
Abstract
Background and objectives
Shwachman Diamond syndrome (SDS) is an inherited bone marrow failure syndrome (IBMFS) associated with pancreatic insufficiency, neutropenia, and skeletal dysplasia. Biallelic pathogenic variants (PV) in SBDS account for >90% of SDS. We hypothesized that the SDS phenotype varies based on genotype and conducted a genotype-phenotype correlation study to better understand these complexities.
Methods
We reviewed records of all patients with SDS or SDS-like syndromes in the National Cancer Institute’s (NCI) IBMFS study. Additional published SDS cohorts were reviewed and compared with the NCI cohort.
Results
PVs in SBDS were present in 32/47 (68.1%) participants. Biallelic inheritance of SBDS c.258 + 2T > C and c.183_184TA > CT was the most common genotype in our study (25/32, 78.1%) and published cohorts. Most patients had the SDS hallmark features of neutropenia (45/45, 100%), pancreatic insufficiency (41/43, 95.3%), and/or bony abnormalities (29/36, 80.6%). Developmental delay was common (20/34, 58.8%). Increased risk of hematologic malignancies at young ages and the rarity of solid malignancies was observed in both the NCI cohort and published studies.
Conclusions
SDS is a complex childhood illness with a narrow genotypic spectrum. Patients may first present to primary care, gastroenterology, orthopedic, and/or hematology clinics. Coordinated multidisciplinary care is important for diagnosis and patient management.
Clinical trial registration
ClinicalTrials.gov Identifier: NCT00027274.
Impact
-
The clinical and genetic spectrum of Shwachman Diamond Syndrome was comprehensively evaluated, and the findings illustrate the importance of a multidisciplinary approach for these complex patients.
-
Our work reveals:
-
1.
a narrow genotypic spectrum in SDS;
-
2.
a low risk of solid tumors in patients with SDS;
-
3.
patients with SDS have clinical manifestations in multiple organ systems
-
1.
Similar content being viewed by others

Introduction
Shwachman Diamond syndrome (SDS) is an inherited bone marrow failure syndrome (IBMFS) characterized by three hallmark features: neutropenia, exocrine pancreatic insufficiency, and bony abnormalities.1,2,3,4 Failure to thrive and gastrointestinal (GI) complications, such as steatorrhea and malabsorption, are commonly observed and occur secondary to exocrine pancreatic insufficiency and associated nutritional difficulties. Patients often experience a long and complicated diagnostic journey seeing multiple providers before a conclusive diagnosis is made. Patients with SDS are frequently evaluated for cystic fibrosis, celiac disease, Crohn’s disease, and other diseases prior to suspicion of SDS because the clinical presentation can vary widely and it is often underrecognized.5 Recognition of SDS by pediatric providers is critical for patient identification and management.
Patients with SDS often experience recurrent infections due to neutropenia and in some patients hypogammaglobulinemia.1 Other common complications include eczematous skin rashes, failure to thrive, and mild-severe developmental delay.1 The spectrum of developmental delay and other psychological diagnoses are becoming more widely recognized in patients with SDS.6,7,8,9,10 There is also a high lifetime risk of cancer, particularly hematological malignancies such as myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML).1
Between 90 and 95% of patients with SDS have autosomal recessive (AR) disease with pathogenic variants (PV) in the Shwachman Bodian Diamond Syndrome (SBDS) gene located on chromosome 7.1,11 The most common variants in SBDS are the splice site variant, c.258 + 2T > C, and a two-base pair inversion variant involving the SBDS pseudogene, c.183_184TA > CT.1 Boocock et al.12 established in a cohort of 158 families of which 89% of patients had at least one allele as a result of the gene conversion with the pseudogene, and 60% of patients had at least two of these converted alleles (the cis/trans orientation of the alleles were not determined). These authors noted that 50% of families in that cohort were compound heterozygotes for c.258 + 2T > C and c.183_184TA > CT. Patients who are homozygous for the c.183_184TA > CT have not been identified, implying that some partial SBDS function is required for life, and this observation holds true in several model organisms.11 Individuals with homozygous inheritance of c.258 + 2T > C have been identified, as well, patients with homozygous c.258 + 2T > C with a c.183_184TA > CT allele.12 Other genes associated with an SDS-like phenotype include EFL1 (AR), DNAJC21 (AR), and SRP54 (autosomal dominant).13,14,15,16,17,18,19 A small percentage of patients with an SDS phenotype have no identifiable genetic cause.5,11
The SBDS protein plays a crucial role in many cellular processes including maturation of the 60S ribosomal subunit, stromal microenvironment maintenance, mitosis, and DNA repair.20,21,22,23,24,25 Specifically, SBDS cooperates with EFL1 to catalyze the removal of eIF6 from the pre-60S subunit. This mechanism requires GTP binding and hydrolysis of EFL1, resulting in the release of eIF6. Release of eIF6 is essential for the assembly of the 80S ribosomal subunit.20,26 This process can be inhibited by decreased GTP-ase signaling activity in the presence of SRP54 variants.14,20 SBDS loss leads to accumulation of 40S and 60S subunits in the cytoplasm and fewer 80S are assembled.27 Similarly, DNAJC21 is associated with ribosome biogenesis and the release of maturation factors required for 80S formation.28,29 Decreased levels of the SBDS protein have been shown to contribute to chromosomal instability at the mitotic spindle during mitosis in addition to negatively affecting the DNA repair mechanism.23,24,25
To better understand the complexities of SDS, we conducted a genotype-phenotype correlation study comparing patients with the common PVs SBDS c.258 + 2T > C and c.183_184TA > CT with all other cases of SDS in the National Cancer Institute’s (NCI) IBMFS cohort study. We hypothesized that genotype-phenotype correlations may occur in SDS, similar to observations reported in other IBMFS.30 We asked whether the clinical features of SDS are different in patients with the rarer variants compared with the most common combination of the biallelic SBDS PVs, c.258 + 2T > C and c.183_184TA > CT.31,32,33,34 We also reviewed published cohorts and compared data across cohorts.5,35,36,37,38,39
Methods
Participants
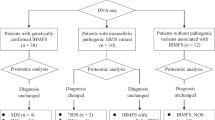
The IBMFS study at the NCI is an ongoing natural history and retrospective/prospective longitudinal cohort study approved by the NCI Institutional Review Board with more than 500 families enrolled (www.marrowfailure.cancer.gov, ClinicalTrials.gov Identifier: NCT00027274).3,40 Affected individuals and their unaffected family members complete detailed family history and medical history questionnaires. Fifty-nine families with SDS or an SDS-like phenotype are currently enrolled. Forty-two families with 47 affected participants with SDS were included in this genotype-phenotype correlation study. SDS was defined as neutropenia plus pancreatic insufficiency with positive genetic testing or negative/absent genetic testing. Seven participants from five families were grouped as SDS-like. These patients had suspicion of SDS by the referring provider and a phenotype suggestive of the disease but did not meet full SDS criteria. SDS-like patients had a hypocellular bone marrow but negative/absent genetic testing with either neutropenia or pancreatic insufficiency (Fig. 1). SDS-like participants were not included in the genotype-phenotype correlation study, but clinical data was extracted and reviewed (Supplementary Tables 1 and 2). Clinical data of twelve families were excluded from this report due to missing or insufficient data. For the purposes of this study, negative genetic testing was defined as an absence of germline pathogenic variant(s) in all the known SDS genes (SBDS, EFL1, DNAJC21, and SRP54).

Genetic testing methods included whole exome sequencing, targeted long-read PacBio sequencing of SBDS, EFL1, DNAJC21, and SRP54, or low-pass long-read whole genome sequencing. aSDS-like participants did not have available samples for sequencing and/or external negative genetic testing results.
Clinical data extraction
We reviewed medical records including, but not limited to, clinic notes and laboratory, pathology and radiology reports. We relied on self-reported information and medical records from participants. We used a comprehensive data collection process, and all organ systems were included in the medical record review. Date of SDS or SDS-like diagnosis was based on physician report when available. The genetic testing report date was used to establish a date of diagnosis if no earlier medical records were available. The age at last follow-up was obtained for all subjects with the date of death serving as the age at last follow-up for those who were deceased.
Laboratory values at diagnosis and the most recently reported values as of October 31, 2020 or last follow-up were extracted. At both time points, absolute neutrophil count (ANC), platelet count, white blood cell count, hemoglobin, and mean corpuscular volume data were evaluated. Neutropenia was defined by an ANC of less than 1500 cells/μL, thrombocytopenia by a platelet count of less than 150,000 cells/μL, and anemia by a hemoglobin level of less than 10 g/dL. Participants with severe bone marrow failure requiring hematopoietic cell transplantation (HCT) and those with malignancies had additional laboratory and system evaluations. The following laboratory evaluations were used to determine pancreatic insufficiency: fecal elastase, serum trypsinogen, pancreatic isoamylase, and 72-h fecal fat levels (normal values were based on reference standards set by Ip et al.).41 For this study, severe neutropenia was defined as ANC < 500 at the time of SDS diagnosis. We also recorded whether patients underwent chronic or intermittent treatment with granulocyte colony stimulating factor (GCSF) and/or intravenous immunoglobulins (IVIG), and/or had a history of recalcitrant infection requiring hospitalization. IVIG treatment administered post-HCT was not included.
The first reported and/or most severe bone marrow biopsy results were examined. Severity of bone marrow findings were determined by lowest cellularity, myeloid to erythroid (M:E) ratio, hematopoietic cellular morphological abnormalities, flow cytometry results when available, degree of cytogenetic abnormalities, and/or greatest number of somatic mutations. Patients were classified has having MDS per 2016 WHO guidelines for refractory cytopenia of childhood – a bone marrow biopsy report with dysplasia in at least one lineage, at least one peripheral cytopenia with any clonal cytogenetic change.42 AML cases were confirmed by medical records and as defined by referring institution.
Genetic evaluation
Many patients had clinical genetic testing prior to enrollment (40/54, 74%). Exome sequencing was performed on probands and available family members as previously described if they did not have PV(s) in a known SDS-associated gene.43 Targeted long-read PacBio gene sequencing (N = 11) and low-pass long-read whole genome PacBio sequencing (N = 1) were completed for the full genomic sequences surrounding SBDS, EFL1, DNAJC21, and SRP54 if exome sequencing did not uncover the genetic cause of disease. Standard manufacturer protocol was followed for PacBio preparation and sequencing methods with genomic DNA extracted from whole blood of probands and family members enrolled in the study. Sequencing data were processed, and raw reads were obtained. Targeted long-read PacBio gene sequencing data was processed using circular consensus reads. All sequence reads were aligned to human genome build 19 (GRCh37) and structural variants were called using pbsv default parameters.
Review of published SDS cohorts
A comprehensive literature review was performed using the National Library of Medicine’s PubMed database with the following search terms: “Shwachman Diamond syndrome AND cohort” or “Shwachman Diamond syndrome AND registry” to study the clinical presentation and genotypes in other SDS cohorts (last accessed January 11, 2021). Individual case reports and those not published in English were excluded. Studies which only reported clinical features without patients’ genotypes were also excluded. Data were compared across cohorts.
Results
NCI IBMFS SDS cohort
Forty-two families with 47 affected participants with SDS were included in our genotype-phenotype correlation study. Demographics of the participants and their phenotypes are described in Table 1. Almost half of patients with SDS in the NCI IBMFS cohort (21/47, 44.7%) were seen at the National Institutes of Health (NIH) Clinical Center (Bethesda, MD) and received formal evaluations of all major organ systems. We relied on self-reported data and medical records in the remaining participants (26/47, 55.3%). Seven participants (five families) with an SDS-like phenotype were not included in the genotype-phenotype correlation analyses, but general clinical and hematology data were extracted and summarized in Supplementary Tables 1, 2, respectively.
Patients were grouped into SDS with a known genetic cause (SBDS c.258 + 2T > C and c.183_184TA > CT OR c.258 + 2T > C and other single nucleotide variant [SNV] or copy number variant [CNV]) or SDS with unknown genetic cause (Table 1). Some phenotype data were not available for all participants. The noted values are calculated based on number of participants with data for that specific phenotype. The median age at diagnosis for SDS was 1.78 years (range, 0.12–41.82). The median age at last follow-up was 12.59 years (range 1.24–70.55). The prevalence of SDS was slightly lower in males than females at a ratio of 0.74 to 1 (Table 1). This slightly decreased prevalence of SDS in males compared to females is not consistent with previous reports of a male to female ratio of 1.5 to 1.4
Hallmark SDS features were seen in most patients; pancreatic insufficiency (41/43, 95.3%) and a history of neutropenia (45/45, 100%) and were the most common findings across all participants. Forty percent of these patients (10/25) had severe neutropenia (ANC < 500) at time of SDS diagnosis (ANC range 52–476). Failure to thrive, often secondary to pancreatic insufficiency and feeding difficulties, was common across all participants (31/36, 86.1%). MDS (N = 3) and AML (N = 2) were only observed in patients with a known genetic cause. Two patients in our cohort developed solid tumors, one had ovarian cancer and one had breast cancer (Table 1).
At least one skeletal dysplasia was observed in all groups of participants (29/36, 80.6%) (Table 1). Bony abnormalities of the extremities including short arms, short legs, small hands, and/or other skeletal dysplasia(s) in the extremities were the most common findings (20/29, 69%) and present in SDS patients with a known and unknown genetic cause. Thoracic and rib cage abnormalities were also a common finding in all participant groups (17/31, 54.8%). Metaphyseal dysostosis and scoliosis were respectively observed at 45.8% (11/24) and 33.3% (9/27) in all SDS patients.
Gastrointestinal manifestations were observed at high rates among all groups of participants (36/42, 85.7%); elevated transaminases (16/18, 88.9%) and malabsorption (25/29, 86.2%) were the most common findings. Patients often had a history of gastroesophageal reflux disease (11/14, 78.6%) and/or steatorrhea (19/27, 70.4%). Liver steatosis, cirrhosis, and/or unspecified liver disease was only seen in participants with a known genetic cause for disease (6/25, 24%). Fifteen patients among all groups received supplemental nutrition via tube feedings (12/15, 80%) or parental nutrition (3/15, 20%) (Table 1).
Most participants with SDS had a history of recurrent infections (34/37, 92%), particularly otitis media (30/32, 94%) and various other respiratory infections (27/39, 69%). The reason for these recurrent infections is unclear but it is possible that some of these patients had some level of immune dysregulation, and several patients needed intervention. Thirteen patients (54.2%) received GCSF injections (5 intermittently and 8 chronically). Eleven patients (45.8%) were treated with IVIG (5 intermittently and 3 chronically). Six of these participants (25%) received both GCSF injections and IVIG treatment. History of infection requiring hospitalization and intravenous antibiotics was identified in 20.5% (8/39) of participants (Table 1).
Attention deficit disorders (8/34, 23.5%) and delay in speech development (4/34, 11.8%) were the most common psycho-developmental phenotypes reported. Although structural neurological findings were not prevalent in our SDS cohort, there were two cases of type 1 Chiari malformation and both participants also presented with developmental delay (c.258 + 2T > C and c.183_184TA > CT, and SDS without a genetic cause of disease). According to available medical records, the remaining patients with SDS and psycho-developmental problems had no observed neurological abnormalities. Microcephaly was observed most often in SDS patients with a known genetic cause for disease (8/22, 36.4%) but was not observed in SDS participants without a known genetic variant (Table 1).
Cardiac malformations and abnormalities were present in almost half of all participants (10/23, 43.5%), but most cardiac anomalies were reported as mild and/or resolving at a young age. Patent foramen ovale was the most common cardiac abnormality (4/10, 40%). Other cardiac findings included cardiomegaly (3/10, 30%), which was noted to co-occur with cardiac anomalies such as ventricular septal defect, patent ductus arteriosus, patent foramen ovale, atrial septal defect, Kawasaki disease, tricuspid or pulmonary valve regurgitation, bicuspid aortic valve, aortic regurgitation, or aortic dilation.
At least one dermatologic finding was observed among nearly all participants with SDS (29/30, 96.7%), with eczema (15/24, 62.5%) and café au lait spots (11/20, 55%) being the most common. Areas of hypopigmentation (7/15, 46.7%) and hyperpigmentation (3/14, 21.4%) were observed in both groups of participants with a known genetic variant (Table 1).
Genetic profile of SDS patients in NCI IBMFS cohort
All SDS patients in the NCI cohort with a known genetic cause for disease (N = 32) have compound heterozygous variants in SBDS (Fig. 2). The genetic cause has not yet been identified in fifteen affected individuals with SDS and seven affected individuals with an SDS-like phenotype despite extensive genetic evaluation or due to a lack of available samples. See Supplementary Table 3 for details of genetic testing performed on those without a known genetic variant. There are no participants with a homozygous genotype or mutations in non-SBDS genes (EFL1, SRP54, and DNAJC21) in our cohort. The most common cause of SDS in our cohort is biallelic inheritance of the SBDS pathogenic variants, c.258 + 2T > C and c.183_184TA > CT (25/32, 78.1%). Clinical genetic testing or exome sequencing and deletion analyses identified seven unrelated SDS patients with biallelic inheritance of c.258 + 2T > C and a different, rare deleterious variant in SBDS (Table 2). Low-pass whole genome long-read (PacBio) sequencing was used, to identify one patient with a novel 19 kb deletion removing part of intron 4, exon 5, and the 3’UTR of SBDS. Targeted long-read panel sequencing identified another patient with an 872 base pair deletion removing exon 3 and portions of the surrounding intronic regions. All patients with an SDS-like phenotype (N = 7) have an unknown genetic cause for disease (Fig. 2 and Supplementary Table 3).

Left chart shows the full cohort with the three genetic groups: c.258+2T>C and c.183_184TA>CT (dark gray), c.258+2T>C and other SNV or CNV (light gray), unknown genetic cause (medium gray). Right chart is the subgroup of those with an unknown genetic cause: SDS with negative genetic testing (white), SDS with no samples available (light gray), SDS-like with no samples available (medium gray).
Hematologic findings in NCI IBMFS SDS cohort
Patients with SDS had a high prevalence of cytopenias with 84.6% (33/39) having neutropenia and 25% (7/28) and 23.1% (6/26) having with thrombocytopenia and anemia, respectively (Table 3) at time of diagnosis.
Most patients had results from at least one bone marrow aspirate and biopsy, summarized in Table 3. Most were hypocellular for age, but cellularity varied ranging from severely hypocellular to hypercellular. Several had cytogenetic abnormalities as noted. Three patients had more than one cytogenetic abnormality with no reported morphologic dysplasia. Deletion of the q-arm of chromosome 20 was the most common cytogenetic abnormality observed in both the first and most severe bone marrow aspirate and biopsy reports.
Four patients (all were c.258 + 2T > C and c.183_184TA > CT) underwent HCT; two for severe bone marrow failure, one for MDS that transformed to AML, and one for unknown reasons due to missing data. One patient underwent HCT with a matched sibling donor and two were transplanted with unrelated donors, donor type for the fourth patient was unknown. Two patients were transplanted with a myeloablative regimen; the details on the preparative regimen for two patients were not available.
Hematologic malignancies in NCI IBMFS SDS cohort
There were three cases of MDS and two cases of AML in the NCI IBMFS SDS cohort of 47 participants. Data on four of these patients have been previously reported.3,44 All five patients had confirmed biallelic variants in SBDS (Table 4). The three patients with MDS and one patient with AML had a compound heterozygous genotype of the pathogenic variants, c.258 + 2T > C and c.183_184TA > CT. The genotype for the second AML case was c.258 + 2T > C with c.123delC. Cytogenetic abnormalities observed in MDS cases included del(20q), and del(7q). Monosomy 5 and monosomy 7 were observed in one patient with AML. In our cohort, there was poor prognosis in SDS patients diagnosed with a hematologic malignancy with 33% (1/3) of MDS patients deceased and both AML patients deceased at date of last follow-up. An additional patient with SDS was diagnosed with AML, after the end of data collection date (October 31, 2020). The patient was 30 years old and had biallelic pathogenic variants in SBDS- a large deletion and c.258 + 2T > C. This patient’s AML had a complex karyotype including monosomy 21, trisomy 9, and a somatic TP53 mutation. The patient passed away from disease progression and infectious complications.
SDS cohort comparisons
In addition to reviewing our own genotype data, we compared this data with six published SDS cohort studies.5,35,36,37,38,39 Genetic variation of SDS in other cohorts was consistent with the compound heterozygous genotype in SBDS of c.258 + 2T > C and c.183_184TA > CT being the most common across all groups (Fig. 3). Compound heterozygous genotypes in SBDS with c.258 + 2T > C on one allele and another SNV on the other allele was also commonly observed. Homozygous incidence of c.258 + 2T > C was observed, with some groups reporting homozygous incidence of c.258 + 2T > C in combination with the allele, c.183_184TA > CT. Reports of novel variants were identified in four cohorts.5,36,38,39

Number of patients with the different SBDS genotypes listed, in the literature cohorts including the NCI IBMFS cohort (black with white dots), the Italian SDS registry (gray lines), the Greek SDS registry (gray dots), the Japanese SDS cohort (black lines), the North American SDS registry (dark gray), the French National cohort of SDS patients (light gray), the Canadian IBMFS cohort (black).
The NCI SDS cohort data and published SDS cohorts emphasize the variable clinical features of SDS and its range in severity. Each study used its own diagnostic criteria, making it difficult to quantitate the phenotypic features. Supplementary Table 4 summarizes the phenotypes of SDS reported from five cohorts in comparison with the NCI IBMFS cohort.5,35,36,37,39
Discussion/conclusion
We assessed the full spectrum of SDS genotypes, phenotypes, and their potential association(s) in a large cohort of patients with SDS (47 patients, 42 families) from the NCI IBMFS study. We hypothesized that patients with the less common variants (i.e., not c.258 + 2T > C and c.183_184TA > CT) would have different clinical features of SDS. However, our findings revealed a narrow genotypic spectrum, not significantly associated with phenotype.
Only seven patients out of 32 with a known genotype had variants other than the biallelic variants c.258 + 2T > C and c.183_184TA > CT. These seven patients also had the splice site variant c.258 + 2T > C as their second pathogenic SBDS allele. Two patients with SDS in the NCI IBMFS cohort had biallelic genotypes made up of a large genomic deletion in SBDS on one allele and c.258 + 2T > C on the other allele. These large deletions were only identified by long-read sequencing technology. One patient had a novel 19 kb deletion removing part of intron 4, exon 5, and the 3’UTR of SBDS. To our knowledge, this is the first report of such a large deletion in SBDS. The other patient with a large deletion had a deletion of exon 3; deletions in this region have been previously reported.45,46 Our findings suggest investigation for large structural variants in patients with a single known SBDS PV without a second pathogenic allele may be required in order to identify the genetic etiology of SDS.
Literature review determined that the high prevalence of biallelic variants c.258 + 2T > C and c.183_184TA > CT in our cohort was consistent with other large cohort studies of patients with SDS. There were no SDS patients in the NCI IBMFS cohort with a homozygous genotype. Other SDS cohort studies have reported c.258 + 2T > C homozygosity; it is important note some of these participants may be included in multiple cohort studies (e.g., the North American SDS Registry and the Canadian IBMF Study).5,35,36,38 This narrow genotypic spectrum of SDS in our cohort and other large studies limits the ability to assess potential genotype and phenotype correlations. It is possible that the variable clinical features seen in our and other SDS cohorts may be due to various modifiers including genetic, epigenetic, environmental, and/or inflammatory. Larger collaborative studies would allow for such research. The apparent lack of genomic diversity in large cohorts could be a result of unreported SDS genes at the time those studies were completed. Follow-up studies in these cohorts will be important to identify the presence of pathogenic variants in other SDS-associated genes, such as EFL1, DNAJC21, and SRP54. In addition, excluding case reports from our cohort review may have contributed to the lack of genomic diversity seen across SDS. This narrow genotypic spectrum contrasts with the other IBMFS where a wide genetic diversity is seen across the disorders.30,32 In other IBMFS such as Fanconi anemia and dyskeratosis congenita disease-associated variants are scattered across the gene, with only a few known founder mutations. In contrast, in SDS, we and others have observed that patients have only a few recurrent variants as described in international cohorts.
The hallmark features of SDS (exocrine pancreatic insufficiency, neutropenia, and skeletal dysplasia) were observed at comparable rates among patients with a known genetic cause for disease in the NCI IBMFS cohort. Consistent with other studies, gastrointestinal complications such as feeding difficulties and steatorrhea were often present at young ages and improved with age and pancreatic enzyme supplementation.47 The median ANC at diagnosis reflected a high prevalence of neutropenia in all groups of participants at the time of diagnosis. Both groups of patients with a known genetic cause for disease had a lower median ANC at diagnosis than those with an unknown genetic cause for disease. Recurrent infections, particularly otitis media and respiratory infections, likely due to chronic and/or variable neutropenia, were common complications for SDS patients over their lifetimes. Skeletal dysplasia varied in its severity with most patients having bony abnormalities of the extremities and/or rib cage/thoracic region. Congenital cardiac abnormalities observed in the NCI SDS cohort were often minor and/or resolved in childhood. There is limited understanding of the full spectrum of neuropsychiatric manifestations in patients with SDS.7,10 The presence of developmental delay in the NCI SDS cohort is consistent with previous studies of the behavioral phenotype of school-age children with SDS, which reported significant differences in attention span and ability when completing tasks in comparison with normative samples and cystic fibrosis control subjects.8,10 The majority of large cohort studies and other literature report a slightly increased prevalence of SDS in males compared to females. Our cohort showed an unexpected female predominance (male:female ratio 0.74:1), which may be due to a small cohort size (Table 1).
We observed a relatively high prevalence of MDS and AML among SDS patients in both groups with a known genetic cause for disease in our cohort. Those without a known pathogenic gene in our cohort presented with a hematologic phenotype, such as neutropenia and hypocellular bone marrow, but have not yet developed a hematologic malignancy. Our findings on the hematological parameters, age and rates of MDS and AML are consistent with those recently reported by the North American SDS Registry.48
Patients with SDS have high lifetime risks of cancer at rates 8.5-fold higher than the general U.S. population.3 Solid malignancies in the SDS population are rarer with the highest lifetime risks being hematological malignancies (O/E: 202).3 There were two cases of solid malignancies in the NCI cohort. One case of ovarian cancer was reported in an SDS patient (41.8 years) with the most common biallelic variants, c.258 + 2T > C and c.183_184TA > CT. There was also one case of breast cancer in a 69-year-old postmenopausal woman in our cohort. She had negative germline genetic testing. The association between SDS and these malignancies is difficult to establish given the older age at cancer onset in the patients with solid malignancies.
The rarity of solid malignancies in the NCI IBMFS SDS cohort is consistent with the literature. Ikuse et al.39 was the only SDS cohort study reporting incidence of a non-hematologic, solid malignancy, a pancreatoduodenal carcinoma in a young adult (died, age 24 years). Bou Mitri et al.49 reported three incidences of solid malignancies over the age of 50 years (breast, ovarian, and esophagus) in a large cohort of SDS patients (N = 155), these low rates of solid tumors are consistent with the NCI IBMFS SDS cohort. This is in contrast to other IBMFS, particularly Fanconi anemia and telomere biology disorders, which have high lifetime risks of both hematological and solid malignancies, notably head and neck squamous cell carcinoma.3 Osteosarcoma has not been reported in SDS in contrast to Diamond Blackfan anemia, the other ribosome-related IBMFS which has a significant risk of osteosarcoma.3,21
Limitations of this study include missing data from patients evaluated externally and, in the literature, and the inability to do robust statistical analyses due to cohort size. Strengths of the study includes thoroughly curated cohort data, formal comprehensive evaluation for those patients seen at the NIH Clinical Center, and a large cohort for a rare disorder.
SDS may be underdiagnosed because of its array of clinical features and variable phenotypes. Many patients, in our cohort and others, report a long diagnostic journey and/or delayed diagnosis.50 This delay is likely multifactorial including variable clinical features, the need to see multiple subspecialities and lack of suspicion of the syndrome on behalf of medical providers. Prior to SDS diagnosis, many patients in the NCI IBMFS cohort were also assessed for a wide variety of diseases. Pediatricians and pediatric subspecialists need to be knowledgeable about SDS and include it in the differential diagnosis of children with one or more of neutropenia, gastrointestinal problems, failure to thrive or skeletal abnormalities. Clinicians should consider SDS in patients with suspicion for cystic fibrosis, celiac disease, or Crohn’s disease (particularly if they have neutropenia, even if borderline). Awareness of SDS may save a lengthy work-up and get patients to the needed subspecialities for care and management. Figure 4 outlines the pediatric specialties who may be the first to see these patients in clinic, and the multiple signs and symptoms that may be observed in a patient with suspected SDS. Genetic testing should be pursued if there is clinical suspicion of SDS and can be directed to the four SDS-associated genes. Most patients can be diagnosed with Sanger or next generation sequencing focused on SNV identification. Other technologies can be employed to identify both small and large deletions if two pathogenic SNVs are not identified. Genetic testing is critical for family planning, and donor choice if hematopoietic cell transplantation is considered. Patients with SDS should be monitored for the development of MDS and/or AML.44 Thus far, no solid tumors have been recurrently reported and there is no evidence-based guideline for solid tumor surveillance. Using a multidisciplinary approach, we can help patients with SDS get prompt diagnosis, and the needed treatment and management.

These providers can assist patients to get the full diagnostic work-up including genetic testing as noted in the boxes. US ultrasound, DEXA dual-energy X-ray absorptiometry.
References
Nelson, A. S. & Myers, K. C. Diagnosis, treatment, and molecular pathology of Shwachman-diamond syndrome. Hematol./Oncol. Clin. North Am. 32, 687–700 (2018).
Burwick, N., Shimamura, A. & Liu, J. M. Non-diamond Blackfan anemia disorders of ribosome function: Shwachman diamond syndrome and 5q- syndrome. Semin. Hematol. 48, 136–143 (2011).
Alter, B. P., Giri, N., Savage, S. A. & Rosenberg, P. S. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30–39 (2018).
Shimamura, A. & Alter, B. P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 24, 101–122 (2010).
Myers, K. C. et al. Variable clinical presentation of shwachman-diamond syndrome: update from the North American Shwachman-diamond syndrome registry. J. Pediatr. 164, 866–870 (2014).
Kerr, E. N. in 9th International Congress on Shwachman-Diamond Syndrome. (Houston, Texas, 2018).
Perobelli, S. et al. Diffuse alterations in grey and white matter associated with cognitive impairment in Shwachman-diamond syndrome: evidence from a multimodal approach. Neuroimage Clin. 7, 721–731 (2015).
Perobelli, S., Nicolis, E., Assael, B. M. & Cipolli, M. Further characterization of Shwachman-diamond syndrome: psychological functioning and quality of life in adult and young patients. Am. J. Med. Genet. A 158A, 567–573 (2012).
Dror, Y. et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-diamond syndrome. Ann. N. Y Acad. Sci. 1242, 40–55 (2011).
Kerr, E. N., Ellis, L., Dupuis, A., Rommens, J. M. & Durie, P. R. The Behavioral phenotype of school-age children with Shwachman diamond syndrome indicates neurocognitive dysfunction with loss of shwachman-bodian-diamond syndrome gene function. J. Pediatr. 156, 433–438 (2010).
Warren, A. J. Molecular basis of the human ribosomopathy Shwachman-diamond syndrome. Adv. Biol. Regul. 67, 109–127 (2018).
Boocock, G. R. et al. Mutations in Sbds are associated with Shwachman-diamond syndrome. Nat. Genet. 33, 97–101 (2003).
Bellanne-Chantelot, C. et al. Mutations in the Srp54 gene cause severe congenital neutropenia as well as Shwachman-diamond-like syndrome. Blood 132, 1318–1331 (2018).
Carapito, R. et al. Mutations in signal recognition particle Srp54 cause syndromic neutropenia with Shwachman-diamond-like features. J. Clin. Invest. 127, 4090–4103 (2017).
D’Amours, G. et al. Refining the phenotype associated with biallelic Dnajc21 mutations. Clin. Genet. 94, 252–258 (2018).
Dhanraj, S. et al. Biallelic mutations in Dnajc21 cause Shwachman-diamond syndrome. Blood 129, 1557–1562 (2017).
Tan, Q. K. et al. Further evidence for the involvement of Efl1 in a Shwachman-diamond-like syndrome and expansion of the phenotypic features. Cold Spring Harb. Mol. Case Stud. 4, 1–12 (2018).
Stepensky, P. et al. Mutations in Efl1, an Sbds partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in ashwachman-diamond like syndrome. J. Med. Genet. 54, 558–566 (2017).
Bezzerri, V. & Cipolli, M. Shwachman-diamond syndrome: molecular mechanisms and current perspectives. Mol. Diagn. Ther. 23, 281–290 (2019).
Finch, A. J. et al. Uncoupling of Gtp hydrolysis from Eif6 release on the ribosome causes Shwachman-diamond syndrome. Genes Dev. 25, 917–929 (2011).
Wegman-Ostrosky, T. & Savage, S. A. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br. J. Haematol. 177, 526–542 (2017).
Raaijmakers, M. H. et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature 464, 852–857 (2010).
Austin, K. M. et al. Mitotic spindle destabilization and genomic instability in Shwachman-diamond syndrome. J. Clin. Invest. 118, 1511–1518 (2008).
Orelio, C., Verkuijlen, P., Geissler, J., van den Berg, T. K. & Kuijpers, T. W. Sbds expression and localization at the mitotic spindle in human myeloid progenitors. PLoS ONE 4, e7084 (2009).
Morini, J. et al. Radiosensitivity in lymphoblastoid cell lines derived from Shwachman-diamond syndrome patients. Radiat. Prot. Dosim. 166, 95–100 (2015).
Weis, F. et al. Mechanism of Eif6 release from the nascent 60s ribosomal subunit. Nat. Struct. Mol. Biol. 22, 914–919 (2015).
Valli, R., Frattini, A. & Minelli, A. Shwachman-diamond syndrome: diagnosis, pathogenesis and prognosis. Expert Opin. Orphan Drugs 5, 753–767 (2017).
Lo, K. Y. et al. Defining the pathway of cytoplasmic maturation of the 60s ribosomal subunit. Mol. Cell 39, 196–208 (2010).
Tummala, H. et al. Dnajc21 mutations link a cancer-prone bone marrow failure syndrome to corruption in 60s ribosome subunit maturation. Am. J. Hum. Genet. 99, 115–124 (2016).
Niewisch, M. R. et al. Disease progression and clinical outcomes in telomere biology disorders. Blood (2021). Online ahead of print.
Gianferante, M. D. et al. Genotype-phenotype association and variant characterization in diamond-blackfan anemia caused by pathogenic variants in Rpl35a. Haematologica 106, 1303–1310 (2021).
Fiesco-Roa, M. O., Giri, N., McReynolds, L. J., Best, A. F. & Alter, B. P. Genotype-phenotype associations in fanconi anemia: a literature review. Blood Rev. 37, 100589 (2019).
Moller, P. et al. Cancer risk and survival in path_mmr carriers by gene and gender up to 75 years of age: a report from the prospective lynch syndrome database. Gut 67, 1306–1316 (2018).
Ryan, N. A. J. et al. Association of mismatch repair mutation with age at cancer onset in lynch syndrome: implications for stratified surveillance strategies. JAMA Oncol. 3, 1702–1706 (2017).
Cesaro, S. et al. A prospective study of hematologic complications and long-term survival of italian patients affected by Shwachman-diamond syndrome. J. Pediatr. 219, 196–201 e191 (2020).
Donadieu, J. et al. Classification of and risk factors for hematologic complications in a French National Cohort of 102 patients with Shwachman-diamond syndrome. Haematologica 97, 1312–1319 (2012).
Delaporta, P. et al. The Greek Registry of Shwachman diamond-syndrome: molecular and clinical data. Pediatr. Blood Cancer 64, 1–4 (2017).
Tsangaris, E. et al. Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population-based cohort and identification of novel mutations. J. Med. Genet. 48, 618–628 (2011).
Ikuse, T. et al. Shwachman-diamond syndrome: nationwide survey and systematic review in Japan. Pediatr. Int. 60, 719–726 (2018).
Alter, B. P. et al. Malignancies and survival patterns in the national cancer institute inherited bone marrow failure syndromes cohort study. Br. J. Haematol. 150, 179–188 (2010).
Ip, W. F. et al. Serum pancreatic enzymes define the pancreatic phenotype in patients with Shwachman-Diamond syndrome. J. Pediatr. 141, 259–265 (2002).
Arber, D. A. et al. The 2016 Revision to the World Health Organization Classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Ballew, B. J. et al. Germline mutations of regulator of telomere elongation helicase 1, rtel1, in dyskeratosis congenita. Hum. Genet. 132, 473–480 (2013).
Myers, K. C. et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia: a multicentre, retrospective, cohort study. Lancet Haematol. 7, e238–e246 (2020).
Costa, E. et al. Identification of a novel alusx-mediated deletion of exon 3 in the Sbds gene in a patient with shwachman-diamond syndrome. Blood Cells Mol. Dis. 39, 96–101 (2007).
Minelli, A. et al. Structural variation in Sbds gene, with loss of exon 3, in two Shwachman-diamond patients. Blood Cells Mol. Dis. 60, 33–35 (2016).
Hill, R. E., Durie, P. R., Gaskin, K. J., Davidson, G. P. & Forstner, G. G. Steatorrhea and pancreatic insufficiency in Shwachman syndrome. Gastroenterology 83, 22–27 (1982).
Furutani, E. et al. Hematologic complications with age in Shwachman-diamond syndrome. Blood Adv. 6, 297–306 (2022).
Bou Mitri, F. et al. Shwachman-diamond syndrome and solid tumors: three new patients from the French Registry for severe chronic neutropenia and literature review. Pediatr. Blood Cancer 68, e29071 (2021).
Church, J. A. A pediatric genetic disorder diagnosed in adulthood. PLoS Med. 3, e15 (2006).
Acknowledgements
We are grateful to the study participants, their families, and referring clinicians for their valuable contributions to this study. The authors thank Lisa Leathwood, RN, BSN, Maureen Risch, RN, BSN and Ann Carr, MS, CGC for assistance with the National Cancer Institute Inherited Bone Marrow Failure syndrome cohort patient data management. The authors thank members of the Cancer Genomics Research Laboratory for assistance with DNA sequencing and bioinformatics. This research was funded by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute and supported by contract HHSN261201700004C with Westat, Inc.
Author information
Authors and Affiliations
Contributions
A.S.T. was responsible for data acquisition, medical record review, data analysis and interpretation, and wrote the first draft of the manuscript. N.G. evaluated patients, performed data analysis, and interpretation in addition to assisting the manuscript revision. D.M.G. assisted with data analysis and interpretation. S.A.S. contributed to study design, data analysis and interpretation, and manuscript revisions. B.P.A. is responsible for study design and patient recruitment to the NCI Inherited Bone Marrow Failure syndrome cohort and assisted with manuscript development. L.J.M. supervised the study and was responsible for study design, data analysis and interpretation, and manuscript drafting and revisions.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Written informed consent was obtained from all study participants or the parent or guardian of participants who were under the age of 18 years old.
Consent for publication
Consent for the publication of personal or clinical details of participants that have the potential to compromise anonymity was obtained from all study participants or the parent or guardian of participants who were under the age of 18 years old.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Thompson, A.S., Giri, N., Gianferante, D.M. et al. Shwachman Diamond syndrome: narrow genotypic spectrum and variable clinical features. Pediatr Res 92, 1671–1680 (2022). https://doi.org/10.1038/s41390-022-02009-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-022-02009-8
