
Abstract
The molecular underpinnings of seromucinous borderline tumor (SMBT) – an uncommon ovarian epithelial neoplasm characterized by association with endometriosis, frequent bilateral ovarian involvement, and occasional progression to invasive carcinoma – remain poorly understood. Here, we sought to comprehensively characterize the mutational landscape of SMBT and elucidate the clonal relationship between bilateral ovarian SMBTs. We also compared the mutational profiles between SMBTs and concurrent invasive carcinomas. Formalin-fixed, paraffin-embedded tissue specimens were retrieved from 28 patients diagnosed with SMBT. Massively parallel sequencing of 409 cancer-related genes was conducted to identify somatic mutations in 33 SMBT samples and four concurrent invasive carcinoma specimens. TERT promoter mutations were assessed by Sanger sequencing, whereas immunohistochemistry was used as a surrogate tool for detecting deletions or epigenetic silencing of relevant tumor suppressor genes. Twenty-six (92.9%) of the 28 patients were diagnosed with stage I SMBTs. Seven (25%) cases showed bilateral ovarian involvement and 13 (46%) had concomitant endometriosis. Concurrent ovarian carcinomas were identified in three patients, whereas one case had a synchronous endometrial carcinoma. Somatic mutations in the KRAS, PIK3CA, and ARID1A genes were identified in 100, 60.7, and 14.3% of SMBT samples, respectively. In contrast, TERT promoter mutations and DNA mismatch repair deficiencies were absent. Sequencing of paired specimens from patients with bilateral SMBT revealed the presence of at least two shared somatic mutations, suggestive of a clonal relationship. Similarly, we identified shared somatic mutations between SMBT samples and concurrent ovarian carcinoma specimens. Taken together, these findings demonstrated a distinct mutational landscape of SMBT in which (1) KRAS is invariably mutated, (2) PIK3CA is frequently mutated, and (3) TERT promoter mutations and DNA mismatch repair deficiencies are absent. Our findings represent the first extensive characterization of this rare ovarian neoplasm, with potential implications for disease classification and molecular diagnostics.
Similar content being viewed by others


Introduction
Seromucinous borderline tumor (SMBT) – also known as atypical proliferative seromucinous tumor and endocervical-type (müllerian) mucinous borderline tumor – is an uncommon and still incompletely characterized ovarian epithelial neoplasm [1,2,3]. Although SMBT was formerly considered as a subtype of mucinous borderline tumor, the clinical presentation of SMBT more closely resembles that of serous borderline tumor – with frequent bilateral involvement of the ovary and sporadic extraovarian implantation [3,4,5,6,7]. Histologically, SMBT shows the hierarchical papillary architecture typical of serous borderline tumor. However, its papillae are generally lined by endocervical-like mucinous epithelial cells and frequently admixed with a variety of müllerian epithelial cells – including serous (ciliated) cells, endometrioid cells, clear cells, squamous cells, and “indifferent cells” with abundant eosinophilic cytoplasm [3,4,5,6]. Approximately one third of SMBTs are associated with endometriosis, which is seldom found in patients with serous or mucinous borderline tumors [3].
Although SMBT generally portends an excellent prognosis (with only a few cases of recurrence and a single death reported in the literature) [8], the neoplasm may display unfavorable histological features − including micropapillary growth pattern, intraepithelial carcinoma, and presence of microinvasion [3,4,5,6]. Furthermore, SMBT may progress toward a form of invasive carcinoma termed seromucinous carcinoma (SMC) by the 2014 World Health Organization (WHO) Classification of Tumors of Female Reproductive Organs [9]. Currently, SMC cannot be considered a histologically well-defined entity but rather shares significant morphological and immunophenotypical overlaps with other types of ovarian tumors – particularly endometrioid carcinoma and low-grade serous carcinoma with mucinous differentiation [10, 11]. Owing to its low morphologic reproducibility and lack of defining genetic characteristics, some pathologists have even proposed to discontinue the term SMC [10].
The molecular underpinnings of SMBT remains poorly understood as well. Mutational analysis of SMBT has only been performed in one study, which solely focused on KRAS and PTEN mutations in 16 samples [12]. KRAS mutations were found to be common (69% of cases), whereas no PTEN mutations were detected – resulting in a genetic profile similar to that of mucinous borderline tumor (of gastrointestinal type) [12]. Another report demonstrated the loss of immunohistochemical expression of ARID1A (a surrogate for ARID1A mutations) in 33% of SMBT samples, i.e., a frequency in line with that of other endometriosis-related ovarian neoplasms (clear cell carcinoma and endometrioid carcinoma) [13]. Here, we used massively parallel sequencing to comprehensively decipher the mutational landscape of SMBT – with a special focus on the clonal relationships between tumors showing a bilateral presentation. In an effort to shed more light on the controversial issue of SMC, we also investigated the genetic characteristics of invasive carcinomas that arose in association with SMBTs.
Materials and methods
Patient identification and retrieval of tissue specimens
We searched our departmental surgical pathology archives and identified 44 patients with ovarian tumors diagnosed as either seromucinous borderline (or borderline seromucinous) tumor (n = 37) or endocervical-type (or -like) mucinous borderline tumor (n = 7) between January 1, 2005 and December 31, 2016. Two pathologists (R.C.W. and S.M.J.) independently reviewed all hematoxylin and eosin-stained slides to identify SMBT specimens. Seven cases were excluded because the tumor histology is not typical for SMBTs. Additional 9 patients were excluded because the tumor sizes were deemed too small for further molecular analysis (atypical proliferative part <5 mm). The final study cohort consisted of 28 patients with a confirmed pathological diagnosis of SMBT (21 unilateral and seven bilateral). Concurrent invasive ovarian carcinoma adjacent to SMBT was evident in three patients, whereas one case had a synchronous uterine endometrioid carcinoma. For the purpose of the study, the following formalin-fixed, paraffin-embedded (FFPE) tissue specimens were retrieved: SMBT (n = 33, including five cases with bilateral disease), SMBT-concurrent carcinoma (n = 4), and matched benign control tissue from lymph nodes, fallopian tubes, or uterus (n = 28). Clinical characteristics included age at diagnosis, disease stage, type of surgery, and survival figures. Tissue specimens were drawn from the tissue bank of the Chang Gung Memorial Hospital (Taoyuan, Taiwan) after ethical approval was granted by the Institutional Review Board of Chang Gung Memorial Hospital (approval number: 201701220B0). Owing to the retrospective nature of the study, the need for informed consent was waived.
Sample processing and DNA extraction
A pathologist (R.C.W.) performed a careful selection of representative FFPE blocks and identified areas of SMBT or invasive carcinomas suitable for macrodissection. A thorough manual dissection of different tumor components was conducted on 10-μM-thick tissue sections to reduce contamination and – in case of invasive carcinoma samples – to rule out the presence of non-invasive tissue. Genomic DNA for mutation analysis was extracted from FFPE samples using a commercially available kit (Qiagen Inc., Valencia, CA, USA) as previously described [14, 15].
Massively parallel sequencing and data analysis
Paired tumor and normal samples were subjected to massively parallel sequencing targeting the coding regions of 409 cancer-related genes. The sequencing procedures and the approach used for data analysis have been previously described in detail [14, 15]. In brief, genomic DNA from each sample (80 ng) was amplified by polymerase chain reaction (PCR) to enrich the coding exons of targeted genes. To this aim, AmpliSeq Comprehensive Cancer Panel primer pools (Thermo Fisher Scientific, Waltham, MA, USA) were used. Amplicons were subsequently ligated with barcoded adaptors, conjugated with sequencing beads, and enriched using Ion Chef (Thermo Fisher Scientific) according to the Ion Torrent protocol. Sequencing was performed on an Ion Proton sequencer using the Ion PI chip (Thermo Fisher Scientific). Raw data from the sequencer were mapped to the hg19 reference genome using the Ion Torrent Suite (v. 4.2). Single nucleotide variants and short insertion/deletions were identified with the Torrent Variant Caller plug-in (v. 4.2). All variants were annotated using Variant Effect Predictor (VEP, release 78) and filtered out when their frequency was <5% or in presence of <50 reads. Variants not identified in matched normal samples were considered as somatic mutations. Further annotation was performed using COSMIC (v. 70), dbSNP (138), and 1000 Genomes (phase 1).
Mutation analysis of the TERT promoter
The promoter region of the TERT gene – which is known to contain two mutation hotspots (chr5: 1,295,228 and 1,295,250; hg19) – was amplified by PCR using the following primers: 5ʼ-M13-CAGCGCTGCCTGAAACTC-3ʼ and 5ʼ-GTCCTGCCCCTTCACCTT-3ʼ, where M13 indicates a universal sequencing primer (5ʼ-GTAAAACGACGGCCAGT-3ʼ). PCR conditions were as follows: 95 °C for 5 min, followed by 45 cycles at 98 °C for 20 s, 60 °C for 15 s, and 72 °C for 1 min, followed by a final extension at 72 °C for 5 min. Amplified PCR products were purified and subjected to Sanger sequencing.
Immunohistochemistry
Immunohistochemical staining of 3-µm-thick paraffin sections was performed using the following antibodies: ARID1A (1:200 dilution; clone HPA005456, MilliporeSigma, St. Louis, MO, USA), ER (1:200; clone 6F11, Leica Biosystems), MLH1 (1:50 dilution; clone GM011, Genemed Biotechnologies, Torrance, CA, USA), Napsin A (1:200 dilution; clone IP64, Leica Biosystems), p53 (1:100 dilution; clone DO7, Leica Biosystems, Buffalo Grove, IL, USA), PAX8 (1:50 dilution; clone BC12, Biocare, Pacheco, CA, USA), PR (1:400 dilution; clone 16, Leica Biosystems), PTEN (1:100 dilution; clone 138G6, Cell Signaling, Danvers, MA, USA), and WT1 (1:200 dilution; clone 6F-H2, Quartett, Berlin, Germany). Immunostaining was performed on a BOND-MAX automated stainer (Leica Biosystems). Heat-induced epitope retrieval was performed at 100 °C using citrate-based pH 6.0 buffer (BOND Epitope Retrieval Solution 1, Leica Biosystem) for Napsin A, and EDTA-based pH 9.0 buffer (BOND Epitope Retrieval Solution 2, Leica Biosystems) for the other antibodies. Immunoreactivity was assessed with a BOND Polymer Refine Detection system (Leica Biosystems). For ER, PR, Napsin A, and PAX8, reactions were interpreted as positive if at least 5% of tumor cells showed expression.
Results
Patient characteristics
The clinical characteristics of the 28 patients with SMBT (unilateral, n = 21; bilateral, n = 7) are summarized in Table 1. The median age at diagnosis was 37 years (range, 25–58 years). In general, patients presented with early-stage disease with 26 (92.9%) diagnosed at stage I and two at stage II, with a median tumor size of 7.7 cm (range, 2.7–15.2 cm). The median length of follow-up was 38.8 months (range, 14.6–240.6 months). Disease recurrence was observed in one case only (S20). The patient initially underwent surgical removal of an SMBT (stage II) involving the left ovary and the pelvic peritoneum. Twenty-seven months thereafter, a right ovarian SMBT was diagnosed and excised. A recurrence of SMBT at the right ovary was observed at 127 months of follow-up. The patient was successfully salvaged with hysterectomy and right salpingo-oophorectomy. At the time of last follow-up, all participants were alive without disease.
Histopathologic characteristics of seromucinous borderline tumors
All SMBTs were characterized by the presence of hierarchical papillary structures lined by endocervical-like mucinous epithelial cells and cilia-bearing serous epithelial cells (Fig. 1a, b). Moreover, variable amounts of endometrioid cells, squamous cells, piling-up clear cells, and “indifferent cells” with ample eosinophilic cytoplasm were observed (Fig. 1b–d). Nuclear atypia in SMBT cells was generally mild-to-moderate. All SMBTs showed, at least focally, prominent intraepithelial and mesenchymal neutrophilic infiltration (Fig. 1c, d). Five cases (17.9%) had evidence of focal intraepithelial carcinoma characterized by exuberant cribriform proliferation over papillary surfaces or cystic linings (Fig. 1e–f). There was no evidence of micropapillary growth pattern or microinvasion in any of the study specimen. Three cases had foci of invasive growth adjacent to SMBT (all >5 mm in their greatest dimension) and their histological and genetic features will be subsequently described in detail. There were two cases in whom SMBT was associated with mature cystic teratoma (n = 1) and synchronous uterine endometrioid carcinoma (n = 1). A total of 13 (46%) patients had concomitant endometriosis (Fig. 2a). All endometriotic lesions were cystic involving ovary in 13 patients.

a Presence of hierarchical papillary structures. b Lined by an admixture of mucinous cells, cilia-bearing cells. c Endometrioid cells, piling-up clear cells. d Piling up squamous cells with mild-to moderate nuclear atypia, (c and d) prominent neutrophilic infiltration was evident. e An SMBT specimen with a region of intraepithelial carcinoma (lower portion of the figure). f Intraepithelial carcinoma characterized by an exuberant cribriform proliferation over papillary surfaces and cystic linings.

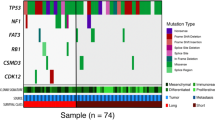
a List of the six most commonly mutated genes (in decreasing order from the top to the bottom). The five cases below the orange bar had bilateral disease (both lesions subjected to sequencing). b A clonal relationship between bilateral SMBTs was evident when the somatic mutations detected in paired samples were compared. The concomitant presence of endometriosis (EMOsis) and intraepithelial carcinoma (IEC) is reported in the first two rows.
Mutational landscape of seromucinous borderline tumors
Massively parallel sequencing identified a median of three somatic mutations (range, 1–10) in each sequenced SMBT specimen. Six genes were found mutated in at least two cases (Fig. 2a; Supplementary Fig. 1). Strikingly, somatic KRAS mutations were invariably identified in all SMBT samples – all of them being hotspot mutations involving codons 12 or 13 (p.G12A, p.G12C, p.G12D, p.G12V, and p.G13D). Somatic mutations of the PIK3CA and PTEN genes were identified in 60.7% (17/28) and 3.6% (1/28) of the SMBT specimens, respectively, in a mutually exclusive fashion. Four (14.3%) patients harbored ARID1A truncating mutations, whereas somatic mutations in the KMT2C, TET2, and ZNF521 genes were identified in two (7.1%) cases. No mutations in genes coding for DNA mismatch repair proteins (i.e., MLH1, MSH2, MSH6, PMS1, and PMS2) were identified. Similarly, Sanger sequencing did not identify TERT promoter mutations. A complete list of somatic mutations detected in SMBT specimens is provided in Supplementary Table 1.
Clonal relationship between bilateral seromucinous borderline tumors
We subsequently focused on cases with bilateral SMBT (n = 7). Of them, we were able to extract sufficient amounts of high-quality DNA from paired samples derived from five patients. Targeted massively parallel sequencing revealed that all paired specimens shared at least two somatic mutations (range: 2–4), suggesting that bilateral SMBTs were clonally related to each other (Fig. 2b). All of the five paired samples subjected to sequencing had an intact capsule without tumor cells over the ovarian surface – a finding that argued against the metastatic nature of the contralateral lesion.
Immunohistochemical analysis
All SMBTs were positive for PAX8 and negative for WT1. ER was expressed in all SMBTs and PR in most (91%) SMBTs. Napsin A was focally expressed (<25%) in two SMBT samples (S02R, S16R). The aforementioned results were in line with previous studies [16, 17], supporting the histological diagnosis of SMBT in our study cohort.
We analyzed the immunohistochemical expression of ARID1A, MLH1, p53, and PTEN in an effort to identify potential epigenetic silencing or large genomic deletions undetectable by targeted sequencing. All of the SMBT specimens were characterized by diffuse nuclear expression of MLH1 (Fig. 3a). These results indicate that epigenetic silencing of MLH1 – which is commonly encountered in endometrioid carcinoma – is a rare event in SMBT. All of the samples showed heterogeneous p53 expression, suggesting that TP53 genetic aberrations are invariably absent in SMBTs (Fig. 3b). Loss of PTEN expression was identified in a single specimen (sample S26R) known to harbor a somatic PTEN mutation (Fig. 3c). ARID1A expression was undetectable in three SMBT samples (S01L, S26R, and S31L), all of them carrying ARID1A mutations (Fig. 3d). Notably, the associated ipsilateral endometriosis also lost ARID1A expression (S01L and S26R). Taken together, the results of immunohistochemistry did not detect molecular aberrations other than those already identified by targeted sequencing.

a An intact MLH1 nuclear expression was evident in all samples. b A heterogeneous, “wildtype” p53 staining pattern was detected in all specimens. c Loss of PTEN expression in an SMBT sample (S26R). d Loss of ARID1A nuclear expression in an SMBT sample (S31L).
Mutational analysis of concurrent gynecologic cancers in patients with seromucinous borderline tumor
Of the 28 study patients, three (S08, S21, and S29) had a concurrent invasive ovarian carcinoma adjacent to the SMBT (Fig. 4a). A fourth case (S06) was diagnosed with synchronous uterine endometrioid carcinoma. The invasive lesions identified in cases S21 and S29 were classified as SMC. In case S21, we observed an admixture of endocervical-like mucinous, endometrioid, and clear cells arranged predominantly in a confluent glandular pattern and focally in a solid pattern. In case S29, variable amounts of endocervical-like mucinous, endometrioid, and “indifferent cells” with abundant eosinophilic cytoplasm were structured in a confluent glandular pattern (Fig. 4b-d). The immunoprofiles of both SMCs were identical to those of the corresponding SMBTs. The invasive component of case S08 showed the typical histological findings of clear cell carcinoma – consisting of tumor cells with clear cytoplasm growing in either a tubulopapillary or solid pattern (Fig. 4e). Immunohistochemically, the clear cell carcinoma was positive for Napsin A and negative for ER and PR, whereas the adjacent SMBT was negative for Napsin A and positive for hormonal receptors.

a A case (S29) of SMBT (left upper part of the image) with an adjacent invasive ovarian carcinoma (right lower part, 4× objective lens). b The invasive ovarian carcinoma diagnosed in case S29 – a seromucinous carcinoma (SMC) – consisted of confluent glandular structures lined by endocervical-like mucinous, endometrioid, and eosinophilic indifferent cells (20× objective lens). c The SMC diagnosed in case S21 was characterized by the presence of glandular structures lined by mucinous, endometrioid, and squamoid cells (20× objective lens). d The SMC diagnosed in case S21 showed focal neoplastic glands lined by cells with clear cytoplasm (20× objective lens). e A case (S08) of ovarian clear cell carcinoma that arose adjacent to SMBT; note the presence of tubulopapillary structures lined by clear cells (20× objective lens). f Oncoplot comparing the mutational landscapes of SMBT and concurrent ovarian carcinomas.
Massively parallel sequencing revealed that all of the three concurrent invasive ovarian carcinomas shared certain somatic mutations with concomitant SMBTs – supporting the existence of a clonal relationship (Fig. 4f). The following three shared mutations were identified in patient S08: KRAS (c.35G>A), PIK3CA (c.1633G>A), and ZNF521 (c.2836C>T). Five shared mutations were detected in patient S21, as follows: KRAS (c.35G>T), PIK3CA (c.3140A>G), NCOA2 (c.1991C>A), NTRK3 (c.446C>T), and USP9X (c.4548_4551delCAAA). A single shared mutation – KRAS (c.38G>A) – was identified in patient S29. The SMC identified in patient S21 also carried a mutation in NTRK1 (c.1486_1487insC) – which was absent in the adjacent SMBT and may theoretically be involved in the progression from SMBT to SMC (Fig. 4f). In contrast, the synchronous uterine endometrioid carcinoma diagnosed in patient S06 did not share any somatic mutation with its coexisting SMBT (Supplementary Table 2).
Discussion
This study is the first comprehensive attempt to shed more light on the molecular underpinnings of SMBT through the application of next-generation sequencing on carefully dissected tumor specimens. Our main results can be summarized as follows. First, SMBT was found to have unique molecular features that set it apart from both other borderline tumors of the ovary and endometriosis-associated neoplasms. Specifically, the SMBT signature consisted of frequent somatic mutations in the KRAS (100%), PIK3CA (60.7%), and ARID1A (14.3%) genes, with TERT promoter mutations and DNA mismatch repair deficiencies being consistently absent. Second, we show that bilateral SMBTs are frequent (25%) and clonally related to each other – as attested by the presence of shared somatic mutations. Third, a similar clonal relationship was identified between SMBT and concurrent ovarian carcinomas – including SMC and clear cell carcinoma. In light of these findings, the presence of KRAS mutations may serve as a genetic hallmark of SMBT – a finding that confirm and expand previous observations [12].
Formerly known as endocervical-type mucinous borderline tumor, SMBT was reclassified as a separate disease category in the latest 2014 WHO Classification of Tumors of Female Reproductive Organs [9]. Because the term “seromucinous” may cause confusion with serous and mucinous borderline tumors, Kurman and Shih have previously recommended the term “mixed müllerian borderline tumor” and emphasized the morphological and immunohistochemical differences between SMBT and serous/mucinous borderline tumor [18]. Our study provides further molecular genetic evidence that distinguishes SMBT from serous/mucinous borderline tumor. Differently from serous borderline tumor – which is characterized by mutually exclusive mutations in KRAS (17–39.5% of cases) and BRAF (23–48% of cases) [19] – our SMBT samples were found to invariably harbor KRAS mutations, with BRAF being rarely mutated (3.6%). Notably, a very high rate of KRAS mutations (92.3%) has been previously reported in mucinous borderline tumor [20] – potentially indicating a shared molecular basis with SMBT. However, PIK3CA mutations – which were identified in 60.7% of our SMBT samples – seem to occur much less frequently in MBT (15%) [20]. Moreover, we did not identify CDKN2A mutations – previously reported in 19.2% of MBT cases [20] – in our SMBT specimens.
Although SMBT is frequently associated with endometriosis (46% of cases in the current study), our data indicate that the mutational landscape of SMBT is distinct from that of other endometriosis-associated neoplasms − including ovarian endometrioid carcinoma and clear cell carcinoma (Table 2) [21,22,23,24,25,26,27,28,29,30]. Differently from ovarian endometrioid carcinoma, CTNNB1 mutations, loss of PTEN expression, or DNA mismatch repair deficiencies were rarely identified in our SMBT specimens [22, 23, 30]. In addition, SMBT was distinct from ovarian clear cell carcinoma, as TERT promoter mutations were not found in SMBT whereas KRAS mutations were ubiquitous in SMBT [24, 25].
Not uncommonly, patients with SMBT show bilateral ovarian involvement at presentation [31]. Here, we demonstrated for the first time that bilateral ovarian SMBTs were clonally related to each other. Two potential explanations for this clonal relationship could be offered, including: (1) metastatic spread of a primary SMBT to the contralateral ovary, or (2) independent onset of contralateral SMBT from clonal endometriotic lesions affecting both ovaries. Notably, a similar clonal relationship has been previously reported for bilateral ovarian serous borderline tumor, which is likely attributable to contralateral ovarian metastasis owing to the frequent presence of ovarian surface involvement by these neoplasms [31]. However, all bilateral SMBTs identified in our study were characterized by an intact capsule and the absence of tumor cells over the ovarian surface. These observations argue against a metastatic origin and support the view that bilateral SMBTs arise independently of each other – most likely from clonally related bilateral ovarian endometriotic lesions harboring driver mutations in KRAS, PIK3CA, or other genes [32, 33].
SMC – a poorly characterized entity of ovarian epithelial cancer introduced in the 2014 WHO Classification of Tumors of Female Reproductive Organs [9] − can exhibit a wide variety of histopathological features consisting of an admixture of different cell types (including endocervical-like mucinous, endometrioid, eosinophilic “indifferent”, hobnail, squamous, signet-ring, and clear cells) [11]. The question as to whether SMC should be regarded as a distinct category of ovarian cancer is still a matter of debate owing to its obvious morphological overlaps with low-grade serous, mucinous, and endometrioid carcinomas [10, 11]. From the perspective of multistep carcinogenesis, it is reasonable to regard as “true” SMC those arising in association with SMBT – which accounted for approximately 50% of cases previously reported in a large series [11]. SMBT-associated SMCs are likely to derive from SMBTs – as shown by the clonal relationship between SMBTs and SMCs identified in our study. They should be possibly regarded as a genetically distinct group of neoplasms in which KRAS and PIK3CA are commonly mutated. However, the differential diagnosis between SMCs without a coexisting SMBT component and other histotypes (especially endometrioid carcinoma with mucinous differentiation) is challenging at best and frequently impossible [10]. Because KRAS is invariably mutated in SMBT, it can be hypothesized that the absence of KRAS somatic mutations may help identify at least certain SMC mimickers. Unfortunately, KRAS mutations are not uncommon in endometrioid carcinoma with mucinous differentiation – ultimately limiting the diagnostic utility of this molecular approach [10, 34].
In our study, we identified one patient with SMBT and concurrent ovarian clear cell carcinoma – with the two lesions being clonally related. To our knowledge, only another similar case has been reported in the literature [35]. While this observation seems to suggest that SMBT may act as a precursor to clear cell carcinoma, such lesions might as well be collision tumors. Indeed, an SMBT and a clear cell carcinoma within a collision tumor may appear clonally related to each other if they arise independently from the same endometriotic cyst that already harbors cancer driver mutations.
In conclusion, our current data represent the first extensive characterization of SMBT in terms of histology, immunohistochemistry, and molecular pathogenesis. If independently confirmed, our findings may have significant implications for disease classification and molecular diagnostics.
References
Rutgers JL, Scully RE. Ovarian mixed-epithelial papillary cystadenomas of borderline malignancy of mullerian type. A clinicopathologic analysis. Cancer. 1988;61:546–54.
Rutgers JL, Scully RE. Ovarian mullerian mucinous papillary cystadenomas of borderline malignancy. A clinicopathologic analysis. Cancer. 1988;61:340–8.
Shappell HW, Riopel MA, Smith Sehdev AE, Ronnett BM, Kurman RJ. Diagnostic criteria and behavior of ovarian seromucinous (endocervical-type mucinous and mixed cell-type) tumors: atypical proliferative (borderline) tumors, intraepithelial, microinvasive, and invasive carcinomas. Am J Surg Pathol. 2002;26:1529–41.
Lee KR, Nucci MR. Ovarian mucinous and mixed epithelial carcinomas of mullerian (endocervical-like) type: a clinicopathologic analysis of four cases of an uncommon variant associated with endometriosis. Int J Gynecol Pathol. 2003;22:42–51.
Rodriguez IM, Irving JA, Prat J. Endocervical-like mucinous borderline tumors of the ovary: a clinicopathologic analysis of 31 cases. Am J Surg Pathol. 2004;28:1311–8.
Dube V, Roy M, Plante M, Renaud MC, Tetu B. Mucinous ovarian tumors of Mullerian-type: an analysis of 17 cases including borderline tumors and intraepithelial, microinvasive, and invasive carcinomas. Int J Gynecol Pathol. 2005;24:138–46.
Yasunaga M, Ohishi Y, Oda Y, Misumi M, Iwasa A, Kurihara S, et al. Immunohistochemical characterization of mullerian mucinous borderline tumors: possible histogenetic link with serous borderline tumors and low-grade endometrioid tumors. Hum Pathol. 2009;40:965–74.
Nagamine M, Mikami Y. Ovarian seromucinous tumors: pathogenesis, morphologic spectrum, and clinical issues. Diagnostics. 2020;10:77.
Kurman RJ, International Agency for Research on Cancer., World Health Organization. WHO classification of tumours of female reproductive organs. 4th ed. Lyon: International Agency for Research on Cancer; 2014. p. 307.
Rambau PF, McIntyre JB, Taylor J, Lee S, Ogilvie T, Sienko A, et al. Morphologic reproducibility, genotyping, and immunohistochemical profiling do not support a category of seromucinous carcinoma of the ovary. Am J Surg Pathol. 2017;41:685–95.
Taylor J, McCluggage WG. Ovarian seromucinous carcinoma: report of a series of a newly categorized and uncommon neoplasm. Am J Surg Pathol. 2015;39:983–92.
Kim KR, Choi J, Hwang JE, Baik YA, Shim JY, Kim YM, et al. Endocervical-like (Mullerian) mucinous borderline tumours of the ovary are frequently associated with the KRAS mutation. Histopathology. 2010;57:587–96.
Wu CH, Mao TL, Vang R, Ayhan A, Wang TL, Kurman RJ, et al. Endocervical-type mucinous borderline tumors are related to endometrioid tumors based on mutation and loss of expression of ARID1A. Int J Gynecol Pathol. 2012;31:297–303.
Chao A, Chang TC, Lapke N, Jung SM, Chi P, Chen CH, et al. Prevalence and clinical significance of BRCA1/2 germline and somatic mutations in Taiwanese patients with ovarian cancer. Oncotarget. 2016;7:85529–41.
Chao A, Wu RC, Jung SM, Lee YS, Chen SJ, Lu YL, et al. Implication of genomic characterization in synchronous endometrial and ovarian cancers of endometrioid histology. Gynecol Oncol. 2016;143:60–7.
Vang R, Gown AM, Barry TS, Wheeler DT, Ronnett BM. Ovarian atypical proliferative (borderline) mucinous tumors: gastrointestinal and seromucinous (endocervical-like) types are immunophenotypically distinctive. Int J Gynecol Pathol. 2006;25:83–9.
Hauptmann S, Friedrich K, Redline R, Avril S. Ovarian borderline tumors in the 2014 WHO classification: evolving concepts and diagnostic criteria. Virchows Arch. 2017;470:125–42.
Kurman RJ, Shih IeM. Seromucinous tumors of the ovary. What’s in a Name? Int J Gynecol Pathol. 2016;35:78–81.
Malpica A, Wong KK. The molecular pathology of ovarian serous borderline tumors. Ann Oncol. 2016;27:i16–9.
Mackenzie R, Kommoss S, Winterhoff BJ, Kipp BR, Garcia JJ, Voss J, et al. Targeted deep sequencing of mucinous ovarian tumors reveals multiple overlapping RAS-pathway activating mutations in borderline and cancerous neoplasms. BMC Cancer. 2015;15:415.
Yamamoto S, Tsuda H, Takano M, Iwaya K, Tamai S, Matsubara O. PIK3CA mutation is an early event in the development of endometriosis-associated ovarian clear cell adenocarcinoma. J Pathol. 2011;225:189–94.
McConechy MK, Ding J, Senz J, Yang W, Melnyk N, Tone AA, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Mod Pathol. 2014;27:128–34.
Stewart CJ, Walsh MD, Budgeon CA, Crook ML, Buchanan DB. Immunophenotypic analysis of ovarian endometrioid adenocarcinoma: correlation with KRAS mutation and the presence of endometriosis. Pathology. 2013;45:559–66.
Kuo KT, Mao TL, Jones S, Veras E, Ayhan A, Wang TL, et al. Frequent activating mutations of PIK3CA in ovarian clear cell carcinoma. Am J Pathol. 2009;174:1597–601.
Wu RC, Ayhan A, Maeda D, Kim KR, Clarke BA, Shaw P, et al. Frequent somatic mutations of the telomerase reverse transcriptase promoter in ovarian clear cell carcinoma but not in other major types of gynaecological malignancy. J Pathol. 2014;232:473–81.
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl J Med. 2010;363:1532–43.
Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–31.
Stewart CJ, Bowtell DD, Doherty DA, Leung YC. Long-term survival of patients with mismatch repair protein-deficient, high-stage ovarian clear cell carcinoma. Histopathology. 2017;70:309–13.
Bennett JA, Morales-Oyarvide V, Campbell S, Longacre TA, Oliva E. Mismatch repair protein expression in clear cell carcinoma of the ovary: incidence and morphologic associations in 109 cases. Am J Surg Pathol. 2016;40:656–63.
Vierkoetter KR, Ayabe AR, VanDrunen M, Ahn HJ, Shimizu DM, Terada KY. Lynch Syndrome in patients with clear cell and endometrioid cancers of the ovary. Gynecol Oncol. 2014;135:81–4.
Sieben NL, Kolkman-Uljee SM, Flanagan AM, le Cessie S, Cleton-Jansen AM, Cornelisse CJ, et al. Molecular genetic evidence for monoclonal origin of bilateral ovarian serous borderline tumors. Am J Pathol. 2003;162:1095–101.
Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noe M, Horlings HM, et al. Cancer-associated mutations in endometriosis without cancer. N. Engl J Med. 2017;376:1835–48.
Anglesio MS, Yong PJ. Endometriosis-associated ovarian cancers. Clin Obstet Gynecol. 2017;60:711–27.
Xiong J, He M, Jackson C, Ou JJ, Sung CJ, Breese V, et al. Endometrial carcinomas with significant mucinous differentiation associated with higher frequency of k-ras mutations: a morphologic and molecular correlation study. Int J Gynecol Cancer. 2013;23:1231–6.
Nakamura E, Sato Y, Moriguchi S, Yamashita A, Higo T, Asada Y. Ovarian seromucinous borderline tumor and clear cell carcinoma: an unusual combination. Case Rep. Obstet Gynecol. 2015;2015:690891.
Acknowledgements
This study was supported by the Chang Gung Medical Foundation, Taiwan (grants CRRPG3F0041/2/3, CMRPG3H1151/2, and CMRPG3H0351/2/3).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wu, RC., Chen, SJ., Chen, HC. et al. Comprehensive genomic profiling reveals ubiquitous KRAS mutations and frequent PIK3CA mutations in ovarian seromucinous borderline tumor. Mod Pathol 33, 2534–2543 (2020). https://doi.org/10.1038/s41379-020-0611-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-0611-3
This article is cited by
-
Ovarian seromucinous carcinoma: an independent epithelial ovarian cancer?
Journal of Ovarian Research (2023)
-
Towards less invasive molecular diagnostics for endometrial cancer: massively parallel sequencing of endometrial lavage specimens in women attending for an office hysteroscopy
Journal of Molecular Medicine (2022)
