
Abstract
Background
Maternal nutrient restriction (MNR) is a widespread cause of fetal growth restriction (FGR), an independent predictor of heart disease and cardiovascular mortality. Our objective was to examine the developmental and long-term impact of MNR-induced FGR on cardiac structure in a model that closely mimics human development.
Methods
A reduction in total caloric intake spanning pregestation through to lactation in guinea pig sows was used to induce FGR. Proliferation, differentiation, and apoptosis of cardiomyocytes were assessed in late-gestation fetal, neonatal, and adult guinea pig hearts. Proteomic analysis and pathway enrichment were performed on fetal hearts.
Results
Cardiomyocyte proliferation and the number of mononucleated cells were enhanced in the MNR–FGR fetal and neonatal heart, suggesting a delay in cardiomyocyte differentiation. In fetal hearts of MNR–FGR animals, apoptosis was markedly elevated and the total number of cardiomyocytes reduced, the latter remaining so throughout neonatal and into adult life. A reduction in total cardiomyocyte number in adult MNR–FGR hearts was accompanied by exaggerated hypertrophy and a disorganized architecture. Pathway analysis identified genes related to cell proliferation, differentiation, and survival.
Conclusions
FGR influences cardiomyocyte development during critical windows of development, leading to a permanent deficiency in cardiomyocyte number and compensatory hypertrophy in a rodent model that recapitulates human development.
Similar content being viewed by others


Introduction
The negative impact of an adverse intrauterine environment on the fetus and its potential for immediate and chronic sequelae has long been appreciated. Barker and colleagues1,2,3 first drew attention to this phenomenon in the late 1980s with their historical cohort studies in the United Kingdom linking ischemic heart disease in adults with low birth weight. Mortality rates were inversely proportional to birth weight, with low-birth-weight infants having a 70% increase in risk of cardiovascular-related mortality in adulthood.3,4,5 Since these early formative studies, the relationship between birth weight and heart disease has been substantiated in numerous populations around the world and expanded to other chronic diseases, including obesity, diabetes, atherosclerosis, and hypertension.6,7,8,9 This programming of disease susceptibility is thought to arise from altered developmental trajectories in response to an adverse intrauterine environment, in turn leading to persistent deficits in organ structure and function. Cardiac function in adulthood is particularly vulnerable to developmental insults as the full complement of cardiomyocytes is determined during critical perinatal and early postnatal periods, with limited proliferative capacity thereafter.10,11
Early growth of the myocardium is due to rapidly proliferating mononucleated cardiomyocytes, which subsequently differentiate into binucleated cells.10,11 This latter phase begins in late gestation and is completed shortly after birth in prenatal developers such as sheep, guinea pigs, and humans.12,13 Binucleated cells are terminally differentiated in that they permanently exit the cell cycle; thus, the postnatal heart accumulates mass primarily through hypertrophic growth. Given the limited regenerative capacity in the adult heart, interference in cardiomyocyte endowment during development may later compromise the ability of the heart to compensate for increases in afterload or recover from ischemic injury. Therefore, the heightened risk of cardiovascular death in low-birth weight adults may be attributable to aberrant cardiomyocyte maturation in utero.
Developmental outcomes of an adverse in utero environment will depend on the nature, timing, and duration of the adversity. To date, the sensitivity of cardiomyocyte development to perturbations in the intrauterine environment has been studied exclusively in the context of fetal hypoxia, a common result of antenatal complications such as placental insufficiency and umbilical cord compression. Animal surgeries used to induce placental insufficiency are often performed in late gestation; thus, the results of these studies reflect acute responses to late-onset fetal hypoxia. Nevertheless, these studies reveal delayed differentiation of cardiomyocytes in fetal sheep subjected to chronic hypoxia.14,15,16 Less is known regarding the cardiac consequences of maternal malnutrition, a predominant cause of fetal growth restriction (FGR) in the developing world. For the current study, we examined developmental and long-term effects in the heart using a model of global maternal nutrient restriction (MNR) spanning pregestation, gestation, and lactation in the guinea pig. Unlike other rodents but similar to humans and sheep, guinea pigs are prenatal developers and have a relatively long gestation. The time course of key developmental steps during guinea pig intrauterine development, such as white and brown adipose tissue, mirror that of humans.17 As well, the extent of trophoblast invasion is more comparable to the human placenta than other widely studied animals such as mice and sheep, making the guinea pig an ideal model for in utero nutrient acquisition and metabolism.17 For these reasons, the guinea pig has proven valuable in studies investigating the influence of FGR on developmental outcomes18,19,20,21,22 and was thus exploited in the present study to test the hypothesis that MNR-induced FGR impairs cardiac development, leading to permanent deficiencies in the adult heart.
Methods
Animal model of nutrient restriction
All protocols and procedures were approved by The University of Western Ontario Animal Use Subcommittee and followed the guidelines of the Canadian Council on Animal Care. Briefly, nulliparous female guinea pigs (Dunkin–Hartley, Charles River) were randomly assigned to either a control group provided a full-calorie ad libitum diet or an MNR group provided 70% of the average food intake per kilogram of body weight of the control cohort beginning 4 weeks prior to conception and continued until mid-gestation (day 35). Thereafter, the MNR cohort was provided a diet consisting of 90% of the average food intake of the control cohort until postpartum day 14 to simulate undernourishment through the lactation period. On day 15 post parturition all sows allowed to deliver were provided a full diet (Guinea Pig Diet 5025, LabDiet, St. Louis, MO).
The breeding strategy and diet used to establish a model of moderate maternal nutrient restriction (MNR) in guinea pigs has been previously described in detail.23 In addition, details regarding litter size and the number of sows/litters for these cohorts have recently been reported.23,24 For the fetal cohorts, 9 control sows produced 31 viable fetuses and 1 stillborn, whereas 12 MNR sows produced 42 viable fetuses and one stillborn. No more than one male and one female fetus from any one pregnancy was selected for further analysis for a total of eight males and eight females in each of the control and MNR–FGR cohorts. For the neonatal and adult cohorts, 16 control sows produced 44 live-born and nine stillborn pups, while 17 MNR sows produced 52 live-born and 6 stillborn pups. For the neonatal cohort, 18 control (8 males and 10 females) and MNR–FGR (9 males and 9 females) were killed at 25 ± 1 d and 26 ± 1 d, respectively. For the adult cohort, 18 control (6 males and 12 females) and 16 MNR–FGR (6 males and 10 females) were killed at 109 ± 1 d and 111 ± 1 d, respectively. For the neonatal and adult cohorts, no more than one male and two females or two males and one female from the same litter were used.
Animal necropsy and tissue preparation
A total of 102 animals were studied, thirty two from the fetal group (60/61 days of gestation), 36 from the neonatal group (25–27 days after birth), and 34 from the adult group.23 Prior to necropsy, animals were weighed and then euthanized with 0.3 cc intraperitoneal injection of sodium pentobarbital (MTC Pharmaceuticals, Cambridge, ON, Canada). At necropsy, visceral organs were harvested, weighed, and stored for further analysis. Whole hearts were placed in 5% potassium chloride solution, bisected through the ventricular myocardium, and segments including both ventricles and the interventricular septum were analyzed. The middle third of the ventricular sections were stored in 4% paraformaldehyde. The upper and lower thirds were flash-frozen in liquid nitrogen for later analysis. The middle sections were then embedded in paraffin and mounted on slides for histological examination.
Cardiomyocyte number, surface area, proliferation, and apoptosis
H&E-stained sections of the left ventricular myocardium were analyzed in five separate high-power (40×) fields (hpf) per sample using an Olympus inverted microscope equipped with a Nikon digital camera system. In each 40× hpf, two blinded observers counted the total number of cardiomyocytes and mononucleated cardiomyocytes with an inter-observer concordance of >90%. To determine the numbers of mononuclear cardiomyocytes, the number of mono- and binucleated cardiomyocytes in five separate hpf per sample were counted and the mean value for each animal was calculated. Mononuclear cardiomyocytes were expressed as a percent of the total cardiomyocytes per hpf. Only cardiomyocytes where striations were clearly visible were counted. Using ImageJ software, the cell area function was used to obtain length and width measurements of cardiomyocytes sliced along their longitudinal axis. In total 20 cells in 3 hpf of the mid-myocardium from each subject were measured, including 10 mono- and 10 binucleated cells, as previously reported.15 Cardiomyocyte length was determined using the longest axis in longitudinally sliced sections. A bisecting perpendicular line across the midline of mononucleated cells determined width measurements for cardiomyocytes and midway between the two nuclei in binucleated cardiomyocytes, as previously reported.15 All results are expressed as μm2.
Cardiac cross sections from fetal and neonatal control and MNR–FGR animals were also stained for Ki-67, a nuclear marker of cell proliferation, and DNA strand breaks (TUNEL), a marker of apoptosis. Following antigen retrieval with citrate buffer, sections were blocked in normal goat serum and incubated with rabbit anti-human Ki-67 (1:3000). Cardiomyocyte apoptosis was determined using the Apoptag peroxidase detection kit (EMD Millipore) according to the manufacturer’s instructions. For quantification, the number of positive cells and total cardiomyocytes in five distinct 40× digital images were analyzed. Counts were performed by two blinded observers with an inter-observer concordance of >95%. Finally, a pathologist who was blinded to group allocations reviewed all slides.
LC–MS/MS analysis
Protein concentration was measured with a BCA protein assay kit (Thermo Scientific) from the lower-third (ventricular myocardium) segment of neonatal hearts (five animals per group, one animal per litter) that were homogenized in ice-cold lysis buffer (RIPA, Thermo Scientific) and protease inhibitor cocktail (Thermo Scientific). Later, 100 μg of protein was reduced with dithiothreitol, alkylated using iodoacetamide, and digested overnight in trypsin. Digested peptides were cleaned using C18 spin columns (Harvard Apparatus), lyophilized, and analyzed on an Orbitrap Fusion tribrid mass spectrometer coupled with an Ultimate 3000 nano-UPLC system (Thermo Scientific). Reconstituted peptide (2 µl) was trapped and washed on a Pepmap 100 C18 trap for 10 min, followed by separation on a Pepman 100 RSLC C18 column using a gradient. Samples were analyzed by data-dependent acquisition in positive mode with an Orbitrap MS analyzer for precursor scan and ion-trap MS analyzer for MS/MS scans. The fragmentation method was used for collision-induced dissociation and raw data processed with Proteome Discoverer (v1.4, Thermo Scientific), and then submitted for SequestHT search against the Uniprot database. Peptide spectrum matching validation was performed with the fixed-value PSM validator algorithm. Parallel reaction monitoring was carried out on the same LC–MS platform and LC elution conditions. For each candidate peptide, one signature fragment was selected to calculate the peak area on the extracted-ion chromatograph for that peptide using Thermo Xcalibur software (ver. 3.0.63, Thermo Scientific). The peak area for each peptide was normalized to the total number of identified peptide sequences across different samples to control for variance. The limma Bioconductor package was used to perform an empirical Bayesian analysis (with settings trend=true, robust=true) of the log-transform counts after quantile normalization to determine differential protein levels between MNR–FGR and control cohorts.
Pathway analysis
The full complement of mouse RefSeq mRNA entries were cross-referenced with the Ingenuity Pathway Analyst knowledgebase for genes expressed in the heart and related to a priori processes of interest. Rather than relying on Uniprot annotation of guinea pig names, which is incomplete, murine mRNA orthology was deemed the most reliable mapping method for Ingenuity Pathway Analysis since IPA primarily relies on human, mouse, and rat gene names. Of the 824 resulting genes, 399 orthologous genes were detected in at least one sample in the proteomics data. All subsequent analysis of pathway enrichment within these 399 genes was performed with the causal network module of Ingenuity Pathway Analysis (Qiagen Inc.).
Pathway analysis was confirmed using RT-PCR and immunohistology. Primer sequences for RT-PCR: PARP1: CAG ACA CTG ACC TGG TAG AT (forward); CTC TGG GGA ACC TAG TAA GG (reverse); S15 (housekeeping): TTC TAC GCT CCT CAA AGA CT (forward); CAT TTC TTT CCG GTT AGC CA (reverse). Total RNA was extracted with Trizol and purified on a spin column (Qiagen). cDNA was generated from 2 μg of RNA using Superscript III (Invitrogen). Real-time PCR was performed with SYBR green on a CFX384 Touch Real-time PCR detection system (Biorad). Samples were run in triplicate. Cardiac cross sections from fetal control and MNR–FGR animals were also stained for PARP1 (Cell Signaling), as described above.
Statistical analysis
Growth characteristics including body and organ weights, growth rates, as well as cardiomyocyte counts and percentages, proliferation markers, and cell surface area were compared using analysis of variance and unpaired Student’s t test as indicated (Prism 7, GraphPad Software, San Diego, CA). For all analysis, statistical significance was assumed for p < 0.05.
Results
Fetal growth restriction impairs the accumulation of heart mass during development
The guinea pig is an excellent model system for understanding the effects of developmental perturbations given its resemblance to human gestation. The guinea pig model of MNR used for the present study was previously developed, and its impact on fetal growth and other visceral organ development has been reported.23 MNR in pregnant guinea pig sows significantly impaired fetal growth and led to a slightly shorter gestational length and profound reduction in offspring birth weight (Fig. 1a, b). On average, the offspring of MNR-induced FGR resulted in a 33% reduction in birth weight compared with control offspring. Consistent with a reduction of maternal caloric intake throughout the lactation period, MNR–FGR pups gained less absolute mass over the first 25 days of life compared with controls (Fig. 1c). However, relative to birth weight, MNR–FGR pups exhibited a rapid growth acceleration phase over the first 25 days after birth, with nearly all pups doubling their birth weight by 3 weeks of life (Fig. 1d). This finding was uncharacteristic of the control cohort. Adult weights did not differ in a second cohort of animals that was allowed to reach maturity (not shown). Although perinatal maternal nutrient restriction results in impaired fetal growth, MNR–FGR offspring experience rapid “catch-up growth” in the early postnatal period to meet their anticipated final weight.

Descriptive characteristics of control and MNR birth cohorts. a, b Number of completed days of gestation at birth (a) and birth weight (b) for the control and MNR neonatal cohorts. c, d Absolute weight gain (grams) (c) and relative weight gain (%) as a percent of birth weight (d) at 4 weeks of life for the control and MNR neonatal cohorts. Bars represent mean ± S.D. Analysis by Student’s t test
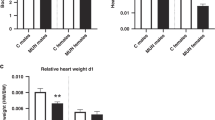
Visceral organs were harvested at 60 days post conception (fetal stage) and 23–27 days postnatal age (neonatal stage) to examine the gross and microscopic effects of MNR on heart development.23 At the fetal stage, the brain-to-total body weight ratio in MNR–FGR fetuses is significantly increased compared with control fetuses (Fig. 2a). In contrast, heart-to-total body weight ratio did not differ between MNR–FGR and control fetuses (Fig. 2b). Despite the significant postnatal “catch-up” somatic growth of the MNR–FGR cohort, heart mass did not increase proportionate to the increase in body mass. In the neonatal cohort, brain-to-total body weight ratio continued to be elevated in MNR–FGR neonates (4-weeks old) compared with the control cohort, but heart-to-total body weight ratio was now significantly reduced in MNR–FGR neonates as compared with control neonates (Fig. 2c, d). Thus, maternal nutrient restriction throughout pregnancy and the lactation phase of early postnatal growth significantly impairs heart development such that relative heart mass is reduced in the neonatal period.

Maternal nutrient restriction preserves brain mass at the expense of heart mass. a–d Comparison of brain weight to total body weight (TBW) ratio (a, c) and heart weight to total body weight ratio (b, d) for control and MNR–FGR fetuses (a, b) and neonates (c, d). Bars represent mean ± S.D. Analysis by Student’s t test
Cardiomyocyte proliferation and morphology are adversely affected by fetal growth restriction
Based on the observation that MNR–FGR neonates exhibit reduced heart mass relative to total body weight, we sought to examine the histologic features of the myocardium at both the fetal and neonatal stages in each cohort. Microscopic examination of H&E-stained sections of the ventricular myocardium from MNR–FGR fetuses and neonates showed cytoplasmic degeneration and diminished cell density, which were noted in the subendocardium and extended into the adjacent mid-myocardium of the left ventricle. Subsequently, we focused on the mid-myocardium to permit blinded quantitative analysis (Fig. 3a). Absolute numbers of cardiomyocytes per high-power field (hpf) were significantly reduced in MNR–FGR fetuses and neonates when compared with control fetuses and neonates, respectively (Fig. 3b). No difference in the absolute number of cardiomyocytes for fetal and neonatal cohorts was observed when data were disaggregated for sex (data not shown). In comparison with control neonates, the relative expansion in cardiomyocyte numbers (per hpf) between MNR–FGR fetal and neonatal cohorts was significantly reduced (Fig. 3c). Interestingly, MNR–FGR neonatal hearts had a slightly but significantly higher percent of mononucleated cardiomyocytes (per hpf) when compared with control neonates, which was not apparent during the fetal stage of development (Fig. 3d).

Maternal nutrient restriction impairs cardiomyocyte development in fetal and neonatal hearts. a Photomicrographs of left ventricular myocardium from control and MNR–FGR fetuses and neonates. Black boxes indicate the area magnified below. *Denotes lumen of the left ventricle. Scale bar equals 200 μm. b Number of cardiomyocytes per 40× hpf in fetal and neonatal control (white bars) and MNR–FGR (black bars) hearts (*p < 0.001). c Relative increase in cardiomyocyte number from fetal to neonatal stage in 40× hpf cross sections of control and MNR–FGR hearts. d Percent of mononucleated cardiomyocytes of total cardiomyocytes per hpf in fetal and neonatal control (white bars) and MNR–FGR (black bars) hearts (*p < 0.05). For (b–d) data represent mean number ± S.D. (n = 16–18 per group). Analysis by two-way ANOVA (b, d) and Student’s t test
Highly proliferative mononucleated cardiomyocytes are common during fetal heart development, but give way to binucleated cardiomyocytes in adult hearts.25 Thus, it is thought that mononuclear cardiomyocytes are a more primitive cell population. To assess cell proliferation within each cohort, we identified Ki-67-positive cardiomyocytes within the left ventricular mid-myocardium. At both the fetal and neonatal stage, MNR–FGR animals exhibit a threefold increase in Ki-67-positive cardiomyocytes (per hpf) when compared with control (Fig. 4a, b). Most strikingly, while only 12% of cardiomyocytes in control fetuses were noted to be Ki-67 positive, 30–40% of cardiomyocytes were Ki-67 positive in MNR–FGR fetuses with some hearts demonstrating >50% Ki-67-positive cardiomyocytes per hpf. Along with increased proliferation, a high level of apoptosis was observed in the MNR–FGR fetal heart, while apoptosis was negligible in the hearts of control fetuses (Fig. 4c, d). Interestingly, the reverse pattern emerged in the neonatal period with greater apoptosis in the control hearts compared with MNR–FGR hearts.

Maternal nutrient restriction increases the cardiomyocyte proliferative index in fetal and neonatal hearts. a Photomicrographs of ventricular myocardium from control and MNR–FGR fetuses and neonates stained with anti-Ki-67 antibody (brown) and counterstained with hematoxylin (blue). Scale bar equals 100 μm. b Quantification of Ki-67-positive cardiomyocytes (%) per 40 × hpf in fetal and neonatal control (white bars) and MNR–FGR (black bars) hearts. Data represent mean number ± S.D. (n = 6–8 per group, *p < 0.001). c Photomicrographs of ventricular myocardium from control and MNR–FGR fetuses and neonates stained with anti-TUNEL antibody (brown) and counterstained with hematoxylin (blue). Scale bar equals 100 μm. d Quantification of TUNEL-positive cardiomyocytes (%) per 40 × hpf in fetal and neonatal control (white bars) and MNR–FGR (black bars) hearts. Data represent mean number ± S.D. (n = 4 per group, one animal per litter, *p < 0.001). Analysis by two-way ANOVA
The prominent effect of fetal growth restriction on proliferation, differentiation, and apoptosis in the ventricular myocardium, as revealed by immunohistochemistry, was substantiated by proteomics analysis. Of the 399 salient protein candidates (see Methods: Pathway analysis), only PARP1 passed the unpaired Student’s t test with a false-discovery rate (FDR) multiple testing to correct p-value < 0.05. This plus eight additional proteins passing the limma test (see Methods: LC–MS/MS analysis) with FDR < 0.05 were analyzed for pathway enrichment in Ingenuity Pathway Analyst (IPA). The strongest predicted causal network as determined from IPA for the nine differentially regulated proteins was identified around genes related to cellular proliferation, differentiation, and survival (Fig. 5a), with a network-bias-corrected p-value of 0.0177. Pathway enrichment was confirmed using RT-PCR and immunohistology. PARP1 transcript expression was modestly, but significantly enhanced in MNR–FGR fetal hearts (Fig. 5b). PARP1 expression on cardiac cross sections was widespread in fetal MNR–FGR hearts, with high concentrations observed in papillary muscles and in the endocardium through to the mid-myocardium. In contrast, PARP1-positive cells were rarely identified in control fetal hearts (Fig. 5c).

Strongest predicted causal network by IPA analysis of neonatal hearts. a Network contains differentially upregulated (green) and downregulated (red) proteins, as well as literature-inferred activation (blue) and inhibition (orange) proteins. Top functions of the genes were related to cellular proliferation, differentiation, and survival. The node shapes denote the following: triangle: kinase, fat diamond: peptidase, tall diamond: enzyme, rhomboid: transporter, oval: transcription regulator, circle: other. n = 5 animals per group (one animal per litter). b Quantification of PARP-1 expression (RT-PCR) in whole hearts from fetal control (white bars) and MNR–FGR (black bars) animals. Data represent mean ± S.E.M. Analysis by Student’s t test. c Photomicrographs of papillary muscle and ventricular myocardium from control and MNR–FGR fetal hearts stained with anti-PARP1 antibody (brown) and counterstained with hematoxylin (blue). Scale bar equals 100 μm
The residual effects of fetal growth restriction on adult hearts
Next, we examined the effects of MNR–FGR on adult hearts. Both control and MNR–FGR offspring reached similar weights by 90 days after birth and did not differ in appearance, activity, or total body weight (not shown). Differences observed in H&E-stained sections from the left ventricular myocardium of MNR–FGR vs. control adults were more pronouncede in fetal and neonatal cross sections. MNR–FGR hearts were relatively atrophic with disorganized and degenerating cardiomyocytes in the inner aspect of the myocardium, including the papillary muscles, which appeared atrophic (Fig. 6a). In contrast, cardiomyocytes were progressively more hypertrophic in the mid and outer myocardium with patchy anisonucleosis noted throughout. The diminished cell density and cytoplasmic degeneration apparent in the earlier MNR–FGR cohorts were more distinct in MNR–FGR adults along with marked cardiomyocyte hypertrophy observed throughout the left ventricular myocardium (Fig. 6b).

Fetal growth restriction impairs heart growth and cardiomyocyte growth in adult hearts. a Low- (20 × ) and high-power (40 × ) photomicrographs of the left ventricular endocardium and epicardium from MNR–FGR adults. Scale bar equals 100 μm. b Photomicrographs of ventricular mid-myocardium from control and MNR–FGR adults. Black boxes indicate the area of myocardium magnified below. *Denotes lumen of the left ventricle. Scale bar equals 200 μm. c Comparison of heart weight to total body weight ratio for control and MNR–FGR adult guinea pigs. d Number of cardiomyocytes per hpf (40 × ) in control and MNR–FGR adult hearts. e Cross-sectional cardiomyocyte area in control and MNR–FGR adult hearts. f Percent of mononuclear cardiomyocytes of total cardiomyocytes per hpf in control and MNR–FGR adult hearts. g Quantification of Ki-67-positive cardiomyocytes (%) per 40 × hpf in control and MNR–FGR hearts. Data represent mean number ± S.D. (n = 16–18 per group). Analysis by Student’s t test
While most visceral organs and brain weights were proportional to total body weight and did not differ between MNR–FGR and control offspring, the heart-to-total body weight ratio was significantly reduced in MNR–FGR offspring as compared with control offspring (Fig. 6c). Similar to our observations in the fetal and neonatal MNR–FGR cohorts, the absolute number of cardiomyocytes (per hpf) was significantly reduced in MNR–FGR adults as compared with control animals (Fig. 6d). Despite the reduction in total cardiomyocyte number in MNR–FGR adults, the mean cross-sectional area of a random sample of cardiomyocytes from each animal was significantly increased in MNR–FGR adults as compared with control adults (Fig. 6e). Mononuclear cardiomyocytes are relatively rare in the mature myocardium of control animals, comprising <5% of the total cardiomyocyte number. In contrast, mononuclear cardiomyocytes were 50% more abundant in the ventricular myocardium of MNR–FGR adult hearts as compared with controls (Fig. 6f). Despite the increase in immature mononuclear cardiomyocytes in MNR–FGR adults, Ki-67-positive cardiomyocytes were rarely identified and did not differ between control and MNR–FGR adults (Fig. 6g).
Discussion
In the setting of nutrient deprivation, intrauterine development slows to conserve energy, leading to low birth weight, premature delivery, and increased fetal and neonatal mortality. The well-documented predisposition of low-birth-weight neonates to cardiovascular disease as adults is likely attributable to aberrant developmental trajectories involving the cardiovascular system. This supposition has been demonstrated largely in rat and sheep models of intrauterine oxygen deprivation, wherein the resultant FGR is accompanied by delayed cardiomyocyte maturation and structural changes in the vasculature.12,13,14,26,27 In the present study, we characterize a novel model of the longitudinal effects of FGR on cardiac development in a rodent species that closely recapitulates human gestation and perinatal nutrition as well as postnatal maturation. Taken together, our results suggest that cardiomyocyte maturation characterizing the transition from intrauterine to extrauterine life is delayed under conditions of MNR, leading to FGR and results in a permanent deficiency in cardiomyocyte number. The exaggerated cardiomyocyte hypertrophy observed later in MNR–FGR adult offspring is suggestive of compensatory growth in response to physiologic blood pressure increases that accompany body growth throughout childhood and adolescence. The guinea pig is especially suited to extrapolating findings with respect to developmental outcomes, given its resemblance to human gestation.
The functional capacity of the heart is largely determined during critical developmental windows. Over the first two-thirds of gestation, the accrual of cardiac mass is driven by rapidly proliferating mononucleated cardiomyocytes.10 These early cardiomyocytes enter a maturation phase wherein they terminally differentiate and permanently exit the cell cycle, a process that is completed in the early postnatal period in precocious developers such as guinea pigs and humans.10,11,28 Indeed, the relative abundance of mononucleated cells decreased from 40% in the late fetal stage to ~5% in the neonatal period in our control guinea pigs, validating the use of this species in studies of cardiac development. After terminal differentiation, the cell population remains stable and further growth of the heart depends on extracellular and intracellular expansion. The decreased number of total cardiomyocytes and increased proportion of mononucleated cells in the neonatal and adult MNR–FGR guinea pig hearts imply an interruption in this maturation process. With a fixed deficit in cardiomyocyte population, postnatal normalization of wall stress to hemodynamic load requires an exaggerated degree of hypertrophy, evident by the increased cardiomyocyte area observed in the hearts of our MNR–FGR guinea pigs. Apart from the normal increase in hemodynamic load that occurs with postnatal body growth, ventricular function in an under-endowed heart would be further compromised by a pathological increase in afterload due to hypertension, which has been reported in IUGR animal models.29 Excessive intracellular hypertrophy following cardiomyocyte reduction during development has been demonstrated in studies using a hypertrophic rat model30 and a knockout mouse model.31 Cellular disorganization, which contributes to contractile dysfunction in heart disease,32 is known to accompany excessive cardiomyocyte hypertrophy and was noted in the adult MNR–FGR guinea pig hearts. It should also be noted that previous studies have reported changes in cardiac shape, reduced stroke volume, and abnormal systolic and diastolic function in children and adolescents who experienced fetal growth restriction.33
There has been little investigation into the role of apoptosis in cardiac development; however, our results in normally grown guinea pigs showing negligible apoptosis in the late fetal period followed by marked apoptosis in the neonatal period suggest pruning to be a normal process in the final stages of cardiac maturation. This process is likely required for remodeling in response to the abrupt redistribution of blood flow that accompanies birth, a theory supported by Fernandez et al.34, who report a surge in apoptosis in the post-birth murine heart that was greater in the right ventricle and subsided at weaning. In contrast to the negligible apoptosis in control fetal guinea pig hearts, apoptosis is a striking feature in the fetal MNR–FGR hearts and likely contributes to the reduction in total cardiomyocyte number that persisted after birth and into adulthood. MNR–FGR neonates did not exhibit postnatal upregulation of apoptosis as observed in control neonates; however, MNR–FGR neonates did maintain a higher proliferative index, which is likely due to the persistence of mononucleated cardiomyocytes. This may reflect an altered developmental trajectory if apoptotic signaling is coupled to the cell cycle or a need for the undergrown heart to restrain apoptosis to maximize its later functional capacity. Alternatively, it is also possible that in MNR–FGR hearts, the physiologic phase of neonatal apoptosis is delinked with maturation, occurs prematurely during the fetal period, and is quantitatively exaggerated as compared to control guinea pigs, resulting in a persistence of mononucleated cardiomyocytes in extrauterine life.
A pre-existing cardiomyocyte shortage may worsen gradually with the normal age-related loss of cardiomyocytes35 and limit the heart’s capacity to overcome mechanical strain or recovery from injury. Indeed, the risk of death from heart disease is elevated in low-birth-weight adults1,2,3 and recovery from ischemic injury was impaired in a rat model of FGR.36 On the other hand, the greater number of residual mononucleated cells found in the adult MNR–FGR heart appeals to the unconventional hypothesis that regenerative capacity is enhanced to some degree. In vitro evidence suggests that mononucleated cells maintain proliferative potential into adulthood37 and the idea that regenerative capacity is entirely lost with maturation has been recently challenged with evidence from several species, including guinea pigs and humans, revealing that the adult myocardium retains certain “niches” of cardiac progenitor and stem cells able to differentiate in response to injury.30,38,39 Therefore, the higher proportion of immature cells in our MNR–FGR adults may serve as a safeguard to protect the myocardium against stress or disease, at least in the early stages. Emerging evidence highlights the potential for gene therapy involving the transcriptional factors GATA-4, Mef2c, and Tbx5 that can be used to reprogram damaged myocardium into functioning cardiomyocytes.40,41,42
In summary, our study reveals that the impact of maternal nutrient restriction on heart development involves a deviation in the normal course of proliferation, differentiation, and apoptosis that follows the transition from fetal to neonatal life. This developmental interference prevents the heart from attaining the full complement of cardiomyocytes, thereby predisposing the heart to excessive hypertrophy and cellular disorganization later in life, a finding that sheds light on the relationship between fetal growth restriction and cardiac mortality in later life.
References
Barker, D. J. & Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1, 1077–1081 (1986).
Barker, D. J., Osmond, C., Golding, J., Kuh, D. & Wadsworth, M. E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. Br. Med. J. 298, 564–567 (1989).
Barker, D. J., Winter, P. D., Osmond, C., Margetts, B. & Simmonds, S. J. Weight in infancy and death from ischaemic heart disease. Lancet 2, 577–580 (1989).
Burton, G. J., Fowden, A. L. & Thornburg, K. L. Placental origins of chronic disease. Physiol. Rev. 96, 1509–1565 (2016).
Jiang, B., Godfrey, K. M., Martyn, C. N. & Gale, C. R. Birth weight and cardiac structure in children. Pediatrics 117, e257–e261 (2006).
Barker, D. J. et al. The early origins of chronic heart failure: impaired placental growth and initiation of insulin resistance in childhood. Eur. J. Heart Fail 12, 819–825 (2010).
Eriksson, J. G., Kajantie, E., Lampl, M. & Osmond, C. Trajectories of body mass index amongst children who develop type 2 diabetes as adults. J. Intern. Med. 278, 219–226 (2015).
Ravelli, G. P., Stein, Z. A. & Susser, M. W. Obesity in young men after famine exposure in utero and early infancy. N. Engl. J. Med. 295, 349–353 (1976).
Stansfield, B. K. et al. Nonlinear relationship between birthweight and visceral fat in adolescents. J. Pediatr. 175, 1550–1559 (2016).
Paradis, A. N., Gay, M. S. & Zhang, L. Binucleation of cardiomyocytes: the transition from a proliferative to a terminally differentiated state. Drug Discov. Today 19, 602–609 (2014).
Zak, R. Cell proliferation during cardiac growth. Am. J. Cardiol. 31, 211–219 (1973).
Botting, K. J. et al. Early origins of heart disease: low birth weight and determinants of cardiomyocyte endowment. Clin. Exp. Pharmacol. Physiol. 39, 814–823 (2012).
Burrell, J. H. et al. Growth and maturation of cardiac myocytes in fetal sheep in the second half of gestation. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 274, 952–961 (2003).
Botting K. J., McMillen I. C., Forbes H., Nyengaard J. R., Morrison J. L. Chronic hypoxemia in late gestation decreases cardiomyocyte number but does not change expression of hypoxia-responsive genes. J. Am. Heart Assoc. 3 (2014) e000531.
Louey, S., Jonker, S. S., Giraud, G. D. & Thornburg, K. L. Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J. Physiol. 580, 639–648 (2007).
Morrison, J. L. et al. Restriction of placental function alters heart development in the sheep fetus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R306–R313 (2007).
Engle, W. A. & Lemons, J. A. Composition of the fetal and maternal guinea pig throughout gestation. Pediatr. Res. 20, 1156–1160 (1986).
Carter, A. M. Current topic: restriction of placental and fetal growth in the guinea-pig. Placenta 14, 125–135 (1993).
Ashwell, M., Purkins, L., Cowen, T. & Day, K. C. Pre- and postnatal development of adipose tissue at four sites in the guinea pig: effect of maternal diet restriction during the second half of pregnancy. Ann. Nutr. Metab. 31, 197–210 (1987).
Kind, K. L. et al. Chronic maternal feed restriction impairs growth but increases adiposity of the fetal guinea pig. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R119–R126 (2005).
Sohlstrom, A. et al. Food restriction alters pregnancy-associated changes in IGF and IGFBP in the guinea pig. Am. J. Physiol. 274, E410–E416 (1998).
Kind, K. L. et al. Restricted fetal growth and the response to dietary cholesterol in the guinea pig. Am. J. Physiol. 277, R1675–R1682 (1999).
Elias, A. A., Ghaly, A., Matushewski, B., Regnault, T. R. & Richardson, B. S. Maternal nutrient restriction in guinea pigs as an animal model for inducing fetal growth restriction. Reprod. Sci. 23, 219–227 (2016).
Nevin C. L., et al. Maternal nutrient restriction in guinea pigs as an animal model for studying growth restricted offspring with post-natal catch-up growth. Am. J. Physiol. Regul. Integr. Comp. Physiol. 219–27 (2018).
Soonpaa, M. H. & Field, L. J. Assessment of cardiomyocyte DNA synthesis during hypertrophy in adult mice. Am. J. Physiol. 266, H1439–H1445 (1994).
Bubb, K. J. et al. Intrauterine growth restriction delays cardiomyocyte maturation and alters coronary artery function in the fetal sheep. J. Physiol. 578, 871–881 (2007).
Briscoe, T. A. et al. Cardiovascular and renal disease in the adolescent guinea pig after chronic placental insufficiency. Am. J. Obstet. Gynecol. 191, 847–855 (2004).
Rolph, T. P., Jones, C. T. & Parry, D. Ultrastructural and enzymatic development of fetal guinea pig heart. Am. J. Physiol. 243, H87–H93 (1982).
Kind, K. L., Simonetta, G., Clifton, P. M., Robinson, J. S. & Owens, J. A. Effect of maternal feed restriction on blood pressure in the adult guinea pig. Exp. Physiol. 87, 469–477 (2002).
Porrello, E. R. et al. Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080 (2011).
Levkau, B. et al. Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation 117, 1583–1593 (2008).
Kooij, V. et al. Profilin modulates sarcomeric organization and mediates cardiomyocyte hypertrophy. Cardiovasc Res. 110, 238–248 (2016).
Crispi, F. et al. Fetal growth restriction results in remodeled and less efficient hearts in children. Circulation 121, 2427–2436 (2010).
Fernandez, E., Siddiquee, Z. & Shohet, R. V. Apoptosis and proliferation in the neonatal murine heart. Dev. Dyn. 221, 302–310 (2001).
Olivetti, G., Melissari, M., Capasso, J. M. & Anversa, P. Cardiomyopathy of the aging human heart. Myocyte loss and reactive cellular hypertrophy. Circ. Res. 68, 1560–1568 (1991).
Xu, Y., Williams, S. J., O’Brien, D. & Davidge, S. T. Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J. 20, 1251–1253 (2006).
Chen, X. et al. Adolescent feline heart contains a population of small, proliferative ventricular myocytes with immature physiological properties. Circ. Res. 100, 536–544 (2007).
Graham, E. & Bergmann, O. Dating the heart: exploring cardiomyocyte renewal in humans. Physiology 32, 33–41 (2017).
Weinberger, F. et al. Cardiac repair in guinea pigs with human engineered heart tissue from induced pluripotent stem cells. Sci. Transl. Med. 8, 363ra148 (2016).
Chen, J. X. et al. Inefficient reprogramming of fibroblasts into cardiomyocytes using Gata4, Mef2c, and Tbx5. Circ. Res. 111, 50–55 (2012).
Ieda, M. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142, 375–386 (2010).
Zhao, Y. et al. High-efficiency reprogramming of fibroblasts into cardiomyocytes requires suppression of pro-fibrotic signalling. Nat. Commun. 6, 8243 (2015).
Acknowledgements
This work was supported by grants from the American Heart Association (B.K.S.); Department of Defense (B.K.S.); Department of Pediatrics, Augusta University, Augusta, GA (E.P.M., A.A.S., and B.K.S.); National Institutes of Health (J.A.T.); and the Children’s Health Research Institute, University of Western Ontario, London, Canada (TRHR, BSR). BSR is the recipient of a Canada Research Chair Tier I in Fetal and Neonatal Health and Development.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Masoumy, E.P., Sawyer, A.A., Sharma, S. et al. The lifelong impact of fetal growth restriction on cardiac development. Pediatr Res 84, 537–544 (2018). https://doi.org/10.1038/s41390-018-0069-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-018-0069-x
This article is cited by
-
The association of low body mass index with neonatal morbidities in preterm infants
Scientific Reports (2021)
-
Changes in miR 21 and 23b expression in postnatal hypertrophic heart derived from gestational diabetes precede dilated cardiomyopathy
Journal of Biosciences (2021)
