
Abstract
Congenital diaphragmatic hernia (CDH)-related deaths are the largest contributor to in-hospital neonatal deaths in children with congenital malformations. Morbidity and mortality in CDH are directly related to the development of pulmonary hypertension (PH). Current treatment consists of supportive measures. To date, no pharmacotherapy has been shown to effectively reverse the hallmark finding of pulmonary vascular remodeling that is associated with pulmonary hypertension in CDH (CDH-PH). As such, there is a great need for novel therapies to effectively manage CDH-PH. Our review aims to evaluate emerging therapies, and specifically focuses on those that are still under investigation and not approved for clinical use by the Food and Drug Administration. Therapies were categorized into antenatal pharmacotherapies or antenatal regenerative therapies and assessed on their method of administration, safety profile, the effect on pulmonary vascular pathophysiology, and overall efficacy. In general, emerging antenatal pharmaceutical and regenerative treatments primarily aim to alleviate pulmonary vascular remodeling by restoring normal function and levels of key regulatory factors involved in pulmonary vascular development and/or in promoting angiogenesis. Overall, while these emerging therapies show great promise for the management of CDH-PH, most require further assessment of safety and efficacy in preclinical models before translation into the clinical setting.
Impact
-
Emerging antenatal therapies for congenital diaphragmatic hernia-induced pulmonary hypertension (CDH-PH) show promise to effectively mitigate vascular remodeling in preclinical models. Further investigation is needed in preclinical and human studies to evaluate safety and efficacy prior to translation into the clinical arena.
-
This review offers a comprehensive and up-to-date summary of emerging therapies currently under investigation in experimental animal models.
-
There is no cure for CDH-PH. This review explores emerging therapeutic options for the treatment of CDH-PH and evaluates their impact on key molecular pathways and clinical markers of disease to determine efficacy in the preclinical stage.
Similar content being viewed by others

Introduction
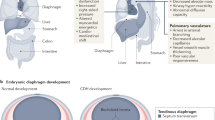
Congenital diaphragmatic hernia (CDH) is a highly morbid birth defect that occurs in ~1 in 3000 live births.1 Despite advancements, the etiology remains unclear in over 50% of patients. CDH consists of a diaphragmatic defect, dextrocardia, lung hypoplasia, and often, pulmonary hypertension (PH) (Fig. 1a). In gestation the diaphragm fails to develop properly, allowing abdominal organs to herniate into the chest cavity (Fig. 1b) and impede normal lung development; furthermore, lung hypoplasia (Fig. 1d) and compromised pulmonary vascular development (Fig. 2) lead to PH, resulting in respiratory failure, right heart failure, and even death.2 As such, pulmonary hypertension in CDH (CDH-PH) is a major determinant of disease-related morbidity and mortality.2,3,4 CDH-related deaths account for the highest number of in-hospital neonatal deaths in infants with congenital malformations,5 with a striking difference in survival rates between CDH infants with and without PH (20% vs. 70%).6 Management of the disease is expensive, with estimated costs ranging from $250–800 million every year.7,8

a Chest X-ray of an infant with congenital diaphragmatic hernia. Intraoperative image of the diaphragmatic defect with b herniated bowel and c subsequent reduction back into the abdomen. d Gross image of severe ipsilateral lung hypoplasia. Images reprinted from Tovar et al.127 under the terms of the Creative Commons CC BY license.

a Masson’s Trichrome staining demonstrating increased wall muscularization in pulmonary arterioles. b Immunofluorescence staining for smooth muscle-alpha actin (green) demonstrates increased pulmonary arterial smooth muscle cell proliferation. Images acquired at ×40.
Current treatment consists of supportive measures such as gentle mechanical ventilation, vasodilators, supplemental oxygen, and extracorporeal life support, which aim to minimize cardiopulmonary symptoms. No available pharmacotherapy effectively reverses the vascular remodeling process that is responsible for the structural vascular alterations observed in CDH-PH.2 Not surprisingly, no available therapy significantly improves CDH morbidity and mortality related to PH.2,9 Given the considerable impact this disease poses on our neonatal population, a novel therapy is clearly necessary. Our review aims to evaluate emerging therapies that are still under investigation and not approved for clinical use by the Food and Drug Administration.
Pathophysiology
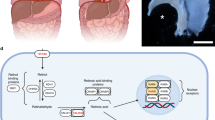
CDH-PH pathogenesis is still not fully understood. A two-hit hypothesis10 proposes that an initial embryological insult disrupts lung development, followed by external compression of the ipsilateral lung from the herniated abdominal organs. This suggests that both intrinsic genetic and molecular defects, as well as extrinsic mechanical forces, are responsible for CDH-PH pathology and the clinical cardiopulmonary changes that occur after birth. CDH infants develop a reduced number of pulmonary vessels, and their remaining vessels are impaired by vascular remodeling processes that induce medial wall thickening and distal muscularization (Fig. 3). An imbalance in vasodilatory and vasoconstrictive mediators further impedes signaling between dysfunctional endothelial and pulmonary arterial smooth muscle cells, resulting in increased pulmonary vascular resistance and elevated pulmonary arterial pressures consistent with PH. Evidence demonstrates that disruptions to key regulatory pathways involved in normal vascular development are associated with CDH-PH pathogenesis.

Reprinted by permission from Springer Nature Customer Service Centre GmbH: Nature. Montalva et al. COPYRIGHT (2019).15
Antenatal CDH-PH pharmacotherapies
To date, all CDH-PH pharmacotherapies are delivered postnatally and only address pulmonary artery vasoconstriction as part of supportive management. Some theorize that interventions are administered too late, as the vascular remodeling begins in utero.11,12 An antenatal approach that addresses vascular changes early in development may mitigate disease before it progresses. Importantly, since lung development in humans continues through at least the second year of life, and per some authors through seven years of life, postnatal interventions are still necessary to achieve satisfactory outcomes. Furthermore, prenatal therapies present an ethical dilemma due to the possibility of unintended side effects for both the mother and the fetus, thus potentially harming two lives instead of just the targeted patient. For this reason, postnatal interventions must continue to be explored, and antenatal therapies appropriately evaluated for maternal risk. Here, we discuss only antenatal therapies (Table 1), and evaluate their effects on molecular pathways.
Nitrofen-induced CDH rodents are the most commonly used animal model to investigate antenatal therapies, as they develop surprisingly similar pathology compared to CDH infants. Much like humans, a portion of the litter will have large diaphragmatic defects containing herniated abdominal organs and have associated bilateral lung hypoplasia.13 Histopathological analysis reveals arterial wall thickening, a hypoplastic vascular bed, and reduced airway branching consistent with human CDH pathophysiology14 (Fig. 4).

Scale bars: 100 µm. Reprinted by permission from Springer Nature Customer Service Centre GmbH: Springer. Makanga et al. COPYRIGHT (2013).128
Therapies targeting NO-cGMP pathways
Nitric oxide (NO) is key for endothelial cell function and vascular regulation. It is produced by endothelial nitric oxide synthase (eNOS) in pulmonary endothelial cells and activates soluble guanylate cyclase to release cyclic guanosine monophosphate (cGMP) within the vascular smooth muscle cells. Activation of cGMP-dependent pathways increases vasodilation, anti-thrombotic activity, and cellular proliferation.2,15 Endothelial-derived NO production increases in response to signaling from vascular endothelial growth factor (VEGF), an angiogenic factor that promotes endothelial cell migration and proliferation,15 and stimulates angiogenesis.16,17 Studies show both increased18 and decreased19,20,21,22 VEGF expression in nitrofen-induced rat lungs. In contrast, phosphodiesterase enzymes (PDE) regulate NO pathway activity via cGMP inactivation.
Animal and human CDH studies report conflicting results regarding molecular changes within the NO pathway.15 Some nitrofen-induced CDH rat models show decreased pulmonary levels of NO23 and eNOS,21,23,24,25 while others show increased eNOS levels.26,27 Similarly, human CDH studies demonstrate decreased28 and increased29 NOS levels in pulmonary vessels. In nitrofen-exposed fetal rat lungs, the pathway is further impaired by increased PDE expression and diminished reactivity to NO and cGMP stimuli.30 This dampened cellular response may be attributed to prolonged NO exposure, as may occur in the setting of increased eNOS or inducible nitric oxide synthase (iNOS) expression because this has been shown to reduce both soluble guanylate cyclase activity and cGMP levels in pulmonary artery smooth muscle cells.31 These findings suggest that pathological downregulation of the NO-cGMP pathway contributes to CDH-PH.
Phosphodiesterase inhibitors
Sildenafil
Sildenafil is a phosphodiesterase type 5 (PDE5) inhibitor used for adult and pediatric PH,32,33,34,35 including CDH-PH, and particularly for NO-refractory disease.26,36,37,38 PDE5 inhibitors improve pulmonary vasodilation, oxygenation,37 cardiac output, and reduce pulmonary vascular resistance in PH patients.22,39 Antenatal administration was first explored in nitrofen-induced CDH rats. The treatment arm received subcutaneous sildenafil from embryonic day (E)11.5 until E20.5.22 Fetuses were delivered at term. Results showed increased pulmonary angiogenesis, improved vasoreactivity, and decreased right ventricular hypertrophy following sildenafil administration. Histology confirmed no adverse visual effects or brain impairment. Comparable results showing attenuated vascular remodeling with reduced arterial muscularization23,26,40,41 and increased vessel density23,42 were reproduced in other nitrofen-induced rat models, and in a surgical diaphragmatic hernia rabbit model.43 Sildenafil-treated rats had increased levels of eNOS,22,23 iNOS,23 and VEGF, with reductions in active PDE5.22 Although VEGF levels were not upregulated in treated rabbits, enhanced vasorelaxation in response to VEGF stimulation was significantly increased.43 This suggests that sildenafil restores pulmonary vasculature and lung morphology via normalization of NO-cGMP-dependent pathways. Interestingly, sildenafil had a negative effect on pulmonary vascular development in healthy control fetal rats26,40 and rabbits.43
In 2016, the first human study was initiated. The SToP PH Trial (EU Clinical Trials Register (2016–002619–17)) is a randomized controlled trial, investigator-blinded, double-armed, parallel-group, phase I/IIb study designed to determine the safety profile of transplacental sildenafil delivery.44 The second arm of the study involving pregnant women with isolated CDH fetuses is currently on hold after significant treatment-related mortality was reported in a recent multicenter, international randomized controlled trial. The Dutch-STRIDER (NCT02277132) trial used antenatal sildenafil to treat early-onset intrauterine fetal growth restriction but was prematurely discontinued in 2018 following high mortality rates in the sildenafil group, including 11 newborn deaths.45 Previous human studies investigating antenatal sildenafil to manage pediatric PH,46 preeclampsia,47 and intrauterine fetal growth restriction48,49 found no severe maternal or fetal adverse events; however, when given at high prenatal doses, an increased risk of fetal toxicity and growth suppression was reported in fetal mice.50 These worrisome findings warrant careful consideration prior to clinical advancement.
Tadalafil
Tadalafil is a PDE inhibitor with greater PDE5 specificity and longer half-life than sildenafil.38,51 Although predominantly used to treat adult PH, tadalafil has been used off-label for pediatric PH.34,52 A surgical lamb model was used to investigate tadalafil for CDH-PH. Pregnant ewes underwent surgical diaphragmatic hernia creation at gestational day 75,38 and then received oral tadalafil postoperatively for up to 7 days. Treated fetal lambs had increased pulmonary vasodilation with improved pulmonary blood flow, and increased cGMP and eNOS levels. The upregulation of NO-cGMP pathway proteins likely contributed to tadalafil’s ability to ameliorate vascular remodeling in utero. Of interest, tadalafil did not significantly improve oxygenation, pulmonary arterial pressures, or right ventricular hypertrophy. The lack of meaningful clinical changes may be due to the insufficient duration of treatment, which must be determined in further studies. No evidence of intrauterine fetal growth restriction or adverse fetal effects were observed. No human CDH studies exist, but positive results in animals and other forms of PH support conducting a trial in CDH patients.34,52
Phosphodiesterase type 3 (PDE3) has also been associated with pulmonary hypertension. Milrinone is a PDE3 inhibitor that is used in the neonatal population to treat persistent PH.53 Currently, there is an ongoing trial (NCT02951130) investigating the use of postnatal milrinone in CDH infants, which is planned to be completed in February 2021. While antenatal administration has yet to be explored, results from this study may support future investigations.
Soluble guanylate cyclase agonists
BAY 41–2272
BAY 41–2272 is a synthetic soluble guanylate cyclase stimulator that directly enhances receptor activity to promote cGMP-mediated vasodilation and anti-thrombotic activity, as well as reduced right ventricular systolic pressure and vascular remodeling in experimental PH models.54,55,56 In a fetal sheep model of the persistent PH of the newborn, BAY 41–2272 improved pulmonary vasodilation and reduced pulmonary vascular resistance.57
Rabbits with surgical diaphragmatic hernias received a single dose of BAY 41–2272 via tracheal instillation on E28,58 and were retrieved at term. Results showed reduced right ventricular pressure and ameliorated vascular remodeling with decreased medial thickening in small arteries and increased capillary formation. Interestingly, in normal pathology, angiogenesis is predominantly regulated by VEGF-mediated activation of the NO-cGMP pathway. However, in this study, treated lungs had increased endothelial cell proliferation and vessel density with increased eNOS expression, but lacked VEGF overexpression. This suggests that BAY 41–2272’s angiogenic effects are mediated via a VEGF-independent pathway, consistent with prior in vitro findings,59 and may present a novel therapeutic pathway. While larger studies are necessary, BAY 41–2272 seems promising with no evidence of maternal or fetal adverse effects.
BAY 60–2770
A soluble guanylate cyclase activator, BAY 60–2770, increases pulmonary vasodilation and reduces pulmonary and systemic arterial pressures in monocrotaline (MCT)-induced PH rats.60 To assess BAY 60–2770 in CDH-PH, neonatal pulmonary arteries were excised from fetal rabbits with surgical diaphragmatic hernias and studied ex vivo.61 Pulmonary arteries were harvested and bathed in phenylephrine solution to keep the vessels contracted. BAY 60–2770, tadalafil, and the NO donor sodium nitroprusside were compared to determine effects on vasodilation. BAY 60–2770 induced potent vasorelaxation in the CDH group in a concentration-dependent manner, while tadalafil had no significant effect. In vitro analysis demonstrated increased levels of NO metabolites in the CDH group versus controls. The CDH group also had decreased arterial relaxation in response to sodium nitroprusside compared to controls, suggesting that reductions in soluble guanylate cyclase bioavailability and cGMP production may contribute to CDH pathology.
Therapies targeting cAMP pathways
The adenylate cyclase enzyme in pulmonary artery smooth muscle cells stimulates the production of cyclic adenosine monophosphate (cAMP). This promotes vasodilation, anti-platelet aggregation, inhibition of inflammatory mediators, and regulation of smooth muscle cell proliferation and vascular development. Prostaglandins are vasodilatory compounds whose effects are mediated by the adenylate cyclase/cAMP pathway.62,63 Within the prostaglandin family, prostacyclin is a particularly potent vasodilator that exhibits anti-inflammatory and anti-thrombotic properties. Patients with CDH-PH have decreased expression of the prostacyclin receptor,64 and small studies show that administration of prostaglandins improves symptoms, cardiac markers, echocardiography, and cardiac catheterization measurements which are all findings consistent with improved PH.65,66 However, prostacyclin’s efficacy is limited by a short half-life that requires continuous administration to achieve therapeutic levels.67 This creates substantial challenges as the risk associated with prolonged vascular access is significant. To address this limitation, synthetic prostacyclin analogs have been engineered and demonstrated variable success in PH.
Prostacyclin agonists
Selexipag
Selexipag (NS-304) and its active compound are oral, long-acting, selective prostacyclin receptor agonists.68 In adults with PH, selexipag reduced PH-related complications and death.69 Similarly, MCT-induced PH rats treated with selexipag demonstrated reduced right ventricular hypertrophy, arterial wall thickening, and improved survival.68 Mous et al.26 were the first to study the targeting of dual vasodilatory pathways using selexipag and sildenafil in nitrofen-induced CDH rats. Treatment cohorts were divided into three groups: selexipag, sildenafil, or a combination of both therapies. Therapies were delivered daily via gastric lavage from E17.4 to E20.5, and fetuses were delivered at term. Isolated and/or combined therapy with selexipag and sildenafil decreased arterial wall thickening, smooth muscle cell proliferation, and right ventricular hypertrophy. Combined therapy also restored prostacyclin receptor expression to near-control levels. However, the combination of the two drugs did not exhibit any added therapeutic benefit, likely due to their competing hepatic metabolism. Administration of selexipag alone reduced PDE3 expression and the downstream target of PDE5, protein kinase G2, but did not impact eNOS. Thus, improvements in vascular remodeling may be partly due to the normalization of key receptors within the prostacyclin pathway. No evidence of maternal or fetal adverse effects was identified. Notable, however, was an unexpected thickening of the medial layer in all treated control groups.
ONO-1301SR
ONO-1301 is a long-acting, synthetic prostacyclin agonist. In MCT-induced PH rats, serial ONO-1301 decreased pulmonary arterial wall thickening and mitigated right ventricular systolic pressure elevation.70 To improve drug delivery, a slow-release formulation (ONO-1301SR) was created and showed comparable effectiveness.19,71 Umeda et al.19 administered ONO-1301SR to nitrofen-exposed rats via a single, subcutaneous injection on E9.5. Fetuses were retrieved at term. Results demonstrated positive effects on pulmonary hypoplasia, which we will not discuss here, as well as improved arterial remodeling evidenced by decreased arterial wall thickness and increased capillary formation. Molecular studies found increased gene expression of VEGF, hepatocyte growth factor, and stromal cell-derived factor in the treated lungs. Protein levels of VEGF were also normalized with ONO-1301SR. Thus, the upregulation of important growth factors likely promotes pro-angiogenic pathways to mitigate vascular remodeling. The authors also hypothesize that arterial wall thinning, as observed in the treatment group, may be due to modifications to the prostacyclin pathway versus an indirect result of the drug’s anti-thrombotic properties. The exact mechanism remains unclear. Safety analysis following single and serial doses demonstrated no significant adverse effects; however, since antenatal ONO-1301SR is given early during organogenesis and is known to target multiple organs, evaluation of off-target toxicity is necessary. This study performed a very limited assessment of accessory organs.
Therapies targeting the endothelin pathway
Endothelin-1 (ET-1) is the primary ligand of the endothelin pathway and is a strong vasoconstrictor produced by the endothelin converting enzyme.72 ET-1 binds to two receptors to regulate vascular tone: endothelin receptor type A (ETA) induces vasoconstriction by promoting the release of cytosolic calcium within pulmonary artery smooth muscle cells, and endothelin receptor type B (ETB) primarily promotes vasodilation via upregulation of NO and prostacyclin.72 In nitrofen-induced pups, upregulation of ET-1 and both ET receptors is observed,23,64,73,74 along with a heightened vasoconstrictive response to ET-1. CDH infants have elevated levels of ET-1 in plasma and lungs,75 and exhibit significant ET-1 receptor dysfunction with a more pronounced increase in ETA compared to ETB.64 The imbalance in ETA and ETB expression is primarily responsible for the shift in vascular tone towards persistent vasoconstriction.
Endothelin receptor antagonists
Bosentan
Bosentan is a dual ETA and ETB antagonist that increases vasodilation, reduces pulmonary vascular resistance, and has been shown to successfully treat newborn PH.76 To explore its role in CDH-PH, nitrofen-exposed rats were divided into three treatment groups: oral sildenafil, oral bosentan, or combined therapies. Treatments were administered via gastric lavage from E16 to E20.41 Animals were harvested on E21. Bosentan alone did not improve vascular remodeling; however, CDH rats that received sildenafil or combined therapies demonstrated a significant reduction in medial wall thickening indicating attenuated vascular cellular proliferation. These data suggest that sildenafil is predominantly responsible for the changes observed in vascular remodeling.
Therapies targeting the tyrosine kinase pathway
Receptor tyrosine kinases, such as platelet-derived growth factor (PDGF) receptors, have been strongly associated with pulmonary vascular remodeling.15 PDGF ligands and their associated receptors induce smooth muscle cell proliferation and migration and regulate angiogenesis in experimental animal models.77 Elevated lung PDGF levels in PH patients indicate its likely involvement in the development of human disease.78
Tyrosine kinase receptor inhibitors
Imatinib
Imatinib is a selective inhibitor of c-Kit and BCR-ABL tyrosine kinase receptors,79 including the PDGF receptor. Animal models and case reports of adults with severe PH demonstrate that imatinib effectively prevents and/or reduces pulmonary vascular pathophysiology.80,81 A randomized controlled trial in adults with medically refractory PH found decreased pulmonary vascular resistance and increased cardiac output in imatinib-treated patients.82 Similarly, a case report of a CDH neonate with intractable PH showed decreased pulmonary arterial pressures and clinical improvement following imatinib administration.83
Nitrofen-exposed rats received oral imatinib via gastric lavage from E17 to E20.84 Fetuses were retrieved at term. Treated CDH lungs had improved vascular remodeling with a reduction in medial arterial wall thickness, number of fully muscularized arteries, vascular cell proliferation, and restoration of the luminal area. Molecular analysis showed a trend toward the downregulation of the PDGF-β ligand and its receptors, which normally promote smooth muscle cell proliferation,85 apoptosis, and vessel maturation.86 Contrarily, when this experiment was repeated by Burgos et al.,87 no significant therapeutic effect was observed in the treated group. This discrepancy may be attributable to different measurement criteria between the groups, but raises concerns about therapeutic efficacy.
Improvement in vascular remodeling in the treated rats likely corresponds to the inhibition of vascular cell proliferation via PDGF downregulation and induction of apoptosis in apoptotic-resistant smooth muscle cells.77,84 Further studies are needed to understand the mechanism. Of concern is imatinib’s known teratogenic effects at high doses, which were required to ameliorate vascular remodeling in preclinical studies.77 Although low doses in the nitrofen-exposed rats were largely safe, unanticipated arterial wall thinning and an increased number of muscularized vessels were found in the treated controls.84 Such abnormal findings within the therapeutic range are worrisome and mandate evaluation. Of interest, two other receptor tyrosine kinase inhibitors, nilotinib and dasatinib, showed positive in vivo results in this study. However, case reports of adults treated with dasatinib for chronic myeloid leukemia reported severe PH as a notable adverse effect.88,89
Therapies targeting pro-inflammatory pathways
Peroxisome proliferator-activated receptor-gamma (PPAR-γ) is a ligand-activated transcription factor involved in angiogenesis and pulmonary artery smooth muscle cell proliferation.90 PPAR-γ regulates inflammatory processes that induce vascular remodeling in many types of human and experimental PH models.91,92 It inhibits expression of important inflammatory mediators, including monocyte chemoattractant protein-1 (MCP-1)91,92,93,94 and interleukin 6 (IL-6).93,94 MCP-1 promotes monocyte perivascular infiltration, endothelial cell dysfunction, and smooth muscle cell proliferation.95,96,97,98,99 IL-6 is a pro-inflammatory cytokine highly associated with PH pathogenesis. Additionally, IL-6 is also activated by an upstream inflammatory cytokine known as the macrophage migration inhibitory factor, which further propagates the development of a chronic inflammatory immune response.100 Patients with PH have elevated plasma IL-698,101 and MCP-1102 levels, and in cases of severe disease, reduced PPAR-γ.103 In CDH-PH, human and experimental models show increased pulmonary vascular levels of inflammatory markers,101,104 including MCP-1,96 while simultaneously demonstrating decreased levels of PPAR-γ.105 Thus, a heightened inflammatory state induces abnormal smooth muscle cell function and proliferation that contributes to vascular remodeling in CDH-PH.
PPARY-γ agonists
Rosiglitazone
Gosemann et al.97 investigated rosiglitazone, a PPAR-γ agonist, in a nitrofen-induced CDH rat model. Rats received daily intraperitoneal injections for 2 days (E18 to E19). Treated CDH fetuses harvested at term showed reduced arterial wall thickening, MCP-1 protein expression, and monocyte perivascular infiltration. The reduction in MCP-1 suggests that rosiglitazone’s effect occurs via stimulation of the PPAR-γ pathway. However, the efficacy of MCP-1 in CDH-PH requires greater evaluation. The therapeutic safety profile of rosiglitazone was not assessed in this study.
Macrophage migration inhibitory factor inhibitors
ISO-92
ISO-92, a synthetic macrophage migration inhibitory factor inhibitor, was tested in nitrofen-induced rats.9 An osmotic device was implanted within the subcutaneous tissue of pregnant rats at E10 or E11, and continuously administered ISO-92 until term delivery. ISO-92 mitigated migration inhibitory factor activity but did not alter its expression or secretion. Treated mice had reduced medial wall thickness and lower right ventricular systolic pressure, suggestive of improved arterial remodeling and cardiovascular physiology, which are both essential to PH treatment. If applied clinically, failure to alter migration inhibitory factor expression may prove to be advantageous; since migration inhibitory factor is physiologically required for the newborn innate immune response, it is rational to suspect that downregulation of the cytokine may be potentially harmful. One downside with ISO-92 is that it is not widely available for use by other investigators or clinicians. Another obstacle is the drug’s short half-life; thus, an invasive pump was required to continuously deliver therapy to the rodents. In the future, ISO-92 will need to be tested in a survival model to assess safety markers.
HMG-CoA reductase inhibitors
Simvastatin
Inhibitors of 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase, known as statins, support endothelial cell function and promote vasorelaxation through upregulation and activation of eNOS, activation of phosphoinositide 3-kinase/Akt pathway,106 and inhibition of Rho GTPases.107,108 Rho GTPases induce vasoconstriction in response to ET-1 stimulation.109 When used to treat patients with atherosclerotic disease, statins promote anti-inflammatory, anti-proliferative, and immunosuppressive properties that improve cardiovascular outcomes. In experimental PH models, simvastatin reduced neointimal hyperplasia, pulmonary arterial pressures, right ventricular hypertrophy, and effectively reversed PH.110,111
To investigate simvastatin in CDH-PH, nitrofen-exposed rats were randomized to three oral treatment arms: simvastatin, sildenafil, or placebo for 10 days (E11 to E21),23 and then harvested fetuses at term. Simvastatin-treated CDH fetuses had decreased medial wall thickness in resistance level pulmonary arteries and a trend toward increased vascular density. Modifications in the endothelin pathway involved reduced gene expression of the ET-1 precursor, preproendothelin-1, and decreased ET-1 protein levels in treated lungs compared to the nitrofen-only group; furthermore, ETA gene expression was restored to control levels. Simvastatin-treated groups demonstrated a trend towards normalized iNOS and eNOS gene expression and higher NO lung levels compared to nitrofen-only groups. Additionally, simvastatin restored pro-apoptotic mechanisms evidenced by an increased ratio of Bax to Bcl-2. These findings suggest that simvastatin improves vascular remodeling by modulating the endothelin pathway, normalizing eNOS function and NO bioavailability, and promoting normal levels of smooth muscle cell apoptosis. The cumulative effect of restoring these pathways may help re-establish vasoreactivity and normal function and phenotype of pulmonary artery smooth muscle cells. Notably, simvastatin-treated CDH rats had reduced body weight potentially signifying intrauterine fetal growth restriction, but morbidity related to growth retardation was not evaluated. Although human studies have shown safe administration in pregnancy,112 these findings warrant further investigation.
Antenatal regenerative therapies for CDH-PH
Arrested lung and vascular development leads to pulmonary hypoplasia and PH in CDH infants. Regenerative therapies that can reverse these processes and enhance pulmonary development are desirable for the management of CDH-PH. Mesenchymal stem cells (MSCs) have been investigated in many types of chronic lung diseases,113 and findings show that they inhibit inflammation, enhance immunity, and stimulate lung growth.113 In chronic hypoxia and MCT-induced PH experimental models, both intravascular and intra-tracheal MSC administration inhibited PH.114,115 Angiogenic MSCs also restored alveolar and vascular morphology and promoted lung development in experimental models of other neonatal pulmonary diseases.116
In CDH, MSCs have been primarily explored to address lung hypoplasia,117 but it is rational to suspect that they may also be effective in CDH-related PH, as airway and vascular development are closely intertwined.118 Promising results in experimental models support this theory. Yuniartha et al.119 administered lung tissue MSCs from donor adult rats to nitrofen-exposed rats via a single uterine vein injection and found that in addition to alleviating pulmonary hypoplasia, the treated CDH group also had reduced medial wall thickening compared to the nitrofen-only group. Similarly, Takayama et al.120 demonstrated decreased wall muscularization in nitrofen-exposed rats following intra-amniotic injection of human MSCs. An ex vivo study examined explanted nitrofen-exposed rat lungs, which were conditioned in amniotic fluid-derived MSC media, and found increased VEGF and fibroblast growth factor expression compared to controls.121 Though the exact mechanism is not fully understood, these findings suggest that therapeutic effects are likely due to MSC-mediated release of various paracrine factors including pro-angiogenic growth factors: VEGF, hepatocyte growth factor, fibroblast growth factor, and angiopoietin.117 Additional secretion of cytokines and chemoattractant factors may further induce regulatory mechanisms, which enhance vascular modifications. No significant maternal or fetal complications were reported in these studies. However, previous studies show that intravascular administration of MSCs is associated with immunodeficiency, microvascular embolism, and inflammation.122,123 To our knowledge, MSCs have never been explored to specifically address CDH-related PH, but given their success in experimental models, they certainly warrant further investigation.
Additionally, MSC-derived exosomes have also demonstrated promising findings when used to treat neonatal lung diseases such as bronchopulmonary dysplasia.124 There is also evidence to suggest that exosomes are effective in treating vascular remodeling in other models of PH.125,126 However, their application in CDH-specific PH has not been investigated. Given their previous success in both neonatal diseases and other forms of PH, exosomes present a likely suitable treatment approach that should be further explored.
Conclusion
Current therapies for CDH-PH are supportive and predominantly target vasodilation in the postnatal period. These interventions are likely too late as the disease is established in fetal development. Thus, the development of novel antenatal therapies that address pulmonary vascular remodeling before the disease progresses is logical. Amongst the emerging antenatal therapies discussed in this review, variable efficacy has been demonstrated in the attenuation of vascular remodeling. While many therapies show isolated therapeutic promise, no single agent effectively addresses the complex and multifactorial etiologies responsible for CDH-PH. The proposed antenatal therapies are largely non-specific, which poses an increased risk of systemic adverse effects both to the mother and fetus. Off-target organ injury in neonates could be particularly detrimental given the vulnerable period in which patients receive the therapy. Instead, the ideal therapeutic approach should use a drug delivery vehicle that specifically targets the diseased pulmonary vasculature and directly releases a therapeutic to the area of interest. Techniques such as regenerative stem cells or versatile nanoparticles, which are currently being investigated in other types of PH, should be strongly considered in the management of CDH-PH to further optimize antenatal options. Furthermore, a combination of synergistic therapies addressing the many molecular pathways involved in pulmonary vascular remodeling will likely be necessary to effectively mitigate pulmonary hypertension associated with congenital diaphragmatic hernia.
References
Dingeldein, M. Congenital diaphragmatic hernia: management & outcomes. Adv. Pediatr. 65, 241–247 (2018).
Harting, M. T. Congenital diaphragmatic hernia-associated pulmonary hypertension. Semin Pediatr. Surg. 26, 147–153 (2017).
Dillon, P. W. et al. The relationship of pulmonary artery pressure and survival in congenital diaphragmatic hernia. J. Pediatr. Surg. 39, 307–312 (2004).
Thebaud, B. & Tibboel, D. Pulmonary hypertension associated with congenital diaphragmatic hernia. Cardiol. Young-. 19, 49–53 (2009).
Centers for Disease Control. Prevention Hospital stays, hospital charges, and in-hospital deaths among infants with selected birth defects-United States, 2003. MMWR Morb. Mortal. Wkly Rep. 56, 25–29 (2007).
Coughlin, M. A. et al. Prenatally diagnosed severe CDH: mortality and morbidity remain high. J. Pediatr. Surg. 51, 1091–1095 (2016).
Raval, M. V., Wang, X., Reynolds, M. & Fischer, A. C. Costs of congenital diaphragmatic hernia repair in the United States-extracorporeal membrane oxygenation foots the bill. J. Pediatr. Surg. 46, 617–624 (2011).
Lally, K. P. Congenital diaphragmatic hernia - the past 25 (or so) years. J. Pediatr. Surg. 51, 695–698 (2016).
Perveen, S. et al. MIF inhibition enhances pulmonary angiogenesis and lung development in congenital diaphragmatic hernia. Pediatr. Res. 85, 711–718 (2019).
Keijzer, R. et al. Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am. J. Pathol. 156, 1299–1306 (2000).
Taira, Y., Yamataka, T., Miyazaki, E. & Puri, P. Comparison of the pulmonary vasculature in newborns and stillborns with congenital diaphragmatic hernia. Pediatr. Surg. Int. 14, 30–35 (1998).
Sluiter, I. et al. Premature differentiation of vascular smooth muscle cells in human congenital diaphragmatic hernia. Exp. Mol. Pathol. 94, 195–202 (2013).
Mortell, A., Montedonico, S. & Puri, P. Animal models in pediatric surgery. Pediatr. Surg. Int. 22, 111–128 (2006).
Montedonico, S., Nakazawa, N. & Puri, P. Congenital diaphragmatic hernia and retinoids: searching for an etiology. Pediatr. Surg. Int. 24, 755–761 (2008).
Montalva, L., Antounians, L. & Zani, A. Pulmonary hypertension secondary to congenital diaphragmatic hernia: factors and pathways involved in pulmonary vascular remodeling. Pediatr. Res. 85, 754–768 (2019).
Pierro, M. & Thebaud, B. Understanding and treating pulmonary hypertension in congenital diaphragmatic hernia. Semin. Fetal Neonatal Med. 19, 357–363 (2014).
Thébaud, B. et al. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: Evidence that angiogenesis participates in alveolarization. Circulation 112, 2477–2486 (2005).
Oue, T. et al. Increased vascular endothelial growth factor peptide and gene expression in hypoplastic lung in nitrofen induced congenital diaphragmatic hernia in rats. Pediatr. Surg. Int. 18, 221–226 (2002).
Umeda, S. et al. Enhanced pulmonary vascular and alveolar development via prenatal administration of a slow-release synthetic prostacyclin agonist in rat fetal lung hypoplasia. PLoS ONE 11, e0161334 (2016).
Hara, A., Chapin, C. J., Ertsey, R. & Kitterman, J. A. Changes in fetal lung distension alter expression of vascular endothelial growth factor and its isoforms in developing rat lung. Pediatr. Res. 58, 30–37 (2005).
Burgos, C. M. et al. Gene expression analysis in hypoplastic lungs in the nitrofen model of congenital diaphragmatic hernia. J. Pediatr. Surg. 45, 1445–1454 (2010).
Luong, C. et al. Antenatal sildenafil treatment attenuates pulmonary hypertension in experimental congenital diaphragmatic hernia. Circulation 123, 2120–2131 (2011).
Makanga, M. et al. Prevention of pulmonary hypoplasia and pulmonary vascular remodeling by antenatal simvastatin treatment in nitrofen-induced congenital diaphragmatic hernia. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L672–L682 (2015).
Zhaorigetu, S. et al. Perturbations in endothelial dysfunction-associated pathways in the nitrofen-induced congenital diaphragmatic hernia model. J. Vasc. Res. 55, 26–34 (2018).
Karamanoukian, H. L. et al. Decreased pulmonary nitric oxide synthase activity in the rat model of congenital diaphragmatic hernia. J. Pediatr. Surg. 31, 1016–1019 (1996).
Mous, D. S. et al. Treatment of rat congenital diaphragmatic hernia with sildenafil and NS-304, selexipag’s active compound, at the pseudoglandular stage improves lung vasculature. Am. J. Physiol. Lung Cell Mol. Physiol. 315, L276–L285 (2018).
Hofmann, A. et al. Imbalance of caveolin-1 and eNOS expression in the pulmonary vasculature of experimental diaphragmatic hernia. Birth Defects Res. B Dev. Reprod. Toxicol. 101, 341–346 (2014).
Shehata, S. M. K., Sharma, H. S., Mooi, W. J. & Tibboel, D. Pulmonary hypertension in human newborns with congenital diaphragmatic hernia is associated with decreased vascular expression of nitric-oxide synthase. Cell Biochem. Biophys. 44, 147–155 (2006).
Sood, B. G. et al. Expression of eNOS in the lungs of neonates with pulmonary hypertension. Exp. Mol. Pathol. 90, 9–12 (2011).
Vukcevic, Z., Coppola, C. P., Hults, C. & Gosche, J. R. Nitrovasodilator responses in pulmonary arterioles from rats with nitrofen-induced congenital diaphragmatic hernia. J. Pediatr. Surg. 40, 1706–1711 (2005).
Scott, W. S. & Nakayama, D. K. Sustained nitric oxide exposure decreases soluble guanylate cyclase mRNA and enzyme activity in pulmonary artery smooth muscle. J. Surg. Res. 79, 66–70 (1998).
Galie, N. et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 119, 2894–2903 (2009).
Mourani, P. M., Sontag, M. K., Ivy, D. D. & Abman, S. H. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J. Pediatr. 154, 379–384, 384 e371–372 (2009).
Cohen, J. L. et al. Sildenafil use in children with pulmonary hypertension. J. Pediatr. 205, 29–34 e21 (2019).
Baquero, H. et al. Oral sildenafil in infants with persistent pulmonary hypertension of the newborn: a pilot randomized blinded study. Pediatrics 117, 1077–1083 (2006).
Kipfmueller, F. et al. Continuous intravenous sildenafil as an early treatment in neonates with congenital diaphragmatic hernia. Pediatr. Pulmonol. 53, 452–460 (2018).
Bialkowski, A., Moenkemeyer, F. & Patel, N. Intravenous sildenafil in the management of pulmonary hypertension associated with congenital diaphragmatic hernia. Eur. J. Pediatr. Surg. 25, 171–176 (2015).
Shue, E. H. et al. Antenatal maternally-administered phosphodiesterase type 5 inhibitors normalize eNOS expression in the fetal lamb model of congenital diaphragmatic hernia. J. Pediatr. Surg. 49, 39–45 (2014).
Noori, S. et al. Cardiovascular effects of sildenafil in neonates and infants with congenital diaphragmatic hernia and pulmonary hypertension. Neonatology 91, 92–100 (2007).
Mous, D. S. et al. Clinically relevant timing of antenatal sildenafil treatment reduces pulmonary vascular remodeling in congenital diaphragmatic hernia. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L734–L742 (2016).
Lemus-Varela Mde, L. et al. Antenatal use of bosentan and/or sildenafil attenuates pulmonary features in rats with congenital diaphragmatic hernia. World J. Pediatr. 10, 354–359 (2014).
Kattan, J., Céspedes, C., González, A. & Vio, C. P. Sildenafil stimulates and dexamethasone inhibits pulmonary vascular development in congenital diaphragmatic hernia rat lungs. Neonatology 106, 74–80 (2014).
Russo, F. M. et al. Transplacental sildenafil rescues lung abnormalities in the rabbit model of diaphragmatic hernia. Thorax 71, 517–525 (2016).
Russo, F. M. et al. Antenatal sildenafil administration to prevent pulmonary hypertension in congenital diaphragmatic hernia (SToP-PH): study protocol for a phase I/IIb placenta transfer and safety study. Trials 19, 524 (2018).
Pels, A. et al. STRIDER (Sildenafil TheRapy in dismal prognosis early onset fetal growth restriction): an international consortium of randomised placebo-controlled trials. BMC Pregnency Childbirth 17, 440 (2017).
Unegbu, C. et al. Pulmonary hypertension therapy and a systematic review of efficacy and safety of PDE-5 inhibitors. Pediatrics 139, e20161450 (2017).
Samangaya, R. A. et al. A randomised, double-blinded, placebo-controlled study of the phosphodiesterase type 5 inhibitor sildenafil for the treatment of preeclampsia. Hypertens. Pregnency 28, 369–382 (2009).
von Dadelszen, P. et al. Sildenafil citrate therapy for severe early-onset intrauterine growth restriction. Bjog 118, 624–628 (2011).
Sharp, A. et al. Mortality in the UK STRIDER trial of sildenafil therapy for the treatment of severe early-onset fetal growth restriction. Lancet Child Adolesc. Health 3, e2–e3 (2019).
Abou-Tarboush, F. M., Abdel-Samad, M. F. & Al-Meteri, M. H. Developmental toxicity of orally administered sildenafil citrate (Viagra) in SWR/J mice. Saudi J. Biol. Sci. 18, 135–139 (2011).
Montani, D. et al. Phosphodiesterase type 5 inhibitors in pulmonary arterial hypertension. Adv. Ther. 26, 813–825 (2009).
Shiva, A. et al. Oral tadalafil in children with pulmonary arterial hypertension. Drug Res. 66, 7–10 (2016).
Samiee-Zafarghandy, S. et al. Safety of milrinone use in neonatal intensive care units. Early Hum. Dev. 91, 31–35 (2015).
Stasch, J.-P. et al. NO-independent regulatory site on soluble guanylate cyclase. Nature 410, 212–215 (2001).
Deruelle, P. et al. BAY 41-2272, a direct activator of soluble guanylate cyclase, reduces right ventricular hypertrophy and prevents pulmonary vascular remodeling during chronic hypoxia in neonatal rats. Biol. Neonate. 90, 135–144 (2006).
Evgenov, O. V. et al. Soluble guanylate cyclase activator reverses acute pulmonary hypertension and augments the pulmonary vasodilator response to inhaled nitric oxide in awake lambs. Circulation 110, 2253–2259 (2004).
Deruelle, P., Grover, T. R. & Abman, S. H. Pulmonary vascular effects of nitric oxide-cGMP augmentation in a model of chronic pulmonary hypertension in fetal and neonatal sheep. Am. J. Physiol. Lung Cell Mol. Physiol. 289, L798–L806 (2005).
Vuckovic, A. et al. Antenatal BAY 41-2272 reduces pulmonary hypertension in the rabbit model of congenital diaphragmatic hernia. Am. J. Physiol. Lung Cell Mol. Physiol. 310, L658–L669 (2016).
Pyriochou, A. et al. Soluble guanylyl cyclase activation promotes angiogenesis. J. Pharm. Exp. Ther. 319, 663–671 (2006).
Pankey, E. A. et al. Pulmonary and systemic vasodilator responses to the soluble guanylyl cyclase activator, BAY 60-2770, are not dependent on endogenous nitric oxide or reduced heme. Am. J. Physiol. Heart Circ. Physiol. 300, H792–H802 (2011).
Rojas-Moscoso, J. A. et al. The soluble guanylyl cyclase activator BAY 60-2770 potently relaxes the pulmonary artery on congenital diaphragmatic hernia rabbit model. Pediatr. Surg. Int. 30, 1031–1036 (2014).
Abman, S. H. & Stenmark, K. R. Changes in lung eicosanoid content during normal and abnormal transition in perinatal lambs. Am. J. Physiol. 262, L214–L222 (1992).
Cassin, S. et al. Effects of prostacyclin on the fetal pulmonary circulation. Pediatr. Pharm. 1, 197–207 (1981).
Mous, D. S. et al. Changes in vasoactive pathways in congenital diaphragmatic hernia associated pulmonary hypertension explain unresponsiveness to pharmacotherapy. Respir. Res. 18, 187 (2017).
Lawrence, K. M. et al. Use of prostaglandin E1 to treat pulmonary hypertension in congenital diaphragmatic hernia. J. Pediatr. Surg. 54, 55–59 (2019).
Olson, E. et al. Short-Term Treprostinil Use in Infants with Congenital Diaphragmatic Hernia following Repair. J. Pediatr. 167, 762–764 (2015).
Ali, F. Y. et al. Role of prostacyclin versus peroxisome proliferator-activated receptor beta receptors in prostacyclin sensing by lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 34, 242–246 (2006).
Kuwano, K. et al. 2-[4-[(5,6-diphenylpyrazin-2-yl)(isopropyl)amino]butoxy]-N-(methylsulfonyl)acetam ide (NS-304), an orally available and long-acting prostacyclin receptor agonist prodrug. J. Pharmacol. Exp. Ther. 322, 1181–1188 (2007).
Sitbon, O. et al. Selexipag for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 373, 2522–2533 (2015).
Kataoka, M. et al. A long-acting prostacyclin agonist with thromboxane inhibitory activity for pulmonary hypertension. Am. J. Respir. Crit. Care Med. 172, 1575–1580 (2005).
Obata, H. et al. Single injection of a sustained-release prostacyclin analog improves pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 177, 195–201 (2008).
Galié, N., Manes, A. & Branzi, A. The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 61, 227–237 (2004).
Okazaki, T., Sharma, H. S., McCune, S. K. & Tibboel, D. Pulmonary vascular balance in congenital diaphragmatic hernia: enhanced endothelin-1 gene expression as a possible cause of pulmonary vasoconstriction. J. Pediatr. Surg. 33, 81–84 (1998).
Hirako, S. et al. Antenatal saireito (TJ-114) can improve pulmonary hypoplasia and pulmonary vascular remodeling in nitrofen-induced congenital diaphragmatic hernia. Phytother. Res. 30, 1474–1480 (2016).
Keller, R. L. et al. Congenital diaphragmatic hernia: endothelin-1, pulmonary hypertension, and disease severity. Am. J. Respir. Crit. Care Med. 182, 555–561 (2010).
Nakwan, N. et al. Successful treatment of persistent pulmonary hypertension of the newborn with bosentan. Acta Paediatr. 98, 1683–1685 (2009).
Schermuly, R. T. et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 115, 2811–2821 (2005).
Perros, F. et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 178, 81–88 (2008).
Capdeville, R., Buchdunger, E., Zimmermann, J. & Matter, A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat. Rev. Drug Discov. 1, 493–502 (2002).
Ghofrani, H. A., Seeger, W. & Grimminger, F. Imatinib for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 353, 1412–1413 (2005).
Balasubramaniam, V. et al. Role of platelet-derived growth factor in vascular remodeling during pulmonary hypertension in the ovine fetus. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L826–L833 (2003).
Ghofrani, H. A. et al. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am. J. Respir. Crit. Care Med. 182, 1171–1177 (2010).
Frenckner, B., Broome, M., Lindstrom, M. & Radell, P. Platelet-derived growth factor inhibition-a new treatment of pulmonary hypertension in congenital diaphragmatic hernia? J. Pediatr. Surg. 43, 1928–1931 (2008).
Chang, Y. T. et al. Antenatal imatinib treatment reduces pulmonary vascular remodeling in a rat model of congenital diaphragmatic hernia. Am. J. Physiol. Lung Cell Mol. Physiol. 302, L1159–L1166 (2012).
Garcia-Olivas, R., Vilaro, S., Reina, M. & Castel, S. PDGF-stimulated cell proliferation and migration of human arterial smooth muscle cells. Colocalization of PDGF isoforms with glycosaminoglycans. Int J. Biochem. Cell Biol. 39, 1915–1929 (2007).
Armulik, A., Abramsson, A. & Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 97, 512–523 (2005).
Burgos, C. M. et al. Lung function and pulmonary artery blood flow following prenatal maternal retinoic acid and imatinib in the nitrofen model of congenital diaphragmatic hernia. J. Pediatr. Surg. 53, 1681–1687 (2018).
Mattei, D. et al. Reversible dasatinib-induced pulmonary arterial hypertension and right ventricle failure in a previously allografted CML patient. Bone Marrow Transplant. 43, 967–968 (2009).
Hennigs, J. K. et al. Multi tyrosine kinase inhibitor dasatinib as novel cause of severe pre-capillary pulmonary hypertension? BMC Pulm. Med. 11, 30 (2011).
Hansmann, G. et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Investig. 118, 1846–1857 (2008).
Hansmann, G., Calvier, L., Risbano, M. G. & Chan, S. Y. Activation of the metabolic master regulator PPARγ: a potential pioneering therapy for pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 62, 143–156 (2019).
Humbert, M. et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur. Respir. J. 53, 1801887 (2019).
Savale, L. et al. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 10, 6 (2009).
Steiner, M. K. et al. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 104, 236–244 (2009).
Itoh, T. et al. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 11, 158–163 (2006).
Okawada, M. et al. Serum monocyte chemotactic protein-1 levels in congenital diaphragmatic hernia. Pediatr. Surg. Int. 23, 487–491 (2007).
Gosemann, J. H. et al. Prenatal treatment with rosiglitazone attenuates vascular remodeling and pulmonary monocyte influx in experimental congenital diaphragmatic hernia. PLoS ONE 13, e0206975 (2018).
Humbert, M. et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 151, 1628–1631 (1995).
Pugliese, S. C. et al. The role of inflammation in hypoxic pulmonary hypertension: from cellular mechanisms to clinical phenotypes. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L229–L252 (2015).
Ahmed, M. & Miller, E. Macrophage migration inhibitory factor (MIF) in the development and progression of pulmonary arterial hypertension. Glob. Cardiol. Sci. Pract. 2018, 14 (2018).
Fleck, S. et al. Fetal production of growth factors and inflammatory mediators predicts pulmonary hypertension in congenital diaphragmatic hernia. Pediatr. Res. 74, 290–298 (2013).
Hagen, M. et al. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L1473–L1479 (2007).
Ameshima, S. et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ. Res. 92, 1162–1169 (2003).
Shima, H. et al. Antenatal dexamethasone suppresses tumor necrosis factor- expression in hypoplastic lung in nitrofen-induced diaphragmatic hernia in rats. Pediatr. Res. 46, 633–637 (1999).
Gosemann, J. H. et al. Increased activation of NADPH oxidase 4 in the pulmonary vasculature in experimental diaphragmatic hernia. Pediatr. Surg. Int. 29, 3–8 (2013).
Kureishi, Y. et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 6, 1004–1010 (2000).
Girgis, R. E. et al. Regression of chronic hypoxic pulmonary hypertension by simvastatin. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L1105–L1110 (2007).
Rikitake, Y. & Liao, J. K. Rho GTPases, statins, and nitric oxide. Circ. Res. 97, 1232–1235 (2005).
Mraiche, F., Cena, J., Das, D. & Vollrath, B. Effects of statins on vascular function of endothelin-1. Br. J. Pharmacol. 144, 715–726 (2005).
Nishimura, T. et al. Simvastatin attenuates smooth muscle neointimal proliferation and pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 166, 1403–1408 (2002).
Nishimura, T. et al. Simvastatin rescues rats from fatal pulmonary hypertension by inducing apoptosis of neointimal smooth muscle cells. Circulation 108, 1640–1645 (2003).
Pollack, P. S. et al. Pregnancy outcomes after maternal exposure to simvastatin and lovastatin. Birth Defects Res. A Clin. Mol. Teratol. 73, 888–896 (2005).
Inamdar, A. C. & Inamdar, A. A. Mesenchymal stem cell therapy in lung disorders: pathogenesis of lung diseases and mechanism of action of mesenchymal stem cell. Exp. Lung Res. 39, 315–327 (2013).
Liang, O. D. et al. Mesenchymal stromal cells expressing heme oxygenase-1 reverse pulmonary hypertension. Stem Cells 29, 99–107 (2011).
Baber, S. R. et al. Intratracheal mesenchymal stem cell administration attenuates monocrotaline-induced pulmonary hypertension and endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 292, H1120–H1128 (2007).
Balasubramaniam, V. et al. Bone marrow-derived angiogenic cells restore lung alveolar and vascular structure after neonatal hyperoxia in infant mice. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L315–L323 (2010).
Di Bernardo, J., Maiden, M. M., Hershenson, M. B. & Kunisaki, S. M. Amniotic fluid derived mesenchymal stromal cells augment fetal lung growth in a nitrofen explant model. J. Pediatr. Surg. 49, 859–864 (2014).
Kang, M. & Thebaud, B. Stem cell biology and regenerative medicine for neonatal lung diseases. Pediatr. Res. 83, 291–297 (2018).
Yuniartha, R. et al. Therapeutic potential of mesenchymal stem cell transplantation in a nitrofen-induced congenital diaphragmatic hernia rat model. Pediatr. Surg. Int. 30, 907–914 (2014).
Takayama, S. et al. An intra-amniotic injection of mesenchymal stem cells promotes lung maturity in a rat congenital diaphragmatic hernia model. Pediatr. Surg. Int. 35, 1353–1361 (2019).
Pederiva, F. et al. Amniotic fluid stem cells rescue both in vitro and in vivo growth, innervation, and motility in nitrofen-exposed hypoplastic rat lungs through paracrine effects. Cell Transplant. 22, 1683–1694 (2013).
Vanover, M., Wang, A. & Farmer, D. Potential clinical applications of placental stem cells for use in fetal therapy of birth defects. Placenta 59, 107–112 (2017).
Furlani, D. et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc. Res. 77, 370–376 (2009).
Yeung, V. et al. Paving the road for mesenchymal stem cell-derived exosome therapy in bronchopulmonary dysplasia and pulmonary hypertension. Stem Cell Based Therapy Lung Dis. 131–152 (2019).
Zhang, M. et al. Exosomal 15-LO2 mediates hypoxia-induced pulmonary artery hypertension in vivo and in vitro. Cell Death Dis. 9, 1022 (2018).
Zhang, S. et al. Mesenchymal stromal cell-derived exosomes improve pulmonary hypertension through inhibition of pulmonary vascular remodeling. Respiratory Res. 21, 71 (2020).
Tovar, J. A. Congenital diaphragmatic hernia. Orphanet J. Rare Dis. 7, 1 (2012).
Makanga, M. et al. Downregulated bone morphogenetic protein signaling in nitrofen-induced congenital diaphragmatic hernia. Pediatr. Surg. Int. 29, 823–834 (2013).
Burgos, C. M. et al. Improved pulmonary function in the nitrofen model of congenital diaphragmatic hernia following prenatal maternal dexamethasone and/or sildenafil. Pediatr. Surg. 80, 577–585 (2016).
Yamamoto, Y., Thebaud, B., Vadivel, A., Eaton, F., Jain, V., Hornberger, L. K., Doppler parameters of fetal lung hypoplasia and impact of sildenafil. Am J Obstet Gynecol. 211, 263.e1–263.e8 (2014).
Acknowledgements
We would like to thank Deb Hepp (Department of Surgery, University of North Carolina) for her administrative assistance with preparing and submitting this manuscript.
Funding
K.M. received support from the National Institutes of Health/National Institute of General Medical Sciences UNC-Duke Collaborative Clinical Pharmacology Postdoctoral Training Program (T32 GM086330-08).
Author information
Authors and Affiliations
Contributions
Conception or design of the work: K.M., N.D.T., S.E.M., and M.R.K. Data collection: K.M. Data analysis and interpretation: K.M., S.E.M., and M.R.K. Drafting the article: KM. Critical revision of the article: K.M., N.D.T., S.E.M., and M.R.K. Final approval of the version to be published: K.M., N.D.T., S.E.M., and M.R.K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Patient consent
Patient consent was not required.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Marulanda, K., Tsihlis, N.D., McLean, S.E. et al. Emerging antenatal therapies for congenital diaphragmatic hernia-induced pulmonary hypertension in preclinical models. Pediatr Res 89, 1641–1649 (2021). https://doi.org/10.1038/s41390-020-01191-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-01191-x
