
Abstract
Background
Dobutamine is particularly suited to treatment of haemodynamic insufficiency caused by increased peripheral vascular resistance and myocardial dysfunction in the preterm infant. Knowledge of the elimination half-life is essential to estimate the steady state when its efficacy/safety can be evaluated.
Methods
Analysis of pharmacokinetic data in ten preterm newborns treated with a new neonatal formulation of dobutamine (IMP) after screening for haemodynamic insufficiency within the first 72 h from birth. Blood samples were withdrawn at the end of IMP infusion and at a random time after the end of infusion (5 min, 15 min, 45 min, 2 h and 6 h). IMP concentration in each sample was measured by ultra-high performance liquid chromatography with electrochemical detection.
Results
Median duration of IMP infusion was 37.7 h (IQR 21.2). Calculated IMP half-life ranged between 3.06 and 36.1 min (median 10.6 min), leading to a time to reach the steady-state concentration between 15 min and >2 h. Adverse events were not related to IMP.
Conclusions
The wide variability in dobutamine metabolism in preterm infants requires awareness about the risk of under- or overtreatment. A delay of up to 3 h might be required before drawing blood samples to evaluate the effective dose.
Impact
-
Small trials suggest dobutamine as the optimal drug in the preterm infant with haemodynamic insufficiency after birth.
-
Age-related differences in drug pharmacokinetics may result in suboptimal treatments.
-
The lack of formal studies in preterms results in inadequate data on efficacy and safety.
-
This study provides data on the variability of the elimination half-life of dobutamine in the very preterm infant during transitional circulation.
-
There is a wide variation in the time to reach the plasma concentration corresponding to steady state, the moment when its efficacy/safety can be reliably evaluated.
-
This information is crucial for planning future trials on cardiovascular support.
Similar content being viewed by others


Introduction
Many medicines used in children, especially in neonates, have never been tested to the level seen in adult healthcare and are used unlicensed or off-label,1 resulting in insufficient data on efficacy and safety. The European Union Drug Regulations require the industry to provide age-appropriate formulations and strengths of medicines in neonates through to adolescents. Cardiovascular drugs are in the priority list.
Circulatory failure during transitional circulation is commonly seen in preterm infants and is associated with adverse outcomes.2,3,4,5,6 Shortly after birth there is an increase in systemic vascular resistance and a decrease in pulmonary vascular resistance resulting from the severing of the umbilical vessels, the inflation of the lungs, and the increase in arterial oxygen content.7 Small-scale observational studies and randomised clinical trials have suggested dobutamine as first-line inotrope for circulatory failure during transitional circulation in the preterm infant.8,9,10,11 Dobutamine increases myocardial contractility due to combined beta 1- and alpha 1-adrenergic receptor stimulation in the myocardium.7,12 This results in increased stroke volume and restoration of tissue perfusion.
The Neocirculation Consortium (FP7; HEALTH-2011.4.2-1 [Investigator-driven clinical trials on off-patent medicines for children]) developed a programme of work in a Paediatric Investigation Plan (PIP) to study the efficacy and safety of a new age-appropriate formulation of dobutamine for the treatment of circulatory failure during transitional circulation in preterm infants. Firstly, a dobutamine formulation with components selected from the currently established range of excipients used in paediatric products, based on guidance by EMA and adapted to the requirement for variable dose administration, was developed.13 Secondly, a therapeutic observational study (NeoCirc-001) to optimise the design of a subsequent placebo-controlled confirmatory trial was started. The intent was for NeoCirc-001 to: (1) provide an estimate of the elimination half-life and time to reach steady state of the new dobutamine formulation in the preterm infant (NeoCirc-001A sub-study); (2) explore relationships between inclusion criteria, treatment and outcomes; (3) develop a population pharmacokinetic and pharmacodynamic model in the target population (NeoCirc-001B sub-study). The trial stopped prematurely after completing the first of the three goals mentioned above, as it was recognised that a randomised allocation to placebo or dobutamine, combined with a more standardised up-titration schedule, were prerequisites for reliably exploring relationships looking at efficacy and safety; and for conducting the population PK-PD analysis.14
This paper mainly reports results from the first goal. That is, pharmacokinetic investigations of a new dobutamine formulation administered to preterm infants with haemodynamic insufficiency in the first 72 h of life. The elimination half-life of dobutamine is essential to estimate the time it reaches steady state, which in turn represents the moment when its efficacy/safety can be evaluated. As a general pharmacokinetics (PK) principle, steady state is reached when the time elapsed since the beginning of a given treatment (or dose adjustment) is equal to five times the elimination half-life.15
Methods
Inclusion criteria
Preterm infants between 24 + 0 and 32 + 6 weeks of gestation were prospectively screened for early haemodynamic insufficiency at two clinical sites (La Paz University Hospital, Spain and Brighton and Sussex University Hospital, UK). Haemodynamic insufficiency was defined as: either two or more of (i) mean blood pressure (MBP) < gestational age (GA)−1 mmHg (invasive/non-invasive, two readings 15 min apart), (ii) echocardiographic assessment of superior vena cava (SVC) flow5,7,10 <51 ml/kg/min, (iii) capillary refill time (CRT) > 4 s, (iv) lactate > 4 mmol/l, (v) base excess < −9 mmol/l; or MBP < GA −5 mmHg (invasive/non-invasive, two readings 15 min apart) alone. Eligible infants were excluded if: considered non-viable, severe congenital hydrops or malformations likely to affect cardiovascular adaptation, surgical treatment within 72 h of birth, chromosomal anomalies, or lack of signed informed consent.
Intervention—investigational medicinal product
Infants who met the definition of haemodynamic insufficiency and whose clinicians decided to prescribe dobutamine received a new neonatal formulation of dobutamine (investigational medicinal product, IMP), solely or in addition to other treatments. The IMP formulation was a concentrate for dilution for infusion contained in a 10-ml glass vial containing 12.5 mg of dobutamine hydrochloride per ml (Proveca Limited, Daresbury, UK) (Table 1). The sterile dobutamine solution was added to the infusion solution (5% dextrose or sodium chloride) immediately prior to administration.
Pharmacokinetics
Two blood samples were withdrawn per neonate: the first one at the end of dobutamine infusion (T1); the second one at a random time after the end of infusion (T2), among the following possibilities: 5 min, 15 min, 45 min, 2 h and 6 h. The randomisation of sampling time was done online by the data management centre.
For each neonate, elimination half-life (T1/2) was calculated as follows:
-
T1/2 = Ln2/Ke, and
-
Ke = (Ln(C1) – Ln(C2))/(T2 − T1),
where Ln is the natural logarithm, Ke the elimination rate constant of dobutamine, and C1 and C2 being two concentrations of dobutamine measured at their respective post-infusion time T1 and T2.
Plasma clearance of dobutamine was calculated from the infusion rate and the plasma concentration at T1, providing the former had remained constant for five times the elimination half-life according to the equation:
-
Clearance (l/h/kg) = infusion rate (μg/kg/h)/concentration al at steady state (ng/ml).
Strict standardisation of the dobutamine administration system was followed to minimise systematic errors due to differences in infusion set-ups, infusion lines and connectors to administer i.v. fluids. The effective start (t0) and end (te) of the infusion was calculated as the time at which the infusion pump is switched on or off plus the empirical value for the interval arising from dead space (td), as follows:
where volume refers to the catheter lumen plus the stopcock dead space, and velocity refers to the summation of the infusion rates of the various infusions delivered through the line. Dobutamine infusion joined the system at the stopcock closest to the catheter entry. Before dobutamine was connected to the stopcock, the whole system (including bionector) was fed with the dobutamine solution and the pump was switched on for a few minutes to ensure that no pulling out effect was added at start. The pump was switched off and the system was then connected to the stopcock. Finally, the pump was switched on and the time was registered (tswitch on). To calculate te, the time when dobutamine infusion was stopped was registered (tswitch off). The time corresponding to the first blood sample (T1) had to be equal or slightly later than te. A pre-analytical test, using methylene blue as a model drug, was conducted to confirm the replicability of calculations according to the equation and standards of procedures defined in this study.
Samples’ handling was performed according to the lab handbook that was developed by one of the researchers (INSERM) based on the results of the short- and long-term stability study of dobutamine in plasma. Accordingly, blood samples had to be centrifuged and plasma frozen at −80 °C within 6 h, and kept at −80 °C until analysis for a maximum period of 12 months.
Dobutamine assay
Dobutamine concentration in each sample was measured by ultra-high performance liquid chromatography with electrochemical detection (UPLC-EC) method that was validated over the concentration range of 2–256 ng/ml that was expected to include the concentrations obtained during continuous infusion of dobutamine. Consequently, the accuracy and precision of the method was uncertain for a concentration <2 ng/ml (lower limit of quantification, LLOQ). Validation results for this concentration range satisfied all criteria requested by the European Medicines Agency guidelines.16
Assessment of treatment response
Positive pharmacodynamic response of each parameter that triggered treatment with dobutamine (or other drugs) included a margin of effect, defined as attainment of: MBP > GA mmHg, SVC flow > 55 ml/kg/min, CRT < 4 s, lactate < 2 mmol/l or base excess > −9 mmol/l, respectively, for those who had low blood pressure, low SVC flow, prolonged CRT, or high serum lactate or low base excess at study entry. Short-term outcome of biomarkers also included urine output and echocardiographic assessment of right ventricular output.
Starting dose, dose escalation or need for additional cardiovascular treatment was guided by clinical judgement relating to any of the criteria by which the baby could become eligible for the study. Down-titration strategy and duration of treatment was at the discretion of the attending physician. Prospective data collection continued until 38 ± 4 weeks of gestation or discharge.
Severe adverse events (SAE) were defined as any untoward medical occurrence or effect in a clinical trial subject receiving the IMP, that at any dose results in death, is life threatening, requires hospitalisation or prolongation of hospitalisation, or results in persistent or significant disability or incapacity. There does not need to be a causal relationship between the occurrence and the study or any treatment administered.
Statistics
Calculated sample size for the neonatal dobutamine half-life sub-study reported in this paper was ten patients. This sample size was established because no PK data were available in this population and preliminary PK studies are supposed to be performed in a restricted number of subjects.17 The use of random sampling times was necessary as the elimination half-life was unknown and because only two blood samples could be drawn per infant, due to ethical reasons. Two measured blood concentrations are needed to calculate Ke and half-life.
Descriptive analysis of event rates, with 95% confidence intervals, median (IQR) or mean values and standard deviation are reported. The main concern regarding haemodynamic insufficiency early after birth is altered blood flow patterns to the brain and subsequent brain injury. Based on this, the most relevant clinical outcomes were selected as candidate endpoints to assess efficacy. Accordingly, for this observational study, treatment failure was defined if one of the following was true: neonate dies or intraventricular haemorrhage (IVH) grades 2–4, or cystic and non-cystic periventricular leukomalacia (PVL), or porencephalic cysts, ventriculomegaly, or cerebellar haemorrhage.18,19,20
Results
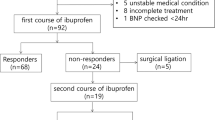
During the 16-month period, 72 patients were screened. Fifteen patients received cardiovascular support (catecholamines or hydrocortisone), starting at a median postnatal age of 5 h (IQR 4.8) (Fig. 1). Of them, two infants received commercial dobutamine and 13 infants were treated with the IMP, who are the study population.

Study flow chart.
Among the 13 infants treated with the IMP, only 10 were included in the dobutamine half-life analysis (Fig. 1). Reasons for exclusion were: unreliable data possibly related to mistaken labelling (concentration of dobutamine in the second blood sample was higher than in the first blood sample) (1 case); blood sample withdrawn while the IMP infusion was still ongoing (2 cases).
Of note, the concentrations measured in the four samples drawn 120 min or more after the first sample were less than the LLOQ of the analytical assay, and were consequently not taken into account for the calculation of T1/2. The results are displayed in Table 2.
Calculated T1/2 ranged between 3.06 and 36.1 min (median = 10.6 min), which corresponded to time to steady state ranging between 15 and 180 min (median 52 min). The mean weight-normalised clearance obtained in this study was 9.43 (l/h/kg). Of note, in one patient (case 7), the duration of dobutamine infusion was four half-lives when the first sample was drawn, instead of the expected duration of five half-lives in order to reach the steady state. However, since 94% of the steady state is reached after four elimination half-lives, the bias on the calculated clearance was considered negligible in this patient.
The following results on clinical outcomes, treatment and safety are presented as feasibility observations to inform the planning of subsequent trials on cardiovascular support:
-
Six out of 13 (46%, 95% Wald confidence interval: 19%; 73%) of the babies were classified as treatment failures at the follow-up assessment. Clinical data are displayed in Table 3.
Table 3 Demographics, clinical outcomes, and cardiovascular treatment of the study population. -
Median duration of treatment with dobutamine was 37.7 h (IQR 21.2), ranging between 19.5 and 181.0 h. There were two patients who received dobutamine for more than 160 h. Treatment duration of all other patients was below 70 h.
-
Six SAEs occurred during the trial in three babies and one baby died (Table 4). The SAEs were not considered to be related to the study drug. Three adverse events were reported for only one patient (case #10), consisting of transient chylothorax, hypothyroidism and peripheral arterial vasospasm related to canalisation causing left foot ischaemia at 26, 31 and 30 days of postnatal age, respectively. A further SAE was reported for this baby at death.
Table 4 Serious adverse events report.
Discussion
The results of NeoCirc-001A address an important knowledge gap, by providing information on the required waiting time between the start of treatment with dobutamine and the moment its efficacy/safety can be reliably evaluated in the very preterm infant during transitional circulation. This information is crucial for the planning of future trials on routine cardiovascular drugs where there is a lack of formal studies in this population. Age-related differences in drug absorption or metabolism result in drugs being started at doses which turn out to be suboptimal. Pragmatic adjustments are then made to improve the target outcome, but the evidence-base for these decisions is weak.
The maximal effect of a given dose is obtained when plasma concentration reaches the steady-state plateau at 5−6 times the elimination half-life.15 Our results thus evidence that the time needed for a true effect of a given dobutamine dose varies greatly within subjects, with values ranging from 15 min to >2 h. This raises a methodological issue when evaluating the response to dobutamine in this vulnerable preterm population. Waiting 2 h to evaluate the effect of a given dose in order to maximise the probability to be at the steady state will cause an unnecessary delay before an eventually needed dose adjustment in infants with short dobutamine half-life. On the other hand, adjusting the dose before steady state will possibly lead to dobutamine over-dosage in infants with long half-life.
Given that the elimination half-life of dobutamine was unknown, we decided to start our work plan with an exploratory PK study on a limited number of subjects. Because a too narrow time-interval between the two blood samples increases the uncertainty of the calculated half-life value, we decided to have different time intervals investigated from a subject to another. These time intervals were chosen to investigate half-lives ranging between approximately 1 min and 1 h (i.e. the delay between the two samples corresponding to 5−6 times the elimination half-life), which seemed likely based on adult data. In human adults, the elimination half-life of dobutamine is 2−3 min. Dobutamine is mainly eliminated via the formation of a 3-O-methyl metabolite produced by catechol-O-methyl transferase, which is expressed in most, if not all tissues in the body.21 This metabolite is subsequently glucuronoconjugated and excreted in urine and bile. Dobutamine can also be sulfoconjugated. Conjugates of dobutamine and 3-O-methyldobutamine are excreted mainly in urine and to a minor extent in faeces.
There are no available studies in juvenile animals. In a study in 13 critically ill newborns (gestational age of 27−42 weeks)22 in which dobutamine was administered starting at 2.5 µg/kg/min and increasing to 5 and then to 7.5 µg/kg/min, the mean calculated threshold value, or the minimum plasma concentration necessary for a change in cardiac output, was 39 ± 8 ng/ml. The mean plasma clearance rate was 90 ± 38 ml/min/kg and was most consistent with first-order kinetics over the range of dosages studied. In neonates, no association was found between plasma clearance and birth weight or gestational age.
In other studies conducted in the paediatric population (age 2 days to 9 years or from 0.13 to 16.6 years), dobutamine dose ranged from 2 to 15 µg/kg/min.23,24 In one study23 the serum concentrations varied from 6.4 to 347 ng/ml and the clearance varied from 32 to 625 ml/kg/min. Clearance of dobutamine was not affected by age or the added infusion of dopamine.23 Another study24 found the steady-state plasma concentration of dobutamine (infusion rate corrected to 5.0 µg/kg/min) ranged between 3.79 and 400 ng/ml and the clearance rate was 151.1 ± 47.5 ml/kg/min. Age, body weight and sex were not related to the value of the pharmacokinetic parameters. Most post-infusion time−concentration data were best fit to a bi-exponential function suggestive of a two-compartment model and the concomitant administration of dopamine-altered dobutamine’s pharmacokinetics.24 The pharmacokinetics of dobutamine in children is linear within the investigated infusion rates (0.5−20 µg/kg/min).25,26,27 The important variability could be related to differences in sulfoconjugation.28
Our mean observed weight-normalised clearance is consistent with the model recently developed by Hallik et al. in neonates.29 Indeed, when incorporating the mean gestational age and body weight of our patients within their equation for dobutamine clearance calculation, the obtained calculated value of 8.67 l/h/kg is very close to our mean observed value of 9.43 l/h/kg.
Of note, in our study, all four samples drawn 120 min or more after the first samples corresponded to concentrations less than the LLOQ. A possible explanation is that the elimination half-life in these four patients was below 24 min (120/5). We believe the data obtained from the infants with the longest time intervals cannot be considered as useless. Indeed, they confirm that dobutamine half-life is likely <24 min in most infants, which is consistent with the half-lives that could be measured in the elder population. This should be taken into account when defining the time window of further pharmacokinetic samples in future trials.
In light of these and our own results, it can be anticipated that the dobutamine plasma concentration obtained in a given neonate with a given infusion rate cannot be predicted. Despite first-order kinetics indicating non-saturability of the elimination mechanisms within the dose range studied, inter-individual differences in dobutamine pharmacokinetics are the reason that predictable pharmacodynamic responses to dobutamine in the individual patient are not guaranteed.
The new, age-appropriate drug formulation for dobutamine used in this study involved the development and preparation of sterile dobutamine hydrochloride solution for dilution for infusion with a focus on keeping the formulation as simple as possible without introducing additional, potentially unnecessary excipients, containing half the quantity of anti-oxidant (sodium metabisulfite) compared to other marketed formulations. The new formulation has been used in preclinical studies by members of the consortium.30 In this study, six serious adverse events occurred during the trial in five infants, and two infants died. The adverse events were not considered to be related to the study drug.
In summary, the wide time range observed for reaching the plasma concentration plateau corresponding to steady state, among the preterm infant population during transitional circulation, suggests a pragmatic approach for conducting PK/PD studies embedded within clinical efficacy trials. Based on our calculated range of dobutamine half-lives, a maximum delay to reach steady state of 180 min can be expected. A delay of 3 h, however, is not compatible with the standards of care, and maintaining it would eventually lead to the frequent use of rescue therapy which would preclude the evaluation of dobutamine´s efficacy. So far, the delay until a dose increase should be based on clinical data. Attending physicians should be aware about clinical signs indicating that faster up-titration is needed, or eventual signs of overtreatment. However, a delay of at least 2−3 h should be respected before drawing the blood sample for the efficient dose (if any) in order to have almost all children at steady state for the second blood sample (first sample being drawn at the pharmacodynamics evaluation of the first dose). Finally, a facultative third blood sample could be drawn 15–30 min after the end of the last infusion in order to have an estimation of the elimination half-life in the population.
References
Ruggieri, L. et al. Successful private-public funding of paediatric medicines research: lessons from the EU programme to fund research into off-patent medicines. Eur. J. Pediatr. 174, 481–491 (2015).
Batton, B. et al. Blood pressure, anti-hypotensive therapy, and neurodevelopment in extremely preterm infants. J. Pediatr. 154, 351–357 (2009).
Faust, K. et al. Short-term outcome of very-low birth weight infants with arterial hypotension in the first 24 h of life. Arch. Dis. Child Fetal Neonatal Ed. 100, F388–F392 (2015).
Kluckow, M. & Evans, N. Low superior vena cava flow and intraventricular haemorrhage in preterm infants. Arch. Dis. Child Fetal Neonatal Ed. 82, F188–F194 (2000).
Kluckow, M. Low systemic blood flow and pathophysiology of the preterm transitional circulation. Early Hum. Dev. 81, 429–437 (2005).
Osborn, D. A. et al. Low superior vena cava flow and effect of inotropes on neurodevelopment to 3 years in preterm infants. Pediatrics 120, 372–380 (2007).
Kluckow, M. & Seri, I. in Hemodynamics and Cardiology. Neonatology Questions and Controversies, 2nd edn. (eds Kleinman, C. S. & Seri, I.) 237−267 (Elsevier Saunders, Philadelphia, 2012).
Osborn, D., Evans, N. & Kluckow, M. Randomized trial of dobutamine versus dopamine in preterm infants with low systemic blood flow. J. Pediatr. 140, 183–191 (2002).
Bravo, M. C. et al. Randomized, placebo-controlled trial of dobutamine for low superior vena cava flow in infants. J. Pediatr. 167, 572–578 (2015).
Roze, J. C. et al. Response to dobutamine and dopamine in the hypotensive very preterm infant. Arch. Dis. Child 69(1 Spec No), 59–63 (1993).
Hentschel, R. et al. Impact on blood pressure and intestinal perfusion of dobutamine or dopamine in hypotensive preterm infants. Biol. Neonate 68, 318–324 (1995).
Ruffolo, R. R. Jr. The pharmacology of dobutamine. Am. J. Med. Sci. 294, 244–248 (1987).
European Medicines Agency. Committee for medicinal products for human use. Reflection paper: formulations of choice for the paediatric population. EMEA/CHMP/PEG/194810/2005.
European Medicines Agency. Opinion of the Paediatric Committee on the acceptance of a modification of an agreed Paediatric Investigation Plan. EMEA-001262-PIP01-12-M02. London, 14 October 2016.
Rowland M., Toze T. N. Clinical Pharmacokinetics: Concepts and Applications, 3rd edn, 83−105 (Williams and Wilkins, Philadelphia, 1995).
European Medicines Agency. Guideline on bioanalytical method validation. EMEA/CHMP/EWP/192217/2009. Rev.1 Corr.2.
European Medicines Agency. Guideline on Pharmacokinetic studies in man. https://www.ema.europa.eu/en/documents/scientific-guideline/pharmacokinetic-studies-man_en.pdf.
Devries, L. S., Eken, P. & Dubowitz, L. M. S. The spectrum of leukomalacia using cranial ultrasound. Behav. Brain Res. 49, 1–6 (1992).
Levene, M. I. Measurement of the growth of the lateral ventricles in preterm infants with real-time ultrasound. Arch. Dis. Child. 56, 900–904 (1981).
Pellicer, A. et al. Cardiovascular support for low birth weight infants and cerebral hemodynamics: a randomized, blinded, clinical trial. Pediatrics 115, 1501–1512 (2005).
Myohanen, T. T. & Mannisto, P. T. Distribution and functions of catechol-O-methyltransferase proteins: do recent findings change the picture? Int. Rev. Neurobiol. 95, 29–47 (2010).
Martinez, A. M., Padbury, J. F. & Thio, S. Dobutamine pharmacokinetics and cardiovascular responses in critically ill neonates. Pediatrics 89, 47–51 (1992).
Banner, W. Jr, Vernon, D. D., Minton, S. D. & Dean, J. M. Nonlinear dobutamine pharmacokinetics in a pediatric population. Crit. Care Med. 19, 871–873 (1991).
Schwartz, P. H., Eldadah, M. K. & Newth, C. J. The pharmacokinetics of dobutamine in pediatric intensice care unit patients. Drug Metab. Dispos. 19, 614–619 (1991).
Habib, D. M. et al. Dobutamine pharmacokinetics and pharmacodynamics in pediatric intensive care patients. Crit. Care Med. 20, 601–608 (1992).
Berg, R. A. et al. Dobutamine pharmacokinetics and pharmacodynamics in normal children and adolescents. J. Pharm. Exp. Ther. 265, 1232–1238 (1993).
Berg, R. A., Donnerstein, R. L. & Padbury, J. F. Dobutamine infusions in stable, critically ill children: pharmacokinetics and hemodynamic actions. Crit. Care Med. 21, 678–686 (1993).
Berg, R. A. & Padbury, J. F. Sulfoconjugation and renal excretion contribute to the interpatient variation of exogenous catecholamine clearance in critically ill children. Crit. Care Med. 25, 1247–1251 (1997).
Hallik, M. et al. Population pharmacokinetics and pharmacodynamics of dobutamine in neonates on the first days of life. Br. J. Clin. Pharm. 86, 318–328 (2020).
Mielgo, V. E. et al. Hemodynamic and metabolic effects of a new pediatric dobutamine formulation in hypoxic newborn pigs. Pediatr. Res. 81, 511–518 (2017).
Richardson, D. K., Corcoran, J. D., Escobar, G. J. & Lee, S. K., for The Canadian NICU Network, The Kaiser Permanente Neonatal Minimum Data Set Wide Area Network, and The SNAP-II Study Group. SNAP-II and SNAPPE-II: simplified newborn illness severity and mortality risk scores. J. Pediatr. 138, 92–100 (2001).
Acknowledgements
The authors thank all participants, parents, and healthcare providers. This work was supported by the European Union Frame Programme Seventh grant number FP7: HEALTH-2011.4.2-1 [Investigator-driven clinical trials on off-patent medicines for children].
Author information
Authors and Affiliations
Consortia
Contributions
A.P., V.J., F.C., and H.R. conceptualised and designed the study; C.G., H.R.-A., A.K. and A.S. supported the study design; A.P., R.F., M.C.B., P.L.O., L.S., and M.Y. assisted with acquisition of the data; A.K. and A.S. analysed the data; A.P. wrote the initial draft; A.P., V.J., C.G., H.R.-A., A.K., A.S. and H.R. provided administrative, technical, and material support. All authors critically reviewed the revised versions of the paper and approved the final draft for submission. All authors had access to the data and contributed to the submitted report. A.P. is the guarantor.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
Ethics approval for this study was obtained from the Ethics Committee for Clinical Research at La Paz University Hospital (HULP 3969) and the NRES Committee South East Coast-Brighton and Sussex (13/LO/1426).
Patient consent
Patient consent was obtained for this study.
Transparency
A.P. affirms that the manuscript is honest, accurate, and transparent account of the study being reported, that no important aspects have been omitted, and that any discrepancies from the study as planned have been explained.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pellicer, A., Fernández, R., Jullien, V. et al. Pharmacokinetic study (phase I−II) of a new dobutamine formulation in preterm infants immediately after birth. Pediatr Res 89, 981–986 (2021). https://doi.org/10.1038/s41390-020-1009-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-1009-0
