
Abstract
Helminths are multicellular ancient organisms residing as parasites at mucosal surfaces of their host. Through adaptation and co-evolution with their hosts, helminths have been able to develop tolerance mechanisms to limit inflammation and avoid expulsion. The study of helminth infections as an integral part of tissue immunology allowed us to understand fundamental aspects of mucosal and barrier immunology, which led to the discovery of a new group of tissue-resident immune cells, innate lymphoid cells (ILC), over a decade ago. Here, we review the intricate interplay between helminth infections and type 2 ILC (ILC2) biology, discuss the host metabolic adaptation to helminth infections and the metabolic pathways fueling ILC2 responses. We hypothesize that nutrient competition between host and helminths may have prevented chronic inflammation in the past and argue that a detailed understanding of the metabolic restraints imposed by helminth infections may offer new therapeutic avenues in the future.
Similar content being viewed by others

Introduction
Parasitic helminths are large multicellular organisms residing at mucosal and non-mucosal tissues of their host. As ancient pathogens, the evolution of helminths is intimately intertwined with human evolution and our ancestors suffered from chronic and recurrent helminth infections1. Rarely seen in Western, industrialized countries nowadays, chronic helminth infections are still endemic in developing countries. However, despite creating tissue damage by migrating through or residing in tissues of the host organism including the muscle, intestine, lung and skin to complete their life cycle, in most cases helminth infections do not result in persistent and uncontrolled inflammation in a well-nourished population2,3,4,5. However, helminth infections can cause severe problems such as stunting in undernourished children6. Lack of overt immune activation and inflammation has been largely attributed to the intimate evolutionary relationship between the host and parasitic helminths, as worms have developed mechanisms to induce tolerance, tissue maintenance and repair1,7. This specific host-pathogen interaction allows the usage of helminth infections to study not only basic concepts of protective tissue immunology but also mechanisms of immune tolerance, tissue maintenance and restoration. In general, infections with tapeworms, flukes or roundworms induce innate and adaptive type 2 immune responses8. While innate type 2 immune responses include the induction of tissue-resident alternatively activated macrophages (AAM), activation of mast cells and eosinophils, the adaptive immune response mainly comprises the generation of adaptive T helper 2 (Th2) cells9. Furthermore, the study of helminth immunology was pivotal in the discovery of a new innate type 2 cell population, type 2 innate lymphoid cells (ILC2) over a decade ago. In this review, we discuss the aspects of basic mucosal immunology that studies using parasitic helminths have brought to the community by focusing on how helminth infections shaped our current understanding of ILC2 biology, metabolism and function.
The discovery of ILC2
To study helminth infections in vivo, mouse models of intestinal helminth infection are mainly based on three model parasites, Nippostrongylus brasiliensis, Heligmosomoides polygyrus bakeri and Trichuris muris, with the gastrointestinal rat nematode N. brasiliensis probably being the most frequently used for studies of mucosal immunology. Both N. brasiliensis and H. polygyrus infect the small intestine of mice10,11,12, while T. muris is found in the caecum and proximal colon13. N. brasiliensis induces a strong type 2 immune response and is rapidly expelled, making it an excellent model for studying protective type 2 immune responses to helminths in mice. In contrast, the natural murine parasite H. polygyrus establishes a chronic infection in C57BL/6 mice in contrast to more resistant BALB/c mice14,15,16. T. muris, depending on the infectious dose, can generate either chronic persistent infections, characterized by a Th1 response and the production of the cytokine interferon (IFN)-γ (low dose infection with ∼25 eggs), or acute infections cleared by a strong Th2 response with the production of interleukin (IL)-5, IL-9, and IL-13 in response to high amounts of eggs (∼150 eggs). This dose-dependent switch in the immune response provides an excellent model to study protective anti-helminth immunity17,18,19.
Before the discovery of ILC2, the production of the type 2 cytokines IL-4, IL-5, IL-9, and IL-13 was mainly attributed to Th2 cells20. However, it became clear several years before the first formal classification and identification of ILC2 as a distinct and separate subset, that a population of innate immune cells must exist21. This initial observation eventually led to the discovery and first characterization of ILC2 as a separate immune cell subset in the mesenteric adipose tissue21 or in the intestine in the context of infections with N. brasiliensis22,23 and T. muris24. In general, ILC have been classified into three distinct subsets, ILC1, ILC2, and ILC3 resembling the discrete T helper cell subsets Th1, Th2 or Th17 cells based on the expression of characteristic transcription factors and signature cytokines25. ILC1 including natural killer (NK) cells express T-bet (encoded by Tbx21) and produce the effector cytokine IFN-γ. ILC1 are implicated in protecting against intracellular pathogens such as Toxoplasma gondii but have also been connected to the pathophysiology of inflammatory bowel disease (IBD)26,27,28. ILC2 express the transcription factor GATA3 and produce the cytokines IL-4, IL-5, IL-9, IL-13, and amphiregulin, mediating anti-helminth immunity13,22,23,29. Besides this protective function, chronic activation and dysregulation of ILC2 may also result in ILC2-driven pathology in the context of allergies and asthma28,30. In contrast, ILC3 are characterized by the expression of the transcription factor RORγt and the cytokine IL-22, mediating anti-bacterial responses28,31,32,33, but also chronic inflammatory conditions such as psoriasis or IBD34,35,36.
The protective function of ILC2 in helminth infections
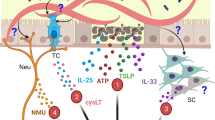
The protective role of ILC2 upon helminth infection has been studied extensively and different mechanisms of ILC2 biology promote and contribute to anti-helminth immunity. In particular, ILC2-derived IL-13 was shown to be essential for protective anti-helminth responses by directly acting on intestinal epithelial and goblet cells, inducing hyperplasia, excessive mucus production and increased muscle contractility, culminating in a “weep and sweep” response assisting worm expulsion37,38 (Fig. 1). In addition to limiting parasite burden, ILC2-derived IL-4 induces class-switching to IgE-producing B cells39,40, while IL-13 and amphiregulin suppress inflammation, promote tissue repair and induce wound healing by acting on epithelial cells and alternatively activated macrophages (AAM)41 (Fig. 1). The maintenance of ILC2 responses is critically dependent on the autocrine survival factor IL-929,42 (Fig. 1), manifesting in aggravated damage and impaired restoration of lung function of IL-9R-deficient mice in response to infections with larval stages of N. brasiliensis traveling through the lung before entering the intestine29. Although administration of neutralizing IL-9 antibodies has been shown to impair T. muris and H. polygyrus bakeri expulsion from the intestine43,44,45,46, the direct effect of ILC2-derived IL-9 in this context remains to be revealed.

Upon intestinal helminth infection, mucosal epithelial barrier sites release the alarmins IL-33 and TSLP, while chemosensory tuft cells are activated by succinate to secrete IL-25 and the leukotriene LTC4 activating ILC2. ILC2 release IL-13 acting on alternatively activated macrophages (AAM) and promoting epithelial, goblet and tuft cell hyperplasia resulting in the “weep and sweep” response and expulsion of intestinal helminths. The ILC2-derived cytokines amphiregulin (Areg) and IL-9 mediate tissue repair or autocrine survival and maintenance of ILC2, respectively. IL-13 further potentiates IL-25 release from tuft cells and IL-4 induces a class switch to IgE producing B cells, while IL-5 triggers eosinophilia.
Activation of ILC2 by helminths
In general, tissue damage created by intestinal infection with helminths triggers the epithelium to produce the alarmin cytokines IL-33, IL-25 and TSLP resulting in the activation of ILC2. Moreover, besides epithelial cells, stromal cells are known as potent producers of IL-3347,48,49,50,51. Mice lacking IL-25 or IL-33 show delayed expulsion of N. brasiliensis, T. muris, T. spiralis and H. polygyrus48,52,53,54,55,56 and IL-25-deficient BALB/c mice are completely unable to expel adult H. polygyrus57, corroborating the importance of these cytokines for initiating a protective anti-helminth immune response. Furthermore, combined IL-25- and IL-33-deficiency completely ablates the expansion of ILC2 consequently leading to severe defects in N. brasiliensis expulsion23. Vice versa, protective anti-helminth immunity can be induced by delivery of recombinant IL-25 to Rag-deficient mice leading to the activation of ILC2 and enhanced production of the effector cytokines IL-5 and IL-1322,57. Other than IL-33 and IL-25, TSLP is involved in the protective function only against T. muris, but not N. brasiliensis or H. polygyrus infections, as TSLP is mainly expressed in the large intestine58,59. Interestingly, only if applied at an early time point after infection with T. muris, exogenous IL-33 administration can promote worm expulsion56, suggesting that early activation of ILC2 may be essential for efficient worm expulsion.
Apart from facilitating the discovery of ILC2, helminth infections proved elemental in the recognition of tuft cells, a rare chemosensory cell type in the intestinal epithelium, as essential initiators of type 2 immunity. Tuft cells provide the long-sought essential source of intestinal IL-25, required for the activation of ILC2 and lack of tuft cells results in abrogated hyperplasia, compromised worm expulsion and defective mucosal type 2 immunity60. Secretion of IL-4 and IL-13 from ILC2 upon exposure to IL-25 is not only inducing goblet cell activation and hyperplasia, but also employs a feed-forward loop by promoting tuft cell differentiation and hyperplasia and thus further production of IL-2561,62. However, this tuft cell-ILC2 axis is mainly described for the murine small intestine, although certain nematodes (T. muris) mainly reside in the caecum or distal colon. Sensing the of the tricarboxylic acid (TCA) cycle intermediate succinate can also activate tuft cells. Succinate binds to the succinate receptor 1 (SUCNR1) initiating a signaling cascade that results in the release of IL-25 in a TRPM5-dependent manner63,64,65. This activation loop appears to be essential for the induction of type 2 immunity in the context of the protist Tritrichomonas, but neglectable for the induction of anti-helminth immunity.
Remarkably, helminths possess mechanisms able to interfere with the activation of ILC2 even in peripheral organs such as the lung66 through the release of excretory-secretory (ES) products. The H. polygyrus-derived protein HpARI (H. polygyrus Alarmin Release Inhibitor) sequesters IL-33 within the nucleus of necrotic cells, preventing its release and thus ILC2 activation67. As a result, delivery of HpARI directly to mice potently inhibited ILC2-mediated allergen responses to Alternaria Alternata and impaired the expulsion of N. brasiliensis67. This concept was then expanded to another ES-protein, H. polygyrus Binds Alarmin Receptor and Inhibits (HpBARI)68. HpBARI binds to the ST2/IL-33 receptor complex preventing IL-33 signaling and thus the induction of allergen-induced airway inflammation68. These findings highlight the propensity that helminths possess to directly modulate immune responses to prevent expulsion.
Neuro-immune control of helminth infections
Besides the release of IL-25, the latest data also suggest an active role of tuft cells in the production of cysteinyl leukotriene LTC4, a known activator of ILC2 resulting in the release of IL-5 and IL-13 to support anti-helminth defense62. The neurotransmitter acetylcholine (ACh) can also be directly synthesized by ILC2 and genetic disruption of ACh synthesis in ILC2 results in reduced immunity to N. brasiliensis infection69. Furthermore, intestinal ILC2 colocalize with cholinergic neurons expressing neuropeptide neuromedin U (NMU) and enteric neurons were shown to release NMU in response to helminth products70. ILC2 express the NMU receptor 1 and stimulation with NMU induces rapid proliferation, activation and production of the effector cytokines IL-5, IL-9, and IL-1370,71. Exogenous supplementation of NMU during N. brasiliensis infection led to accelerated worm expulsion, while NMU-deficient mice displayed increased worm burden and a reduction in ILC2-mediated allergic airway inflammation71. This further emphasizes the essential role of neuronal activation of ILC2 in helminth clearance (Fig. 2). Besides NMU, vasoactive intestinal peptide (VIP) released from neurons in response to feeding, is an important modulator of ILC function. VIP signals through the VIP receptor 2 (VIPR2) on ILC2 and promotes the production of IL-5 and thus the recruitment of eosinophils72. Additionally, VIP acts in concert with IL-33 to activate mammalian target of rapamycin (mTOR), which increases glycolysis, potentiates the production of intestinal ILC2 effector cytokines, and increases resistance to high-dose infection with T. muris73. This may be in part due to increased IL-33 receptor (ST2/IL-33R) expression on ILC2 by VIP, amplifying the responsiveness to activation by IL-3372,74. A similar, potentiating effect is observed for intestinal ILC3 and VIP binding to VIPR2 promoting the release of IL-22 and the anti-bacterial immune response in the gastrointestinal tract75, although one study reported a suppressive effect of VIP on ILC376. In addition to activation of intestinal ILC2, VIP released by pulmonary neurons induces IL-5 release by ILC2 promoting allergic inflammation74. This function seems to be amplified by a direct stimulatory role of IL-5 acting on nociceptors, which accelerated the release of VIP. Thus, the release of VIP and its function on ILC2 may act as an important feed-forward loop in type 2 inflammation. In contrast to the stimulating function of NMU and VIP on ILC2, epinephrine (EPI) signaling through the β2-adrenergic receptor (β2AR) possesses the propensity to repress ILC2 responses77. Intestinal ILC2 colocalize with adrenergic neurons and β2AR deficiency results in accumulation of IL-13 expressing ILC2, aggravated eosinophilia and a concomitant reduction in worm burden upon infection with N. brasiliensis. This putative nervous-immune crosstalk may prevent overactivation of ILC2 and ILC2-mediated pathologies, as ablation of adrenergic signaling augmented airway inflammation induced by intranasal application of IL-33. In contrast, treatment with β2AR agonists impaired the production of ILC2-derived effector cytokines ameliorating airway inflammation77 (Fig. 2).

Intestinal sensory neurons release vasoactive intestinal peptide (VIP), neuropeptide neuromedin U (NMU) or epinephrine (EPI). Feeding-induced VIP potentiates (IL-33-mediated) activation of ILC2, resulting in increased production of IL-5 and accelerated helminth expulsion. In addition, secreted IL-5 induced a feed-forward loop by acting on VIP-producing neurons to further boost the release of VIP. Helminth infections activate cholinergic neurons to produce neuromedin U enabling rapid ILC2 proliferation and release of IL-5, IL-9, and IL-13 enhancing helminth expulsion. Adrenergic neurons release EPI inhibiting ILC2 function, impairing ILC2-mediated anti-helminth immunity.
The intricate interplay of helminths and host metabolism
Helminths are multicellular parasites, and thus require a substantial amount of energy for survival, growth, and replication. A possible link to host metabolism was first extrapolated from epidemiological studies in which a negative correlation between obesity, type 2 diabetes and endemic chronic helminth infections suggested a causal connection78,79. This assumption received experimental support from Locksley and colleagues showing that infection with N. brasiliensis maintained the metabolic health of mice and glucose tolerance through adipose tissue eosinophilia80. Later, based on the knowledge that ILC2 were originally discovered in the mesenteric fat, a direct effect of ILC2 and adipose tissue homeostasis was established81,82. Non-obese adipose tissue-infiltrating immune cells mainly consist of AAM, eosinophils, T regulatory cells (TReg) and ILC280,81,83,84. This homeostasis is perturbed in high-fat diet (HFD)-induced obesity or obese or diabetic humans, where a loss of ILC2 coincides with increases in ILC1, neutrophils and inflammatory macrophages contributing to an inflammatory tissue environment by accelerating adipose tissue fibrogenesis and impairing glycemic tolerance21,84,85. The role of ILC2 in the regulation of host metabolism was further demonstrated by depletion of ILC2 in wild-type or obese Rag-deficient mice, resulting in weight gain and glucose intolerance86,87. Furthermore, treatment of obese mice with IL-33 or IL-25 leads to weight loss and increased glucose tolerance, whereas lack of IL-33 results in weight gain and glucose intolerance82,83,88, demonstrating the pivotal function of ILC2 and ILC2-activating cytokines in maintaining the metabolic fitness of the organism.
Yet, how ILC2 control host metabolism remains somewhat elusive. One proposed mechanism is that ILC2-derived IL-5, and IL-13 induce AAM and eosinophil accumulation, and that both cell types promote the beiging of white adipocytes89,90. Nonetheless, although AAM were initially suggested to directly induce thermogenesis in brown and lipolysis in white adipocytes91, this direct involvement of AAM in adipocyte metabolism was lately challenged89. In addition, the function of IL-13 in this context is not entirely clear, but lack of IL-13 is associated with weight loss, reduced eosinophils and AAM in adipose tissue92. Despite this overwhelming evidence in support of a protective effect of ILC2 preventing metabolic dysfunction, this concept was recently challenged. Rag-Il2rg-deficient mice lacking ILC do not develop obesity and a recent study showed that transfer of intestinal but not adipose ILC2 restored the capacity of these mice to develop HFD-induced obesity93. Although these results may have been influenced by homeostatic expansion of ILC2 following transfer into Rag-Il2rg-deficient mice, this clearly shows that more research is necessary to identify the exact modes of action. In this context, a recent study shed some light on potential neuronal control of adipose tissue ILC2. Sympathetic nerves primed adipose mesenchymal cells to produce glial-derived neurotrophic factor (GDNF) and ablation of GDNF signaling led to a significant defect in ILC2 function shaping host metabolism by reducing glucose tolerance and increasing the propensity for HFD-induced obesity94. Besides the function of IL-5 and IL-13, eosinophil and ILC2-derived IL-4 directly prompted the proliferation and differentiation of adipocyte precursors into beige adipocytes95, characterized by large quantities of mitochondria and expression of uncoupling protein 1 (UCP1)90,96. Upregulation of UCP1 inducing thermogenesis and beiging of white adipose tissue appears to be induced by the release of methionine-enkephalin (MetEnk) from ILC2 in response to IL-33. This leads to increased energy consumption, which prevents obesity and metabolic inflammation83. Alternatively, IL-33 may also be able to directly induce beiging of white adipose tissue by regulating the appropriate splicing of Ucp1 mRNA97. A direct mode of action is supported by the fact that IL-33 can prevent inflammation of adipose tissue by inducing lipolysis98. As both IL-25 and IL-33 modulate host metabolism, their release in the context of helminth infections may be at the center of the observed metabolic alterations. Indeed, H. polygyrus has been shown to attenuate obesity via upregulation of uncoupling protein 1 (Ucp1) increasing energy expenditure and lipolysis in adipose tissue. These effects were mediated through the induction of AAM99 and dependent on norepinephrine100. Besides AAM polarization, the onset of obesity has been further associated with increased nematode induced Th2 and TReg responses in connection to upregulation of UCP1, associated with higher energy expenditure99. However, the involvement of ILC2 in respect to cellular metabolic homeostasis is still unclear.
Metabolic control of ILC2 function
As discussed above, besides their direct immune modulatory capacities, helminth infections can alter the metabolism of the host in general and glucose metabolism in particular101. Consequently, such metabolic changes may impair the protective immune response against infections and a few studies started to investigate the cellular metabolism of ILC2 in helminth infections. Protective ILC2 responses in helminth infection may circumvent competition with helminths for glucose by metabolizing externally acquired fatty acids (FA) for the generation of energy101. Furthermore, malnutrition and vitamin A deficiency is characterized by profound impairment of the adaptive immune system and a selective defect in ILC3102,103. Surprisingly, in vitamin A insufficient mice ILC2 cellularity is increased, and IL-13 cytokine production is boosted to sustain barrier integrity and the defense against helminths102. In the context of intestinal low-dose T. muris infection, ILC2 acquire and utilize fatty acids to boost oxidative phosphorylation to compensate for the loss of the micronutrient vitamin A101. Increased mitochondrial oxidation of FA leads to accelerated proliferation and IL-13 production to maintain anti-parasite responses101. In addition to externally acquired FA, the mobilization of internal FA through autophagy may be essential to maintain the functionality of ILC2, as deletion of the gene autophagy-related gene 5 (Atg5) resulted in accelerated glycolysis but impaired the capacity of ILC2 to oxidize FA in mitochondria104. The important implication of this discovery is that ILC2-mediated tissue repair and anti-helminth immunity are maintained in settings of low glucose availability and malnutrition. Since our ancestors, frequently experienced periods of fluctuating nutrient uptake and chronic helminth infections, a switch to fatty acid metabolism represents a host adaptation to ensure barrier integrity in times of severe dietary restriction to extend host survival. As both, malnutrition and helminth infections are largely absent in the Western World, this evolutionary context of malnutrition and competition for nutrients with multicellular parasites may have been overturned by modern lifestyle nowadays, with unknown consequences for ILC-mediated immune responses. In vivo studies have demonstrated that consumption of excess nutrients in form of a high-fat diet (HFD) leads to allergic asthma105. Interestingly, in airway inflammation, pathogenic ILC2 require increased uptake of both glucose and lipids, which drive extensive proliferation and pathogenicity106. In contrast to the metabolic profile of protective ILC2 in helminth infections101, externally acquired fatty acids are used for anabolic processes to build up cellular membranes106. Accelerated nutrient acquisition was controlled by IL-33 and resulted in the transient storage of externally acquired FA in lipid droplets to build up cellular membranes, a process controlled by the enzyme diacylglycerol o-acyltransferase 1 (DGAT1). This specific metabolic program is under the transcriptional control of peroxisome proliferator-activated receptor gamma (PPARγ) serving as a key transcription factor controlling the activation and lipid metabolism of ILC2. PPARγ is highly expressed in lung and adipose tissue ILC2107 and genetic ablation or pharmacological inhibition of both PPARγ and DGAT1 strongly ameliorated not only ILC2-dependent airway inflammation but also IL-33-mediated colorectal cancer growth106,108. Thus, pathogenic activation might rely on the availability of excess nutrients. In support of this idea, ketogenic diets limiting the availability of glucose provide a potent dietary intervention to treat ILC2-driven pathologies such as allergic asthma106. Expansion of ILC2 in the airways was dramatically impaired, and although this beneficial effect of the ketogenic diet was independent of the microbiota, other modes of action may be involved. Nonetheless, since ketogenic diets mimic a state of fasting and nutrient deprivation this may explain how periods of malnutrition have counterbalanced the development of chronic inflammation in the past and how the increased incidence of immune pathology in the Western World may be driven by increased consumption of carbohydrate and fat106 (Fig. 3).

Helminth infections induce several host metabolic changes resulting in increased glucose tolerance, insulin sensitivity and metabolic health, and an overall reduction in obesity. The effects of such changes on cellular metabolism are not known. However, ILC2 can use the acquisition of external FA either for promotion of OXPHOS and ATP generation to fuel anti-helminth immunity via IL-13 or DGAT1-mediated transient storage in lipid droplets and conversation into phospholipids for proliferation in the context of asthma. PPARγ serves as a key regulator of lipid metabolism in ILC2. In contrast, autophagy generates internal FA used in mitochondria, promoting OXPHOS and ILC2 function. AA and glucose metabolism controlled by mTOR represent additional important metabolic pathways, fueling ILC2-mediated immunity. AA are metabolized via OXPHOS, while glucose converted to lactate fuels glycolysis and IL-13 production. The enzyme ARG-1, metabolizing L-arginine, also supports glycolysis. Helminths favor glucose as their primary nutritional source and alter the amino acid availability of the host, which may result in nutrient competition between host and parasites and could potentially impair ILC2-mediated anti-helminth immunity.
How helminths may impact on ILC2 metabolism
Apart from lipid metabolism, ILC2 function is controlled by amino acid (AA) metabolism and glycolysis regulated by high expression of arginase-1 (Arg1) cleaving L-arginine into urea and ornithine109. Genetic deletion of Arg1 led to reduced ILC2 proliferation and total numbers and dampened airway inflammation109. Contrary, a study used genetic deletion of Arg1 specifically in mature ILC2 and found that neither proliferation nor IL-5 or IL-13 cytokine production was affected during helminth infection110, suggesting that Arg-1 activity might be depending on the specific inflammatory environment and context. Nonetheless, in confirmation of an important function of AA metabolism, human naïve ILC2 require AA for ATP production via OXPHOS111. Upon ILC2 activation biosynthetic demand is higher and branched-chain AA (BCAA) are used to maintain cellular fitness and proliferation111, while mTOR controlled glycolysis promotes ILC2 functionality and the production of IL-13106,111,112. Helminths may exploit this dependence on AA to avoid expulsion. In fact, infections with T. muris increase the amount of AA in the feces113, potentially indicating reduced absorption and depletion of AA as a general mechanism of host metabolic manipulation by helminths (Fig. 3). Moreover, at the chronic stage of liver infections with Opisthorchis felineus causing hepatobiliary disease, metabolomic analysis of the serum revealed a shift towards lipid metabolism accompanied by depletion of AA114. As helminths favor glucose over AA as their primary nutrition source115,116, this might represent an adaptation mechanism limiting the activation and expansion of ILC2 in response to helminth infections to prevent expulsion.
Finally, the observed changes in host metabolism could be a direct effect of competition for locally available nutrients, which may also explain why helminths change the composition of the intestinal microbiome in mice and humans117,118,119,120,121,122,123. Specifically, helminth infections have been associated with increased abundance of carbohydrate-metabolizing Lactobacillaceae121,124,125,126. Improved glucose tolerance and preserved insulin resistance in HFD-fed mice by helminths could be mediated by alterations of the intestinal microbiota78,127. Consequently, helminth infections may prevent pathogenic activation and chronic inflammation by imposing a metabolic restrain on the host. Some of these aspects have been addressed by probing the beneficial effect of helminth infection in the context of allergies. In confirmation of earlier data128,129,130, infection with H. polygyrus protected mice from allergies, an effect abrogated if antibiotics were used to deplete the microbiota131. Furthermore, helminth infections elevate the availability of microbial-derived short-chain fatty acids (SCFAs) by increasing the abundance of Bacteroidetes123,132. This directly results in attenuated allergic airway inflammation by signaling through the cognate SCFA receptor GPR41 (also named free fatty acid receptor 3 (FFAR3))131. Cleary, more research is required to validate this concept and to follow up on this understudied but potentially elemental avenue of helminth research.
Conclusion
Helminths are remarkable in their ability to adapt to their host environment, evading immune activation and expulsion, while being able to feed off and co-exist with their host. Despite having lived with chronic worm infections for centuries, we understand remarkably little about the consequences of untangling such a tight-knit relationship over a relatively short period of time as a consequence of increased hygiene in industrialized countries. A better understanding of this mutualism may be particularly important in regard to the reported alterations in host metabolism implemented by helminths residing at barrier sites. Changes in host metabolism such as increases in glucose tolerance may reflect the need for parasites to gain sufficient access to nutrients, which places intestinal helminths in direct competition with the host and microbiota. Nonetheless, our knowledge of how helminths manipulate host and immune cell metabolism is remarkably limited. Microbiota-derived metabolites produced by intestinal bacteria upon the breakdown of nutrients are emerging as potent activators or inhibitors of cellular function131,133,134. Since we currently lack a complete understanding of the mechanism of how helminth infections can influence host metabolism, it is intriguing to speculate that some of the beneficial effects we observe may be directly mediated by such biologically active compounds. Thus, it may be important to focus future research on if or how helminth infections may alter the immune response by regulating the abundance of microbiota-derived metabolites other than SCFA. Given the regulative burden and public health concerns related to using host-adapted parasites therapeutically, unlocking the immune and metabolism-modulating potential of microbiota-derived metabolites may represent a more feasible approach to stall the negative effects associated with a general absence of helminth infections in the Western World.
References
Amoroso, C. R. & Nunn, C. L. Epidemiological transitions in human evolution and the richness of viruses, helminths, and protozoa. Evol. Med. Public Health 9, 139–148 (2021).
Hagel, I. et al. Helminthic infection and anthropometric indicators in children from a tropical slum: Ascaris reinfection after anthelmintic treatment. J. Trop. Pediatr. 45, 215–220 (1999).
Ranque, J. P., Chippaux, A. & Garcia, S. Follow-up of Ascaris lumbricoides and Trichuris trichiura infections in children living in a community treated with ivermectin at 3-monthly intervals. Ann. Tropical Med. Parasitol. 95, 389–393 (2001).
Hesham Al-Mekhlafi, M. et al. Pattern and predictors of soil-transmitted helminth reinfection among aboriginal schoolchildren in rural Peninsular Malaysia. Acta. Trop. 107, 200–204 (2008).
Saldiva, S. R. M., Carvalho, H. B., Castilho, V. P., Struchiner, C. J. & Massad, E. Malnutrition and susceptibility to enteroparasites: Reinfection rates after mass chemotherapy. Paediatr. Perinat. Epidemiol. 16, 166–171 (2002).
Crompton, D. W. T. & Nesheim, M. C. Nutritional impact of intestinal helminthiasis during the human life cycle. Annu. Rev. Nutr. 22, 35–59 (2002).
Rafaluk-Mohr, C. et al. Microbial protection favors parasite tolerance and alters host-parasite coevolutionary dynamics. Curr. Biol. 32, 1593–1598 (2022). e3.
Inclan-Rico, J. M. & Siracusa, M. C. First Responders: Innate Immunity to Helminths. Trends Parasitol. 34, 861–880 (2018).
Oliphant, C. J., Barlow, J. L. & Mckenzie, A. N. J. Insights into the initiation of type 2 immune responses. Immunology 134, 378–385 (2011).
Rapin, A. et al. Infection with a small intestinal helminth, Heligmosomoides polygyrus bakeri, consistently alters microbial communities throughout the murine small and large intestine. Int J. Parasitol. 50, 35–46 (2020).
Ishiwata, K., Nakao, H., Nakamura-Uchiyama, F. & Nawa, Y. Immune-mediated damage is not essential for the expulsion of Nippostrongylus brasiliensis adult worms from the small intestine of mice. Parasite Immunol. 24, 381–386 (2002).
Inagaki-Ohara, K., Sakamoto, Y., Dohi, T. & Smith, A. L. γδ T cells play a protective role during infection with Nippostrongylus brasiliensis by promoting goblet cell function in the small intestine. Immunology 134, 448–458 (2011).
Hurst, R. J. M. & Else, K. J. Trichuris muris research revisited: a journey through time. Parasitology 140, 1325–1339 (2013).
Valanparambil, R. M., Tam, M., Jardim, A., Geary, T. G. & Stevenson, M. M. Primary Heligmosomoides polygyrus bakeri infection induces myeloid-derived suppressor cells that suppress CD4 + Th2 responses and promote chronic infection. Mucosal Immunol. 10, 238–249 (2017).
Filbey, K. J. et al. Innate and adaptive type 2 immune cell responses in genetically controlled resistance to intestinal helminth infection. Immunol. Cell Biol. 92, 436–448 (2014).
Reynolds, L. A., Filbey, K. J. & Maizels, R. M. Immunity to the model intestinal helminth parasite Heligmosomoides polygyrus. Semin Immunopathol. 34, 829–846 (2012).
Cruickshank, S. M. et al. Rapid dendritic cell mobilization to the large intestinal epithelium is associated with resistance to Trichuris muris infection. J. Immunol. 182, 3055–3062 (2009).
Bancroft, A. J., Else, K. J., Humphreys, N. E. & Grencis, R. K. The effect of challenge and trickle Trichuris muris infections on the polarisation of the immune response. Int J. Parasitol. 31, 1627–1637 (2001).
Else, K. J., Finkelman, F. D., Maliszewski, C. R. & Grencis, R. K. Cytokine-mediated regulation of chronic intestinal helminth infection. J. Exp. Med. 179, 347–351 (1994).
Fallon, P. G. et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 203, 1105–1116 (2006).
Moro, K. et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 463, 540–544 (2010).
Price, A. E. et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA. 107, 11489–11494 (2010).
Neill, D. R. et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 464, 1367–1370 (2010).
Saenz, S. A. et al. IL25 elicits a multipotent progenitor cell population that promotes T(H)2 cytokine responses. Nature 464, 1362–1366 (2010).
Bal, S. M., Golebski, K. & Spits, H. Plasticity of innate lymphoid cell subsets. Nat. Rev. Immunol. 20, 552–565 (2020).
Fuchs, A. et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-γ-producing cells. Immunity 38, 769–781 (2013).
Klose, C. S. N. et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356 (2014).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature 517, 293–301 (2015).
Turner, J. E. et al. IL-9-mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 210, 2951–2965 (2013).
Licona-Limón, P., Kim, L. K., Palm, N. W. & Flavell, R. A. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 14, 536–542 (2013).
Soderquest, K. et al. Genetic variants alter T-bet binding and gene expression in mucosal inflammatory disease. PLOS Genet. 13, e1006587 (2017).
Schroeder, J.-H. et al. T-Bet Controls Cellularity of Intestinal Group 3 Innate Lymphoid Cells. https://doi.org/10.3389/fimmu.2020.623324
Vonarbourg, C. et al. Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt(+) innate lymphocytes. Immunity 33, 736–751 (2010).
Forkel, M. & Mjösberg, J. Dysregulation of Group 3 Innate Lymphoid Cells in the Pathogenesis of Inflammatory Bowel Disease. Curr. Aller. Asthma Rep. 16, (2016).
Zeng, B. et al. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell. Death Dis. 10, (2019).
Bielecki, P. et al. Skin-resident innate lymphoid cells converge on a pathogenic effector state. Nature 592, 128–132 (2021).
Campbell, L. et al. ILC2s mediate systemic innate protection by priming mucus production at distal mucosal sites. J. Exp. Med. 216, 2714–2723 (2019).
Cliffe, L. J. et al. Accelerated intestinal epithelial cell turnover: A new mechanism of parasite expulsion. Science 308, 1463–1465 (2005).
Lebman, D. A. & Coffman, R. L. Interleukin 4 causes isotype switching to IgE in T cell-stimulated clonal B cell cultures. J. Exp. Med. 168, 853–862 (1988).
Drake, L. Y., Iijima, K., Bartemes, K. & Kita, H. Group 2 innate lymphoid cells promote an early antibody response to a respiratory antigen in mice. J. Immunol. 197, 1335–1342 (2016).
Allen, J. E. & Sutherland, T. E. Host protective roles of type 2 immunity: Parasite killing and tissue repair, flip sides of the same coin. Semin Immunol. 26, 329–340 (2014).
Wilhelm, C. et al. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 12, 1071–1077 (2011).
Khan, W. I. et al. Modulation of intestinal muscle contraction by interleukin-9 (IL-9) or IL-9 neutralization: correlation with worm expulsion in murine nematode infections. Infect. Immun. 71, 2430–2438 (2003).
Chow, S. C., Brown, A. & Pritchard, D. Mucosal mast cell responses are not required for protection against infection with the murine nematode parasite Trichuris muris. Parasite Immunol. 22, 21–29 (2000).
Richard, M., Grencis, R. K., Humphreys, N. E., Renauld, J. C. & Snick, J. van Anti-IL-9 vaccination prevents worm expulsion and blood eosinophilia in Trichuris muris-infected mice. Proc. Natl. Acad. Sci. USA. 97, 767–772 (2000).
Hepworth, M. R. et al. Mast cells orchestrate type 2 immunity to helminths through regulation of tissue-derived cytokines. Proc. Natl. Acad. Sci. USA. 109, 6644–6649 (2012).
Mahlakõiv, T. et al. Stromal Cells Maintain Immune Cell Homeostasis in Adipose Tissue via Production of Interleukin-33. Sci. Immunol. 4, (2019).
Martin, A. de et al. IL-33 mediated stromal-myeloid cell crosstalk controls intestinal helminth infestation. J. Immunol. 206, (2021).
Spallanzani, R. G. et al. Distinct immunocyte-promoting and adipocyte-generating stromal components coordinate adipose tissue immune and metabolic tenors. Sci Immunol 4, (2019).
Rana, B. M. J. et al. A stromal cell niche sustains ILC2-mediated type-2 conditioning in adipose tissue. J. Exp. Med. 216, 1999–2009 (2019).
Dahlgren, M. W. et al. Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches. Immunity 50, 707–722 (2019). e6.
Angkasekwinai, P. et al. Interleukin-25 (IL-25) promotes efficient protective immunity against Trichinella spiralis infection by enhancing the antigen-specific IL-9 response. Infect. Immun. 81, 3731–3741 (2013).
Pei, C. et al. Critical Role for Interleukin-25 in Host Protective Th2 Memory Response against Heligmosomoides polygyrus bakeri. Infect. Immun. 84, 3328–3337 (2016).
Owyang, A. M. et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J. Exp. Med. 203, 843–849 (2006).
Hung, L. Y. et al. Cellular context of IL-33 expression dictates impact on anti-helminth immunity. Sci. Immunol. 5, (2020).
Humphreys, N. E., Xu, D., Hepworth, M. R., Liew, F. Y. & Grencis, R. K. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J. Immunol. 180, 2443–2449 (2008).
Smith, K. A. et al. Concerted IL-25R and IL-4Rα signaling drive innate type 2 effector immunity for optimal helminth expulsion. Elife 7, (2018).
Taylor, B. C. et al. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J. Exp. Med. 206, 655–667 (2009).
Massacand, J. C. et al. Helminth products bypass the need for TSLP in Th2 immune responses by directly modulating dendritic cell function. Proc. Natl. Acad. Sci. USA 106, 13968–13973 (2009).
Moltke, J., von, Ji, M., Liang, H. E. & Locksley, R. M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 529, 221–225 (2016).
Gerbe, F. et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 529, 226–230 (2016). 2016 529:7585.
McGinty, J. W. et al. Tuft-Cell-Derived Leukotrienes Drive Rapid Anti-helminth Immunity in the Small Intestine but Are Dispensable for Anti-protist Immunity. Immunity 52, 528–541 (2020). e7.
Nadjsombati, M. S. et al. Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 49, 33–41 (2018). e7.
Schneider, C. et al. A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell 174, 271–284 (2018). e14.
Lei, W. et al. Activation of intestinal tuft cell-expressed Sucnr1 triggers type 2 immunity in the mouse small intestine. Proc. Natl. Acad. Sci. USA. 115, 5552–5557 (2018).
Badr, A. M., Al-Halbosiy, M. M. F. & Ridi, R. E. L. Differential immune responses to excretory–secretory antigens of lung-stage larvae of Schistosoma mansoni in mice and rats. J. Basic Appl. Zool. 69, 26–33 (2015).
Osbourn, M. et al. HpARI Protein Secreted by a Helminth Parasite Suppresses Interleukin-33. Immunity 47, 739–751 (2017). e5.
Vacca, F. et al. A helminth-derived suppressor of ST2 blocks allergic responses. Elife 9, 1–20 (2020).
Roberts, L. B. et al. Acetylcholine production by group 2 innate lymphoid cells promotes mucosal immunity to helminths. Sci. Immunol. 6, (2021).
Cardoso, V. et al. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549, 277–281 (2017).
Wallrapp, A. et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549, 351–356 (2017).
Nussbaum, J. C. et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 502, 245–248 (2013).
Pascal, M. et al. The neuropeptide VIP potentiates intestinal innate type 2 and type 3 immunity in response to feeding. Mucosal Immunol. (2022). https://doi.org/10.1038/S41385-022-00516-9
Talbot, S. et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87, 341–354 (2015).
Seillet, C. et al. The neuropeptide VIP confers anticipatory mucosal immunity by regulating ILC3 activity. Nat. Immunol. 21, 168–177 (2020).
Talbot, J. et al. Feeding-dependent VIP neuron-ILC3 circuit regulates the intestinal barrier. Nature 579, 575–580 (2020).
Moriyama, S. et al. β 2-adrenergic receptor-mediated negative regulation of group 2 innate lymphoid cell responses. Science 359, 1056–1061 (2018).
Khudhair, Z. et al. Gastrointestinal Helminth Infection Improves Insulin Sensitivity, Decreases Systemic Inflammation, and Alters the Composition of Gut Microbiota in Distinct Mouse Models of Type 2 Diabetes. Front. Endocrinol. (Lausanne) 11, (2021).
Rennie, C., Fernandez, R., Donnelly, S. & McGrath, K. C. Y. The Impact of Helminth Infection on the Incidence of Metabolic Syndrome: A Systematic Review and Meta-Analysis. Front. Endocrinol. (Lausanne) 12, (2021).
Wu, D. et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 332, 243–247 (2011).
Molofsky, A. B. et al. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 210, 535–549 (2013).
Hams, E., Locksley, R. M., McKenzie, A. N. J. & Fallon, P. G. Cutting edge: IL-25 elicits innate lymphoid type 2 and type II NKT cells that regulate obesity in mice. J. Immunol. 191, 5349–5353 (2013).
Brestoff, J. R. et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 519, 242–246 (2015).
Feuerer, M. et al. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat. Med. 15, 930–939 (2009).
O’Sullivan, T. E. et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity 45, 428–441 (2016).
Weisberg, S. P. et al. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 112, 1796–1808 (2003).
Okamura, T. et al. ILC2s improve glucose metabolism through the control of saturated fatty acid absorption within visceral fat. Front. Immunol. 12, 2714 (2021).
Wilhelm, C., Masouleh, S. K. & Kazakov, A. Metabolic regulation of innate lymphoid cell-mediated tissue protection-linking the nutritional state to barrier immunity. Front. Immunol. 8, (2017).
Fischer, K. et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med. 23, 623–630 (2017).
Pfeifer, A. & Hoffmann, L. S. Brown, beige, and white: The new color code of fat and its pharmacological implications. Annu. Rev. Pharm. Toxicol. 55, 207–227 (2015).
Nguyen, K. D. et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480, 104–108 (2011).
Duffen, J. et al. Modulation of the IL-33/IL-13 Axis in Obesity by IL-13Rα2. J. Immunol. 200, 1347–1359 (2018).
Sasaki, T. et al. Innate lymphoid cells in the induction of obesity. Cell Rep. 28, 202–217 (2019). e7.
Cardoso, F. et al. Neuro-mesenchymal units control ILC2 and obesity via a brain-adipose circuit. Nature 597, 410–414 (2021).
Lee, M.-W. et al. Activated Type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 160, 74–87 (2014).
Cypess, A. M. et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517 (2009).
Odegaard, J. I. et al. Perinatal Licensing of Thermogenesis by IL-33 and ST2. Cell 166, 841–854 (2016).
Miller, A. M. et al. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ. Res. 107, 650–658 (2010).
Su, C. W. et al. Helminth infection protects against high fat diet-induced obesity via induction of alternatively activated macrophages. Sci. Rep. 8, (2018).
Shimokawa, C. et al. Suppression of obesity by an intestinal helminth through interactions with intestinal microbiota. Infect. Immun. 87, (2019).
Wilhelm, C. et al. Critical role of fatty acid metabolism in ILC2-mediated barrier protection during malnutrition and helminth infection. J. Exp. Med. 213, 1409–1418 (2016).
Spencer, S. P. et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 343, 432–437 (2014).
Veldhoen, M. & Brucklacher-Waldert, V. Dietary influences on intestinal immunity. Nat. Rev. Immunol. 12, 696–708 (2012).
Galle-Treger, L. et al. Autophagy is critical for group 2 innate lymphoid cell metabolic homeostasis and effector function. J. Allergy Clin. Immunol. 145, 502–517.e5. (2020).
Fricke, K. et al. High fat diet induces airway hyperresponsiveness in mice. Sci. Rep. 8, 1–6 (2018).
Karagiannis, F. et al. Lipid-Droplet Formation Drives Pathogenic Group 2 Innate Lymphoid Cells in Airway Inflammation. Immunity 52, 620–634.e6. (2020).
Fali, T. et al. Metabolic regulation by PPARγ is required for IL-33-mediated activation of ILC2s in lung and adipose tissue. Mucosal Immunol. 14, 585–593 (2021).
Ercolano, G. et al. PPARɣ drives IL-33-dependent ILC2 pro-tumoral functions. Nat. Commun. 12, (2021).
Monticelli, L. A. et al. Arginase 1 is an innate lymphoid-cell-intrinsic metabolic checkpoint controlling type 2 inflammation. Nat. Immunol. 17, 656–665 (2016).
Bando, J. K., Nussbaum, J. C., Liang, H.-E. & Locksley, R. M. Type 2 innate lymphoid cells constitutively express arginase-I in the naive and inflamed lung. J. Leukoc. Biol. 94, 877–884 (2013).
Surace, L. et al. Dichotomous metabolic networks govern human ILC2 proliferation and function. Nat. Immunol. 22, 1367–1374 (2021).
Salmond, R. J. et al. IL-33 induces innate lymphoid cell–mediated airway inflammation by activating the mammalian target of rapamycin. J. Allergy Clin. Immunol. 130, 1159 (2012).
Houlden, A. et al. Chronic Trichuris muris Infection in C57BL/6 Mice Causes Significant Changes in Host Microbiota and Metabolome: Effects Reversed by Pathogen Clearance. PLoS One 10, (2015).
Kokova, D. & Mayboroda, O. A. Twenty Years on: Metabolomics in Helminth Research. Trends Parasitol. 35, 282–288 (2019).
Hansen, T. V. A. et al. Glucose absorption by the bacillary band of Trichuris muris. PLoS Negl. Trop. Dis. 10, (2016).
BUEDING, E. Carbohydrate metabolism of schistosoma mansoni. J. Gen. Physiol. 33, 475–495 (1950).
Sweeny, A. R. et al. Supplemented nutrition decreases helminth burden and increases drug efficacy in a natural host-helminth system. Proc. Biol. Sci. 288, (2021).
Cattadori, I. M. et al. Impact of helminth infections and nutritional constraints on the small intestine microbiota. PLoS One 11, (2016).
Barelli, C. et al. Interactions between parasitic helminths and gut microbiota in wild tropical primates from intact and fragmented habitats. Sci. Rep. 11, (2021).
White, E. C. et al. Manipulation of host and parasite microbiotas: Survival strategies during chronic nematode infection. Sci. Adv. 4, (2018).
Peachey, L. E., Jenkins, T. P. & Cantacessi, C. This Gut Ain’t Big Enough for Both of Us. Or Is It? Helminth-Microbiota Interactions in Veterinary Species. Trends Parasitol. 33, 619–632 (2017).
Reynolds, L. A., Finlay, B. B. & Maizels, R. M. Cohabitation in the Intestine: Interactions among Helminth Parasites, Bacterial Microbiota, and Host Immunity. J. Immunol. 195, 4059–4066 (2015).
Martin, I. et al. The effect of gut microbiome composition on human immune responses: An exploration of interference by helminth infections. Front. Genet. 10, (2019).
Holm, J. B. et al. Chronic Trichuris muris infection decreases diversity of the intestinal microbiota and concomitantly increases the abundance of lactobacilli. PLoS. One. 10, (2015).
Walk, S. T., Blum, A. M., Ewing, S. A. S., Weinstock, J. V. & Young, V. B. Alteration of the murine gut microbiota during infection with the parasitic helminth Heligmosomoides polygyrus. Inflamm. Bowel Dis. 16, 1841–1849 (2010).
Reynolds, L. A. et al. Commensal-pathogen interactions in the intestinal tract: Lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes 5, 522–532 (2014).
Pace, F. et al. Helminth infection in mice improves insulin sensitivity via modulation of gut microbiota and fatty acid metabolism. Pharm. Res. 132, 33–46 (2018).
Bashir, M. E. H., Andersen, P., Fuss, I. J., Shi, H. N. & Nagler-Anderson, C. An enteric helminth infection protects against an allergic response to dietary antigen. J. Immunol. 169, 3284–3292 (2002).
McSorley, H. J., Blair, N. F., Robertson, E. & Maizels, R. M. Suppression of OVA-alum induced allergy by Heligmosomoides polygyrus products is MyD88-, TRIF-, regulatory T- and B cell-independent, but is associated with reduced innate lymphoid cell activation. Exp. Parasitol. 158, 8–17 (2015).
Wilson, M. S. et al. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J. Exp. Med. 202, 1199–1212 (2005).
Zaiss, M. M. et al. The intestinal microbiota contributes to the ability of helminths to modulate allergic inflammation. Immunity 43, 998–1010 (2015).
Besten, G. D. E. N. et al. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 54, 2325–2340 (2013).
Yang, W. & Cong, Y. Gut microbiota-derived metabolites in the regulation of host immune responses and immune-related inflammatory diseases. Cell Mol. Immunol. 18, 866–877 (2021).
Chang, Y. L. et al. A screen of Crohn’s disease-associated microbial metabolites identifies ascorbate as a novel metabolic inhibitor of activated human T cells. Mucosal Immunol. 12, 457–467 (2019).
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – EXC2151 – 390873048 and SPP1937 (WI 4554/1-2). We thank Maria Doverman and the Wilhelm lab for critical discussions regarding the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Michla, M., Wilhelm, C. Food for thought – ILC metabolism in the context of helminth infections. Mucosal Immunol 15, 1234–1242 (2022). https://doi.org/10.1038/s41385-022-00559-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-022-00559-y
This article is cited by
-
Lessons from helminths: what worms have taught us about mucosal immunology
Mucosal Immunology (2022)
