both derived from the same biological media on the same day, thus the strong association may be linked to common method variance.
Contrasting the CpG methylation patterns between the placenta and neonatal blood highlighted striking tissue specificity, with the former linked to ISSI and the latter linked to DOI. Furthermore, the differentially methylated genes associated with DOI and ISSI displayed no overlap. Tissue-specific methylation is not necessarily surprising, as CpG methylation is a mechanism of regulating gene expression that results in tissue-specific structure and function.35 However, the lack of any overlap of differentially methylated CpGs between the placenta and neonatal blood was unexpected. This suggests that there may be numerous epigenetically regulated genes that lead to inflammation but that these pathways are differentially regulated based on timing and type of inflammation (DOI versus ISSI) and tissue (neonatal blood versus placenta). Moreover, these results highlight that a single tissue cannot provide a complete epigenetic profile affecting the neonate, particularly as related to inflammation.
This study is among the first to compare and contrast tissue-specific CpG methylation in placenta and blood in relation to neonatal inflammation. However, this study is not without limitations. First, the ELGAN cohort infants are extremely premature, rendering the results of this study less generalizable to infants born at later gestational ages. Additionally, protein-based assessment of neonatal inflammation was only considered for the first 2 weeks of life, potentially not accounting for the role of persistence into later life. Lastly, evaluating the connection between inflammation and other placental -omics signatures (proteomics, transcriptomics) was outside the scope of this study. The integration of these various datasets would be an interesting line of further inquiry. Given that many of the CpGs identified in this study were located in extragenic regions such 5′UTR, 3′UTR, and TSS, and that biologically the location of the methylation mark is associated with differential transcriptional activation, it would also be a worthy line of inquiry to evaluate the relationships between different genomic location of inflammation-associated CpGs and functional effects on gene and protein expression.67 In addition, it would be interesting to evaluate differentially methylated regions and/or epigenetic age in relation to early-life inflammation as well.
In summary, the results from the present study demonstrate the novel finding that placental CpG methylation is a better predictor of ISSI than neonatal day-one blood spot methylation. These results are particularly compelling given the strong body of literature demonstrating that neonatal ISSI conveys excess risk later in life for adverse neurodevelopmental outcomes.2,6,8,39,40,64,65,66 These findings highlight the importance of considering the role of placental CpG methylation in studies assessing the role of early-life inflammation in DOHaD-related outcomes.
References
Bangma, J. T., Hartwell, H., Santos, H. P., O’Shea, T. M. & Fry, R. C. Placental programming, perinatal inflammation, and neurodevelopment impairment among those born extremely preterm. Pediatr. Res 89, 326–335 (2021).
Leviton, A. et al. Systemic inflammation on postnatal days 21 and 28 and indicators of brain dysfunction 2years later among children born before the 28th week of gestation. Early Hum. Dev. 93, 25–32 (2016).
Stoll, B. J. et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics 126, 443–456 (2010).
Kuban, K. C. K. et al. Girls and boys born before 28 weeks gestation: risks of cognitive, behavioral, and neurologic outcomes at age 10 years. J. Pediatr. 173, 69.e1–75.e1 (2016).
Dvir, Y. et al. Psychiatric symptoms: prevalence, co-occurrence, and functioning among extremely low gestational age newborns at age 10 years. J. Dev. Behav. Pediatr. 40, 725–734 (2019).
O’Shea, T. M. et al. Elevated concentrations of inflammation-related proteins in postnatal blood predict severe developmental delay at 2 years of age in extremely preterm infants. J. Pediatr. 160, 395–401.e394 (2012).
O’Shea, T. M. et al. Inflammation-initiating illnesses, inflammation-related proteins, and cognitive impairment in extremely preterm infants. Brain Behav. Immun. 29, 104–112 (2013).
Kuban, K. C. K. et al. Circulating inflammatory-associated proteins in the first month of life and cognitive impairment at age 10 years in children born extremely preterm. J. Pediatr. 180, 116.e1–123.e1 (2017).
Dammann, O. & Leviton, A. Intermittent or sustained systemic inflammation and the preterm brain. Pediatr. Res. 75, 376–380 (2014).
Hansen-Pupp, I. et al. Inflammation at birth is associated with subnormal development in very preterm infants. Pediatr. Res. 64, 183–188 (2008).
O’Shea, T. M. et al. Elevated blood levels of inflammation-related proteins are associated with an attention problem at age 24 mo in extremely preterm infants. Pediatr. Res. 75, 781–787 (2014).
Leviton, A. et al. Antecedents of inflammation biomarkers in preterm newborns on days 21 and 28. Acta Paediatr. 105, 274–280 (2016).
Gude, N. M., Roberts, C. T., Kalionis, B. & King, R. G. Growth and function of the normal human placenta. Thrombosis Res. 114, 397–407 (2004).
Santos, H. P. Jr. et al. Evidence for the placenta-brain axis: multi-omic kernel aggregation predicts intellectual and social impairment in children born extremely preterm. Mol. Autism 11, 97–97 (2020).
Meakin, C. J. et al. Placental CpG methylation of HPA-axis genes is associated with cognitive impairment at age 10 among children born extremely preterm. Horm. Behav. 101, 29–35 (2018).
Tilley, S. K. et al. Placental CpG methylation of infants born extremely preterm predicts cognitive impairment later in life. PLoS ONE 13, e0193271–e0193271 (2018).
Longtine, M. S. & Nelson, D. M. Placental dysfunction and fetal programming: the importance of placental size, shape, histopathology, and molecular composition. Semin. Reprod. Med. 29, 187–196 (2011).
Tomlinson, M. S. et al. Neurocognitive and social-communicative function of children born very preterm at 10 years of age: associations with microorganisms recovered from the placenta parenchyma. J. Perinatol. 40, 306–315 (2020).
Clark, J. et al. Associations between placental CpG methylation of metastable epialleles and childhood body mass index across ages one, two and ten in the Extremely Low Gestational Age Newborns (Elgan) Cohort. Epigenetics 14, 1102–1111 (2019).
Yanni, D. et al. Both antenatal and postnatal inflammation contribute information about the risk of brain damage in extremely preterm newborns. Pediatr. Res. 82, 691–696 (2017).
Gluckman, P. D., Hanson, M. A. & Buklijas, T. A conceptual framework for the developmental origins of health and disease. J. Dev. Orig. Health Dis. 1, 6–18 (2010).
Moore, L. D., Le, T. & Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 38, 23–38 (2013).
Maccani, M. A. & Marsit, C. J. Epigenetics in the placenta. Am. J. Reprod. Immunol. 62, 78–89 (2009).
Meakin, C. J., Martin, E. M., Szilagyi, J. T., Nylander-French, L. A. & Fry, R. C. Inorganic arsenic as an endocrine disruptor: modulation of the glucocorticoid receptor pathway in placental cells via CpG methylation. Chem. Res. Toxicol. 32, 493–499 (2019).
Schmidt, R. J. et al. Self-reported pregnancy exposures and placental DNA methylation in the marbles prospective autism sibling study. Environ. Epigenet. 2, dvw024 (2016).
Martin, E. M. & Fry, R. C. A cross-study analysis of prenatal exposures to environmental contaminants and the epigenome: support for stress-responsive transcription factor occupancy as a mediator of gene-specific CpG methylation patterning. Environ. Epigenet. 2, dvv011 (2016).
Abraham, E. et al. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ. Int. 118, 334–347 (2018).
Monk, C. et al. Distress during pregnancy: epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am. J. Psychiatry 173, 705–713 (2016).
Addo, K. A. et al. Acetaminophen use during pregnancy and DNA methylation in the placenta of the Extremely Low Gestational Age Newborn (Elgan) Cohort. Environ. Epigenet. 5, dvz010 (2019).
Herzog, E. M. et al. The tissue-specific aspect of genome-wide DNA methylation in newborn and placental tissues: implications for epigenetic epidemiologic studies. J. Dev. Orig. Health Dis. 12, 113–123 (2021).
Hannon, E. et al. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 10, 19–19 (2018).
Kochmanski, J., Goodrich, J. M., Peterson, K. E., Lumeng, J. C. & Dolinoy, D. C. Neonatal bloodspot DNA methylation patterns are associated with childhood weight status in the Healthy Families Project. Pediatr. Res. 85, 848–855 (2019).
van Dijk, S. J. et al. DNA methylation in blood from neonatal screening cards and the association with BMI and insulin sensitivity in early childhood. Int. J. Obes. 42, 28–35 (2018).
Hannon, E. et al. Variable DNA methylation in neonates mediates the association between prenatal smoking and birth weight. Philos. Trans. R. Soc. B 374, 20180120 (2019).
Lokk, K. et al. DNA methylome profiling of human tissues identifies global and tissue-specific methylation patterns. Genome Biol. 15, 3248 (2014).
Schroeder, D. I. et al. The human placenta methylome. Proc. Natl Acad. Sci. USA 110, 6037–6042 (2013).
Chu, T. et al. Structural and regulatory characterization of the placental epigenome at its maternal interface. PLoS ONE 6, e14723 (2011).
O’shea, T. et al. The ELGAN study of the brain and related disorders in extremely low gestational age newborns. Early Hum. Dev. 85, 719–725 (2009).
Kuban, K. C. K. et al. Association of circulating proinflammatory and anti-inflammatory protein biomarkers in extremely preterm born children with subsequent brain magnetic resonance imaging volumes and cognitive function at age 10 years. J. Pediatr. 210, 81.e83–90.e83 (2019).
Allred, E. N. et al. Systemic inflammation during the first postnatal month and the risk of attention deficit hyperactivity disorder characteristics among 10 year-old children born extremely preterm. J. Neuroimmune Pharmacol. 12, 531–543 (2017).
Fichorova, R. N. et al. Systemic inflammation in the extremely low gestational age newborn following maternal genitourinary infections. Am. J. Reprod. Immunol. 73, 162–174 (2015).
Leviton, A., Allred, E. N., Yamamoto, H., Fichorova, R. N. & ELGAN Study Investigators. Relationships among the concentrations of 25 inflammation-associated proteins during the first postnatal weeks in the blood of infants born before the 28th week of gestation. Cytokine 57, 182–190 (2012).
Martin, E. et al. Sexual epigenetic dimorphism in the human placenta: implications for susceptibility during the prenatal period. Epigenomics 9, 267–278 (2017).
Onderdonk, A. B. et al. Detection of bacteria in placental tissues obtained from extremely low gestational age neonates. Am. J. Obstet. Gynecol. 198, 110.e1–110.e7 (2008).
Aryee, M. J. et al. Minfi: a flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369 (2014).
Fortin, J.-P., Fertig, E. & Hansen, K. Shinymethyl: interactive quality control of Illumina 450k DNA methylation arrays in R. F1000Res 3, 175 (2014).
Fortin, J.-P., Triche, T. J. & Hansen, K. D. Preprocessing, normalization and integration of the Illumina humanmethylationepic array with Minfi. Bioinformatics 33, 558–560 (2017).
Johnson, W. E., Li, C. & Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 (2007).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The Sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Du, P. et al. Comparison of beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 11, 1–9 (2010).
Salas, L. A. et al. An optimized library for reference-based deconvolution of whole-blood biospecimens assayed using the Illumina humanmethylationepic beadarray. Genome Biol. 19, 64 (2018).
Flowsorted.Blood.Epic: Illumina Epic data on immunomagnetic sorted peripheral adult blood cells v. 1.10.1 (Bioconductor, 2021).
Yuan, V. Placental DNA methylation analysis tools. http://www.bioconductor.org/packages/planet (2021).
McElrath, T. F. et al. Blood protein profiles of infants born before 28 weeks differ by pregnancy complication. Am. J. Obstet. Gynecol. 204, 418.e411–418.e412 (2011).
Dammann, O. & Leviton, A. Brain damage in preterm newborns: might enhancement of developmentally regulated endogenous protection open a door for prevention? Pediatrics 104, 541–550 (1999).
Kuban, K. C. K. et al. Systemic inflammation and cerebral palsy risk in extremely preterm infants. J. Child Neurol. 29, 1692–1698 (2014).
Leviton, A. et al. Early postnatal blood concentrations of inflammation-related proteins and microcephaly two years later in infants born before the 28th post-menstrual week. Early Hum. Dev. 87, 325–330 (2011).
Textor, J., van der Zander, B., Gilthorpe, M. S., Liskiewicz, M. & Ellison, G. T. H. Robust causal inference using directed acyclic graphs: the R package ‘Dagitty’. Int. J. Epidemiol. 45, 1887–1894 (2016).
Santos, H. P. Jr et al. Epigenome-wide DNA methylation in placentas from preterm infants: association with maternal socioeconomic status. Epigenetics 14, 751–765 (2019).
Stekhoven, D. J. & Bühlmann, P. Missforest–non-parametric missing value imputation for mixed-type data. Bioinformatics 28, 112–118 (2012).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 57, 289–300 (1995).
Mansell, G. et al. Guidance for DNA methylation studies: statistical insights from the Illumina Epic array. BMC Genomics 20, 366 (2019).
Illumina. Methylationepic beachchip annotation file. https://support.illumina.com/downloads/infinium-methylationepic-v1-0-product-files.html (2022).
Humberg, A. et al. Preterm birth and sustained inflammation: consequences for the neonate. Semin. Immunopathol. 42, 451–468 (2020).
Korzeniewski, S. J. et al. Elevated protein concentrations in newborn blood and the risks of autism spectrum disorder, and of social impairment, at age 10 years among infants born before the 28th week of gestation. Transl. Psychiatry 8, 115 (2018).
Kuban, K. C. K. et al. Among children born extremely preterm a higher level of circulating neurotrophins is associated with lower risk of cognitive impairment at school age. J. Pediatr. 201, 40–48.e44 (2018).
Rojas, D. et al. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol. Sci. 143, 97–106 (2015).
Liu, Y. et al. Derlin-1 functions as a growth promoter in breast cancer. Biol. Chem. 401, 377–387 (2020).
Mao, M., Zhang, J. & Jiang, J. Overexpression of Derlin-1 is associated with poor prognosis in patients with non-small cell lung cancer. Ann. Clin. Lab. Sci. 48, 29–34 (2018).
Zeng, J. et al. Derlin-1 exhibits oncogenic activities and indicates an unfavorable prognosis in breast cancer. Cell Biol. Int. 44, 593–602 (2020).
Macklin, P. S., McAuliffe, J., Pugh, C. W. & Yamamoto, A. Hypoxia and HIF pathway in cancer and the placenta. Placenta 56, 8–13 (2017).
Bastek, J. A. et al. Biomarkers of inflammation and placental dysfunction are associated with subsequent preterm birth. J. Matern. Fetal Neonatal Med. 24, 600–605 (2011).
McElrath, T. F. et al. Pregnancy disorders that lead to delivery before the 28th week of gestation: an epidemiologic approach to classification. Am. J. Epidemiol. 168, 980–989 (2008).
Meyers, E. A. & Kessler, J. A. TGF-beta family signaling in neural and neuronal differentiation, development, and function. Cold Spring Harb. Perspect. Biol. 9, a022244 (2017).
Aprigliano, R. et al. Increased P53 signaling impairs neural differentiation in Huwe1-promoted intellectual disabilities. Cell Rep. Med. 2, 100240 (2021).
Strickland, M., Yacoubi-Loueslati, B., Bouhaouala-Zahar, B., Pender, S. L. F. & Larbi, A. Relationships between ion channels, mitochondrial functions and inflammation in human aging. Front. Physiol. 10, 158 (2019).
Baines, K. J. et al. Maternal immune activation alters fetal brain development and enhances proliferation of neural precursor cells in rats. Front. Immunol. 11, 1145 (2020).
Smith, S. E., Li, J., Garbett, K., Mirnics, K. & Patterson, P. H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 27, 10695–10702 (2007).
Wu, W. L., Hsiao, E. Y., Yan, Z., Mazmanian, S. K. & Patterson, P. H. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav. Immun. 62, 11–23 (2017).
Spann, M. N., Monk, C., Scheinost, D. & Peterson, B. S. Maternal immune activation during the third trimester is associated with neonatal functional connectivity of the salience network and fetal to toddler behavior. J. Neurosci. 38, 2877–2886 (2018).
Lalonde, R. & Strazielle, C. The Agtpbp1 gene in neurobiology. Gene 809, 146001 (2022).
Siu, M. T. et al. Functional DNA methylation signatures for autism spectrum disorder genomic risk loci: 16p11.2 deletions and Chd8 variants. Clin. Epigenetics 11, 103 (2019).
Aderem, A. Phagocytosis and the inflammatory response. J. Infect. Dis. 187, S340–S345 (2003).
Mezu-Ndubuisi, O. J. & Maheshwari, A. The role of integrins in inflammation and angiogenesis. Pediatr. Res. 89, 1619–1626 (2021).
Funding
This work was funded in part by the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) (R01-HD092374) and Office of The Director, National Institutes of Health (OD) (UH3-OD023348).
Author information
Authors and Affiliations
Contributions
Substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data: L.A.E., A.E.E., H.H., K.R., L.S., K.C.K.K., T.M.O., R.C.F. Drafting the article or revising it critically for important intellectual content: L.A.E., A.E.E., M.C., S.G., W.A.G., W.M.J., E.J., R.M.J., C.J.M., K.R., H.S., J.S.S., D.Y., K.C.K.K., T.M.O., R.C.F. Final approval of the version to be published: L.A.E., A.E.E., M.C., S.G., W.A.G., H.H., W.M.J., E.J., R.M.J., C.J.M., K.R., H.S., J.S.S., L.S., D.Y., K.C.K.K., T.M.O., R.C.F. The authors would like to acknowledge Drs. Raina Fichorova and Doug Ruden for their important contributions to this work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Informed consent was given by all participants in the ELGAN study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Eaves, L.A., Enggasser, A.E., Camerota, M. et al. CpG methylation patterns in placenta and neonatal blood are differentially associated with neonatal inflammation. Pediatr Res 93, 1072–1084 (2023). https://doi.org/10.1038/s41390-022-02150-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-022-02150-4
This article is cited by
-
Association of placental histology and neonatal hematologic outcomes
Journal of Perinatology (2023)
