
Abstract
Cribriform-morular variant of thyroid carcinoma is classically associated with familial adenomatous polyposis but, it can also occur as a sporadic neoplasm. This neoplasm is much more frequently observed in women than in men (ratio of 61:1). In familial adenomatous polyposis patients, tumors are generally multifocal and/or bilateral (multinodular appearance), whereas in the sporadic cases tumors tend to occur as single nodules. The tumors are well delimited, and characteristically show a blending of follicular, cribriform, papillary, trabecular, solid, and morular patterns. Neoplastic cells are tall or cuboidal with the occasional nuclear features of classic papillary thyroid carcinoma. The morules include cells with peculiar nuclear clearing and show positivity for CDX2 and CD10. Angioinvasion and capsular invasion have been described in about 30 and 40% of cases, respectively, with lymph node metastases in less than 10% of patients and distant metastases in 6%. Although this tumor has good prognosis, neuroendocrine and/or poor differentiation have been associated with aggressive behavior. Tumor cells can be focally positive or negative for thyroglobulin, but are always positive for TTF-1, estrogen and progesterone receptors, and negative for calcitonin and cytokeratin 20. Nuclear and cytoplasmic staining for β-catenin is the hallmark of this tumor type; this feature plays a role in fine needle aspiration biopsy. Cribriform-morular variant of thyroid carcinoma has a peculiar endodermal (intestinal-like) type phenotype, activation of the WNT/β-catenin signaling pathway, and belongs to the non-BRAF-non-RAS subtype of the molecular classification of thyroid tumors. Elevated expression of estrogen and progesterone receptors and activation of the WNT/β-catenin pathway may prove useful as putative therapeutic targets in cases that do not respond to conventional therapy. Clinicians should be alerted to the possibility of familial adenomatous polyposis when a diagnosis of cribriform-morular variant of thyroid carcinoma is made. Instead of being considered as a variant of papillary thyroid carcinoma its designation as cribriform-morular thyroid carcinoma seems more appropriate.
Similar content being viewed by others
Introduction
Cribriform-morular variant of thyroid carcinoma is classically associated with familial adenomatous polyposis but, it can also occur as a sporadic neoplasm [1]. Devic and Bussy [2] recognized in 1912 the association of intestinal adenomatous polyposis with extracolonic manifestations, including a “goiter”. Crail [3] reported in 1949 the first case of papillary adenocarcinoma of the thyroid (“with very little colloid”) associated with multiple primary malignancies arising in the rectum and brain but, it was Camiel et al. [4] who suggested the relationship of familial adenomatous polyposis with thyroid carcinoma for the first time in 1968. Later on, in 1990, Chan and Loo [5] described rare papillary thyroid carcinomas with a cribriform pattern, and soon after Harach et al. [6] recognized the familial adenomatous polyposis associated thyroid carcinoma as a distinctive type of follicular cell neoplasm. In 1999 Cameselle-Teijeiro and Chan [7] proposed the name “cribriform-morular variant” of papillary thyroid carcinoma to designate the sporadic morphological counterpart of familial adenomatous polyposis associated thyroid carcinoma.
At present, cribriform-morular variant of papillary thyroid carcinoma is the designation recognized by the WHO [1] for this tumor in both the familiar and the sporadic presentation. Whether this neoplasm is another variant of papillary thyroid carcinoma or a distinct category of thyroid carcinoma remains controversial in the recent WHO classification [1]. We tend to consider it a form of thyroid carcinoma distinct from conventional papillary and follicular carcinoma, with a peculiar primitive endodermal (intestinal-like) phenotype, permanent activation of the wingless (WNT/β-catenin) signaling pathway, and belonging to the non-BRAF-non-RAS subtype of the new molecular classification of thyroid tumors [8]. The goal of this review is to provide a guide for the clinical, pathological and molecular diagnosis of cribriform-morular variant of thyroid carcinoma. We intend to provide arguments in order to demonstrate that this tumor type should not be considered as a variant of papillary thyroid carcinoma but as an independent tumor entity better designated as cribriform-morular carcinoma of the thyroid.
Epidemiology
Cribriform-morular variant seems more common in Asia (65%) and in America (23%), than in the other parts of the world [7, 9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61]. This variant represented 0.2 and 0.5% the cases of two series of thyroid carcinoma [10, 12]. Its prevalence ranged from 0.2 to 6% in different series of consecutive cases of clinically evident papillary thyroid carcinoma [7, 30, 42, 53]. Thyroid cancer among familial adenomatous polyposis patients has been reported to be 0.4 to 2% in older series [13, 15, 16, 62,63,64,65]; the frequency of thyroid cancer among FAP patients has risen from 3 to 12% when ultrasonography was used [36, 66], and up to 16% when ultrasound was combined with fine needle aspiration biopsy [61].
Cribriform-morular variant of thyroid carcinoma is much more frequently observed in women than in men (ratio of 61:1). At diagnosis, the mean age is 26 years (range 8–61 years), both for familial adenomatous polyposis-associated and for sporadic cribriform-morular variant of thyroid carcinoma. In FAP associated cases (about 60% of the total), thyroid carcinoma diagnosis precedes that of familial adenomatous polyposis in up to 40% of the cases [15, 18, 30, 36, 44, 53, 67]. The risk and incidence rate of cribriform-morular variant of thyroid carcinoma in Hispanic patients with familial adenomatous polyposis seems to be higher than in other reported groups [68].
Clinical features
Patients are usually euthyroid, and in most cases (≈50%) the tumor is detected by enlargement of the thyroid gland during a routine physical examination or in screening studies of patients with familial adenomatous polyposis [9, 34, 53]. Less commonly, the lesion is detected during medical examination because of neck pain, hoarseness, dysphagia, and/or dizziness [22]. Sometimes it is incidentally discovered on ultrasonography [54] or because the patient has self-detected a thyroid mass [53]. Because the germline mutation occurs in the same genomic area associated with congenital hypertrophy of the retinal pigment epithelium in patients with cribriform-morular variant of thyroid carcinoma and familial adenomatous polyposis [69], ophthalmoscopy is an effective method to detect patients at risk for developing thyroid cancer and to confirm the APC germinal involvement. Bilateral lesions and lesions with a depigmented halo are the hallmarks of congenital hypertrophy of the retinal pigment epithelium associated with familial adenomatous polyposis [70]. Serum thyroglobulin levels are normal or slightly elevated [71]. In a rare case of familial adenomatous polyposis associated cribriform-morular variant of PTC the patient developed a false positive serum pregnancy test, due to elevated serum β-human chorionic gonadotropin (β-hCG), ectopically produced by the thyroid tumor [56].
Ultrasonography is successfully used for the screening of nodules in patients with familial adenomatous polyposis [11, 36, 66], and it may also be useful to perform fine needle aspiration biopsy in nodules smaller than 1 cm [61]. Cribriform-morular variant of thyroid carcinoma does not usually meet the currently accepted ultrasonographic criteria for a thyroid malignancy, and its sonographic findings are similar to those of follicular tumor or nodular goiter rather than classic PTC [23, 36, 42, 50, 51, 54, 71, 72].
From the pooled data we collected, extrathyroidal extension was described in only five patients [31, 53, 56, 58], and local recurrence in six [10, 15, 17, 56]. Regional lymph node metastases were reported in 13/134 (10%) patients [7, 9, 15, 21, 22, 36, 40, 48, 51, 56, 60], and distant metastases in 6/107 (6%) patients, to lung (5 cases), bone (n = 2) and brain (n = 1) [10, 22, 37, 51, 56, 60, 73]. Death due to the thyroid carcinoma has been reported in 4 patients (3 women and 1 man), whose age ranged from 20 to 42 years (mean, 32). Death occurred in the interval 17 to 360 months (mean = 120 months) after the first surgery [10, 15, 22, 37]. The general indolent behavior of cribriform-morular variant of thyroid carcinoma is also maintained in cases of patients living in radiation-affected regions [74].
Macroscopy
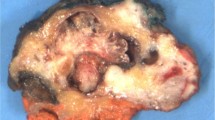
Most tumors are well-circumscribed or encapsulated, white to tan, solid and fleshy, with occasional cystic areas and almost equal distribution in either thyroid lobe [7, 9,10,11, 15, 17, 20, 24, 28, 30,31,32,33,34,35,36,37, 39, 40, 43, 44, 48, 49, 52,53,54, 57,58,59,60,61, 75]. Mean size is 24 mm (median 21, range 2–90 mm), with a smaller mean size in patients with cribriform-morular variant of thyroid carcinoma associated to familial adenomatous polyposis than in sporadic cases (21 vs. 27 mm). In familial adenomatous polyposis the tumors are usually multifocal and/or bilateral, with a multinodular appearance, while sporadic cases usually appear as single nodule.
Histopathology
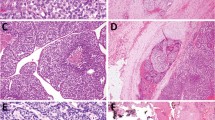
Cribriform-morular variant of thyroid carcinomas are usually circumscribed or encapsulated, and generally divided in lobules by fibrotic septa. Microscopically, they show a blending of follicular, cribriform, papillary, trabecular, and solid patterns with morular (squamoid) formations [6, 7]. Cribriform patterns are formed by anastomosing bars and arches of cells without fibrovascular stroma, often merging with tubular (glandular-like) follicles (Fig. 1). Both cribriform and tubular structures are devoid of colloid and may contain histiocytes. Nonarborizing papillary and pseudopapillary structures are lined by cuboidal or columnar cells. Areas of trabecular arrangement usually show spindle cells mimicking a hyalinizing trabecular tumor pattern. Tumor cells are tall or cuboidal, and display frequent pseudostratification and abundant cytoplasm. The nuclei are generally hyperchromatic, with the occasional presence of nuclear features of classic papillary thyroid carcinoma such as nuclear grooves, clear nuclei, intranuclear pseudoinclusions, and overlapping. In solid areas, tumors cells acquire and/or oval and spindle shape and nodular squamoid whorls (morules). These squamoid nests lack keratinization and include cells with peculiar nuclear clearing of the chromatin. Morules can also appear in trabecular and cribriform areas. Mayer mucicarmine and Alcian blue stains are negative. Psammoma bodies are rare. Necrotic foci can be found in some infrequent cases [15, 17, 40, 76]. Angioinvasion and capsular invasion have been described in about 30 and 40% of cases, respectively [6, 7, 17, 37, 40, 51, 60]. Adenoid cystic carcinoma-like growth pattern due to deposition of basement membrane-like material has been found in a case of cribriform-morular variant of thyroid carcinoma [43]. Adamantinous-like pattern of growth with hyaline globules (thanatosomes) and atypical mitotic figures have been reported in another case with neuroendocrine differentiation and aggressive behavior [37].

Cribriform-morular variant of thyroid carcinoma. Microscopically, cribriform-morular variant of thyroid carcinoma shows a blending of cribriform (a, b), papillary (c), trabecular, and solid patterns with morular (asterisk) formations. The cribriform patterns are formed by anastomosing bars and arches of cells without fibrovascular stroma, often merging with tubular (glandular-like) follicles. Note the lack of colloid (a–c)
Due to the characteristic microscopic appearance of cribriform-morular variant of thyroid carcinoma, clinicians should be alerted to the possibility of familial adenomatous polyposis when a diagnosis of cribriform-morular variant of thyroid carcinoma is made [6, 22, 25, 30, 37]. The few cases of papillary thyroid carcinoma in the setting of familial adenomatous polyposis with no typical cribriform-morular variant of thyroid carcinoma histology [15, 36, 54, 77] either represent tumors still too small to show a characteristic morphology or even result from various interactions among oncogenes and tumor suppressor genes, or perhaps, can simply be sporadic incidental cases. Cases that had no cribriform-morular variant features, even within a familial adenomatous polyposis setting, were not included in this revision.
Immunohistochemistry
Tumor cells can be focally positive or negative for thyroglobulin but are always positive for thyroid transcription factor-1 (TTF-1) and negative for calcitonin. Strong nuclear and cytoplasmic staining for β-catenin is the hallmark of this tumor type [6, 7, 9, 17, 18, 20, 23, 24, 26,27,28,29,30,31,32, 34, 37, 40, 43, 48, 49, 51,52,53,54, 56, 58, 60, 61] (Fig. 2). Because cribriform-morular variant of PTC is the only primary thyroid tumor with nuclear and cytoplasmic positivity for β-catenin, this marker should be used if there are any doubts about the histological diagnosis. Lymphoid enhancer binding factor 1 (LEF-1) is also a sensitive and specific marker for cribriform-morular variant of thyroid carcinoma, especially when used in the setting of a papillary thyroid carcinoma neoplasm [78]. Tumor cells are positive for cytokeratin 7, cytokeratin 19, cytokeratins 5 and 6, cytokeratins cocktail 1, 2, 10, 11, 14, 15, 16, and 19 (clone AE1/AE3), cytokeratins cocktail 1, 5, 10, and 14 (clone 34βE12), cytokeratins cocktail 7 and 8 (clone CAM5.2), epithelial membrane antigen, E-cadherin, vimentin, Hector Battifora mesothelial cell-1 (HBME-1), galectin-3, neuron-specific enolase, bcl-2, Rb, p27, and cyclin D1. Immunohistochemical positivity for β-hCG has been detected in one case [56]. Positivity for chromogranin and synapthophysin in 40% of the tumor cells was reported by our group in a rare case with positivity for TTF-1, and negativity for thyroglobulin and calcitonin [37]. Interestingly, tumor cells in the cribriform-morular variant of thyroid carcinoma are consistently positive for α and β-estrogen receptors, progesterone receptors, and focally positive for androgen receptors, probably as a consequence of cross-talk between WNT/ β-catenin and estrogen signaling pathways [29, 79] (Fig. 2). This peculiar immunophenotype may be associated with the almost exclusive presentation of this tumor in women, both in the sporadic and in the familial adenomatous polyposis-related cases. The cellular proliferation (Ki-67 index) is usually less than 5% but can be up to 60% in cribriform-morular variant of thyroid carcinoma displaying poorly differentiated carcinoma featues [37, 51]. Carcinoembryonic antigen (CEA), CA125, cytokeratin 20, epidermal growth factor receptor, c-kit (CD117), p63, Wilms tumor protein (WT1), and calretinin were always negative.

Cribriform-morular variant of thyroid carcinoma. Tumor cells may be totally negative for thyroglobulin (a), but they are consistently positive for thyroid transcription factor 1 (TTF1) (b). Strong nuclear and cytoplasmic immunoreactivity for β-catenin is the hallmark of cribriform-morular variant of thyroid carcinoma (c). Tumor cells are also positive for β-estrogen receptors (d) and progesterone receptors (e). f Morules lack keratinization and include cells with peculiar nuclear clearing of the chromatin (arrows). The morular structures are positive for β-catenin, CDX2 (g) and CD10 (h), but negative for TTF-1 (b), β-estrogen receptors (d), and progesterone receptors (e)
There is a distinctive immunohistochemical profile in the morular areas of cribriform-morular variant of thyroid carcinoma. Morules are positive for β-catenin, cytokeratins 1, 2, 10, 11, 14, 15, 16, and 19 (clone AE1/AE3), galectin-3, E-cadherin, bcl-2, CA19.9, and cyclin D1, and negative for TTF-1, thyroglobulin, calcitonin, CEA, clone 34βE12, vimentin, and CA125. The peculiar biotin-rich optically clear nuclei, can produce false positive reactions in morular cells when using immunohistochemical systems based on (strep)avidin-biotin affinity [7, 10, 41]. Ultrastructurally, the peculiar biotin-rich nuclear clearings are composed of electron dense fibrils, which are approximately 10 nm in diameter and 100 nm long. These microfilaments run in a quasiparallel manner through an electron-opaque matrix, and are not demarcated by a lining membrane [9, 10, 27, 80, 81]. In both the cribriform-morular variant of thyroid carcinoma and in all biotin-rich optically clear nuclei family tumors, CD10 is a useful tool for identifying morules [82]. The morular structures are also positive for CDX2, an intestine-specific homeobox gene transcription factor [83]. The apparently incongruent CDX2 (along with CD10 and CA19.9) expression in this tumor type fits with the primitive (noncommitted differentiation) intestinal-like phenotype that our group proposed for some thyroid tumors many years ago [84]. Unlike the morular structures, true squamous metaplasia lack nuclear staining for β-catenin, are negative for bcl-2, and are associated with S-100 protein-positive dendritic cells [26].
Differential diagnosis
The cribriform-morular variant of thyroid carcinoma can be confused with breast metastatic carcinoma because its cribriform pattern of growth and positivity for estrogen and progesterone receptors [85]. Its cytoarchitectural pattern can also mimic metastatic colon cancer. But in both cases, positivity for TTF-1 in the cribriform-morular variant of thyroid carcinoma facilitates the differential diagnosis. There is significant microscopic overlap between columnar cell variant of papillary thyroid carcinoma and cribriform-morular variant of thyroid carcinoma (tall cells with marked pseudostratification, darker nuclear chromatin, empty tubular-like follicles, and solid areas); the columnar cell variant usually shows supranuclear and subnuclear cytoplasmic vacuoles simulating early secretory endometrium and is immunoreactive for thyroglobulin. The tall cell variant of papillary thyroid carcinoma also has columnar cells, but unlike cribriform-morular variant of thyroid carcinoma, the nuclei are basally located and the cytoplasm is positive for thyroglobulin. In the cribriform-morular variant of thyroid carcinoma solid areas can mimic poorly differentiated carcinoma, however, the cribriform-morular variant of thyroid carcinoma additionally displays the cribriform pattern with CD10-positive morules, lower mitotic activity and usually negativity for thyroglobulin.
Cytopathology
In patients with cribriform-morular variant of thyroid carcinoma, fine needle aspiration biopsy is effective for diagnostic purposes both for thyroid carcinoma in general [30, 34, 39, 42, 44, 52, 56, 60, 61, 86,87,88], and specifically for cribriform-morular variant recognition of thyroid carcinoma [19, 28, 31, 33, 40, 42, 49, 89]. The samples are usually hypercellular, showing papillary fragments having epithelial-cell lining with nucleomegaly and nuclear stratification along with flat monolayers. There are also tall and columnar cells with abundant spindle cytoplasm, distinct cell borders and obscure ground-glass nuclei. Typical nuclear features of classic PTC (pallor, grooves and pseudoinclusions) are commonly seen. Acinar/cribriform formations lacking colloid and, less commonly, morules can be found (Fig. 3). Hyaline-like globules similar to those seen in salivary gland tumors have been described in one case [89]. Foamy histiocytes and/or hemosiderin-laden macrophages can be seen, but the background is usually clean with no mitosis, necrotic debris, anisonucleosis and psammoma bodies. Because there is much overlap between the cytological features of the cribriform-morular variant of thyroid carcinoma and the conventional thyroid carcinoma, positive nuclear β-catenin immunostaining, plays an essential role in making a diagnosis of cribriform-morular variant of thyroid carcinoma [42, 49, 88].

Cytologic features of the cribriform-morular variant of thyroid carcinoma. The samples are usually hypercellular, showing papillary fragments (a), flat monolayers (b) and/or morular structures (d). There are tall and columnar cells with abundant spindle cytoplasm (a–c), distinct cell borders and obscure ground-glass nuclei. Pseudoinclusions can be seen (b). The background is usually clean (a–d) with no mitosis, necrotic debris, anisonucleosis, and psammoma bodies
Genetic profile
Cribriform-morular variant of thyroid carcinoma is the peculiar thyroid carcinoma that appears in patients with familial adenomatous polyposis. Familial adenomatous polyposis is an autosomal dominant disease secondary to adenomatous polyposis coli (APC) gene germline mutation located on 5q21-22. APC gene encodes the tumor suppressor protein which functions as an antagonist of the WNT/β-catenin signaling pathway. Overall, APC gene somatic mutations can be seen in over 30% of cribriform-morular variant of thyroid carcinoma cases. In patients with germinal APC mutation (familial adenomatous polyposis patients), a somatic APC mutation has been found in 50% of cases (“second hit” of the Knudson’s two-hit model). In patients with FAP and multicentric thyroid tumors, it has been found that each tumor had a different second hit somatic mutation of the APC gene, suggesting that each focus of cribriform-morular variant of thyroid carcinoma developed independently through biallelic inactivation of the APC gene [19, 35]. In one sporadic case of cribriform-morular variant of thyroid carcinoma, a somatic APC gene mutation in exon 15 at codon 1309 (with negative dominant effect) has also been detected in tumor tissue but not in peripheral lymphocytes, supporting a relationship between the morphologic pattern of the cribriform-morular variant of thyroid carcinoma and the APC gene, as well as the existence of this variant as a sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma [20]. In any case, somatic APC mutation, at least in the mutation cluster region, is not always a causal mechanism in sporadic classic papillary thyroid carcinoma [90,91,92]. In physiological conditions, APC protein forms a “destruction complex” with glycogen synthetase kinase 3β (GSK3β), casein kinase 1α (CK1α), and the scaffold protein AXIN, that tightly regulates the cytoplasmic level of β-catenin in the WNT/ β-catenin signaling pathway [93]. APC mutations lead to a truncated APC protein that lacks the β-catenin-binding site and therefore cannot contribute to β-catenin proteosomal degradation. Stabilized β-catenin accumulates in the cytoplasm and is then translocated into the nucleus where it binds to LEF/TCF proteins that lead to constitutive expression of WNT target genes such as MYC, CCND1 (cyclin D1), Axin2 and DKK1, mainly involved in the increase of cell proliferation and loss of differentiation. This accumulation of β-catenin is the hallmark of familial and sporadic forms of cribriform-morular variant of thyroid carcinoma [18, 37]. The existence of cases of this variant of thyroid carcinoma with cytoplasmic and nuclear accumulation of β-catenin, and missense somatic mutations of exon 3 of the β-catenin gene (CTNNB1) without APC gene mutation or loss of heterozygosity (LOH) near the APC gene, confirms that accumulation of mutant β-catenin contributes to cribriform-morular variant of thyroid carcinoma development [24]. As described for the APC gene, each of the two different tumors examined from two patients with multicentric cribriform-morular variant of thyroid carcinoma, had different somatic mutations of CTNNB1 gene, additionally supporting the independent developing of multicentric tumors in the cribriform-morular variant of thyroid carcinoma [24]. More recently, our group first reported two missense AXIN1 somatic mutations in exons 1 and 7 in one sporadic and one familial case of cribriform-morular variant of thyroid carcinoma, respectively [93, 94]. All these molecular findings indicate the key role of the WNT/ β-catenin signaling pathway molecular alterations in both sporadic and familial cribriform-morular variant of thyroid carcinoma.
Clonal characterization of sporadic cribriform-morular variant of thyroid carcinoma showed that although histologic components of cribriform-morular variant of thyroid carcinoma have a common mutational background, additional somatic mutations may be involved in the development of morular structures [95]. With regard to molecular alterations typically found in conventional thyroid tumors [1], RET/PTC rearrangements were described in 8 of 11 (72.7%) familial cases of cribriform-morular variant of thyroid carcinoma [17, 21, 48, 52]. Most of the aforementioned arrangements were the RET/PTC1 isoform, with one tumor showing concurrent RET/PTC1 and RET/PTC3 rearrangement [17]. The prevalence of RET/PTC rearrangement along with the presence of characteristic nuclear features in cribriform-morular variant of thyroid carcinoma suggest that the latter is pathogenetically closer to classic PTC than to follicular carcinoma. BRAFV600E gene mutation, however, the most common molecular alteration in classic papillary thyroid carcinoma, has been consistently negative in all 19 cases studied to date including both sporadic and familial tumors [20, 37, 40, 48, 51, 52, 58, 94, 96,97,98].
PAX8/PPARγ rearrangements and RAS or PIK3CA gene mutations are typically associated with follicular derived tumors with a follicular pattern of growth. In cribriform-morular variant of thyroid carcinoma, PAX8/PPARγ rearrangements were negative in one sporadic and two familial cases [52, 93]. RAS mutations are rarely detected (7.6%); K-RAS mutation was only detected in one familial adenomatous polyposis-associated case [52], and in another sporadic case [93], but no H-RAS nor N-RAS mutations were detected in the 26 cases studied to date [48, 51, 52, 58, 85, 93, 94, 97]. The same PIK3CA gene mutation, in exon 9, was detected in 3 (50%) sporadic cases [58]. Telomerase reverse transcriptase (TERT) promotor mutation was found in one of five sporadic cases [60, 93]. The aggressive clinical behavior observed in the only TERT mutated case [60], supports the role of these mutations as an indicator of poor outcome in differentiated thyroid carcinomas [99].
Genetic susceptibility
In patients with familial adenomatous polyposis and cribriform-morular variant of thyroid carcinoma, about 85% of germline APC mutations have been found in exon 15, in the same genomic area associated with congenital hypertrophy of the retinal pigment epithelium (codons 463 to 1387), with more than 90% of these germline mutations outside the mutation cluster region (MCR) (codons 1286 to 1513) [69]. The majority of the aforementioned mutations occurred before codon 1220 [15, 17, 19, 22, 27, 35, 69]. Codon 1061 has been found to be a hot spot for cribriform-morular variant of thyroid carcinoma and hepatoblastoma [69]. Since in patients with cribriform-morular variant of thyroid carcinoma APC mutations occur predominantly in the 5′ side of the APC gene, between codons 140 and 1513 [35], restriction of the mutational analysis of the APC gene to the mutation cluster region (codons 1286 to 1513) will detect germline and/or somatic mutations in less than 20% of cases [21, 35, 69].
Cribriform-morular variant of thyroid carcinoma usually occurs in patients with familial adenomatous polyposis even in the absence of biallelic inactivation of the APC gene. Some authors have suggested that the WNT/β-catenin signaling pathway, via the β-catenin binding sites (20-amino acid repeats) in the APC protein, is strongly involved in the development of CMV of PTC in familial adenomatous polyposis patients [57]. APC germline mutation confers a genetic susceptibility for thyroid cancer, but other factors are also required for cribriform-morular variant of PTC development [21]. We, along with others [21, 57, 85], postulate that cribriform-morular variant of thyroid carcinoma is the result of germline APC mutations (or functionally equivalent somatic gene mutations in the WNT/β-catenin signaling pathway), combined with somatic mutations typically involved in the development of sporadic thyroid tumors in potential collaboration with hormonal factors such as estrogen and/or progesterone.
In summary, although the precise etiology of cribriform-morular variant of thyroid carcinoma in familial adenomatous polyposis is not yet clearly understood, the initiation of this tumor is related with the constitutive activation of WNT/ β-catenin signaling pathway (with nuclear translocation of β-catenin), through germline APC mutations and additional somatic mutations in APC or in functionally equivalent genes such us CTNNB1 or AXIN1 (biallelic inactivation) (Fig. 4). In the sporadic cases, the tumor may result from a combination of somatic mutations in APC, CTNNB1 and/or AXIN1 genes (phenotypically equivalents). RET/PTC rearrangements, PIK3CA gene mutations or less commonly, RAS gene mutations, typically implicated in the pathogenesis of conventional thyroid tumors, could also act as additional upstream effectors in the WNT/ β-catenin pathway in both familial and sporadic cases of cribriform-morular variant of thyroid carcinoma [100, 101]. Interestingly, the striking predominance of this tumor type in women, suggests the participation of sex hormones in its development, due to overexpression of estrogen and progesterone receptors by the tumor cells themselves and/or interactions [79, 102] between estrogen and progesterone hormones and the WNT/ β-catenin signaling pathway.

Molecular alterations associated with the pathogenesis of the cribriform-morular variant of thyroid carcinoma. In cribriform-morular variant of thyroid carcinoma in familial adenomatous polyposis patients biallelic gene inactivation takes place by means of additional somatic APC gene mutation or as consequence of mutations in functionally equivalent genes (CTNNB1 or AXIN1). In sporadic cases, cribriform-morular variant of thyroid carcinoma result from a combination of somatic mutations in genes that are phenotypically equivalent such as APC, CTNNB1, and/or AXIN1. RET/PTC rearrangements as well as mutations in PIK3CA or RAS genes can act as additional upstream effectors in the WNT/β-catenin pathway in sporadic and familial adenomatous polyposis-associated cribriform-morular variant of thyroid carcinoma. The much greater prevalence of this tumor in women in both settings suggests a role of sex hormones as a tumor growth promoter. As in the other types of follicular carcinoma, TERT mutation seems associated with adverse outcome
Prognosis and predictive factors
This tumor type is generally associated with a relatively indolent clinical course and good prognosis. Cribriform-morular variant of thyroid carcinoma cases with high proliferative index and/or poorly differentiated features can be particularly aggressive [37, 51]. TERT promoter mutations also seem to indicate clinical aggressiveness [60].
In cribriform-morular variant of thyroid carcinoma, follicles are devoid of colloid, immunohistochemical studies are usually weakly positive or totally negative for thyroglobulin, and serum thyroglobulin levels are normal or slightly elevated. In addition, in one case with multiple lymph node and bone metastasis [60] serial post-operative serum thyroglobulin levels were undetectable in the absence of thyroglobulin antibodies. For all these reasons, we agree with Oh et al. [60], that serum thyroglobulin measurement is not generally reliable for following of cases of cribriform-morular variant of thyroid carcinoma patients.
It remains to be determined whether the permanent activation of the WNT/β-catenin pathway and the high expression of estrogen and progesterone receptors may serve as therapeutic targets in cases that do not respond to conventional therapy for differentiated thyroid carcinomas [85].
Conclusions
Cribriform-morular variant of thyroid carcinoma is a rare type of thyroid carcinoma associated with familial adenomatous polyposis, which can also occur as a sporadic neoplasm. Although it shares some features with classic papillary thyroid carcinoma, this tumor deserves to be considered a form of thyroid carcinoma distinct from classic papillary thyroid carcinoma and follicular carcinoma, with a peculiar primitive endodermal (intestinal-like) type phenotype and permanent activation of the wingless (WNT/β-catenin) signaling pathway, belonging to the non-BRAF-non-RAS subtype of the molecular classification of thyroid tumors [8].
References
Rosai J, Albores Savedra J, Asioli S, et al. Papillary thyroid carcinoma. In: Lloyd RV, Osamura RY, Klöppel G, et al. editors. World Health Organization Classification of Tumours. Pathology and genetics of tumours of endocrine organs. 4th edition. Lyon: IARC Press; 2017. p. 81–91.
Bussy DC. Un cas de polypose adenomateuse generalisee a tout l’intestin. Arch Fr Mal Appl Dig. 1912;6:278–89.
Crail HW. Multiple primary malignancies arising in the rectum, brain, and thyroid; report of a case. U S Nav Med Bull. 1949;49:123–8.
Camiel MR, Mulé JE, Alexander LL, et al. Association of thyroid carcinoma with Gardner’s syndrome in siblings. N Engl J Med. 1968;278:1056–8.
Chan JK, Loo KT. Cribriform variant of papillary thyroid carcinoma. Arch Pathol Lab Med. 1990;114:622–4.
Harach HR, Williams GT, Williams ED. Familial adenomatous polyposis associated thyroid carcinoma: a distinct type of follicular cell neoplasm. Histopathology. 1994;25:549–61.
Cameselle-Teijeiro J, Chan JK. Cribriform-morular variant of papillary carcinoma: a distinctive variant representing the sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma? Mod Pathol. 1999;12:400–11.
Yoo SK, Lee S, Kim SJ, et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. 2016;12:e1006239.
Yamashita T, Hosoda Y, Kameyama K, et al. Peculiar nuclear clearing composed of microfilaments in papillary carcinoma of the thyroid. Cancer. 1992;70:2923–8.
Okamoto Y, Kashima K, Daa T, et al. Morule with biotin-containing intranuclear inclusions in thyroid carcinoma. Pathol Int. 1995;45:573–9.
Hizawa K, Iida M, Yao T, et al. Association between thyroid cancer of cribriform variant and familial adenomatous polyposis. J Clin Pathol. 1996;49:611–3.
Mizukami Y, Nonomura A, Michigishi T, et al. Encapsulated follicular thyroid carcinoma exhibiting glandular and spindle cell components. A case report. Pathol Res Pract. 1996;192:67–71.
Bülow C, Bülow S. Is screening for thyroid carcinoma indicated in familial adenomatous polyposis? The Leeds Castle Polyposis Group. Int J Colorectal Dis. 1997;12:240–2.
Cetta F, Olschwang S, Petracci M, et al. Genetic alterations in thyroid carcinoma associated with familial adenomatous polyposis: clinical implications and suggestions for early detection. World J Surg. 1998;22:1231–6.
Perrier ND, van Heerden JA, Goellner JR, et al. Thyroid cancer in patients with familial adenomatous polyposis. World J Surg. 1998;22:738–42.
van der Linde K, Vasen HF, et al. Occurrence of thyroid carcinoma in Dutch patients with familial adenomatous polyposis. An epidemiological study and report of new cases. Eur J Gastroenterol Hepatol. 1998;10:777–81.
Soravia C, Sugg SL, Berk T, et al. Familial adenomatous polyposis-associated thyroid cancer: a clinical, pathological, and molecular genetics study. Am J Pathol. 1999;154:127–35.
Kurihara K, Shimizu S, Chong J, et al. Nuclear localization of immunoreactive beta-catenin is specific to familial adenomatous polyposis in papillary thyroid carcinoma. Jpn J Cancer Res. 2000;91:1100–2.
Miyaki M, Iijima T, Ishii R, et al. Molecular evidence for multicentric development of thyroid carcinomas in patients with familial adenomatous polyposis. Am J Pathol. 2000;157:1825–7.
Cameselle-Teijeiro J, Ruiz-Ponte C, Loidi L, et al. Somatic but not germline mutation of the APC gene in a case of cribriform-morular variant of papillary thyroid carcinoma. Am J Clin Pathol. 2001;115:486–93.
Cetta F, Curia MC, Montalto G, et al. Thyroid carcinoma usually occurs in patients with familial adenomatous polyposis in the absence of biallelic inactivation of the adenomatous polyposis coli gene. J Clin Endocrinol Metab. 2001;86:427–32.
Fenton PA, Clarke SE, Owen W, et al. Cribriform variant papillary thyroid cancer: a characteristic of familial adenomatous polyposis. Thyroid. 2001;11:193–7.
Ng SB, Sittampalam K, Goh YH, et al. Cribriform-morular variant of papillary carcinoma: the sporadic counterpart of familial adenomatous polyposis-associated thyroid carcinoma. A case report with clinical and molecular genetic correlation. Pathology. 2003;35:42–6.
Xu B, Yoshimoto K, Miyauchi A, et al. Cribriform-morular variant of papillary thyroid carcinoma: a pathological and molecular genetic study with evidence of frequent somatic mutations in exon 3 of the beta-catenin gene. J Pathol. 2003;199:58–67.
Chikkamuniyappa S, Jagirdar J. Cribriform-morular variant of papillary carcinoma: association with familial adenomatous polyposis - report of three cases and review of literature. Int J Med Sci. 2004;1:43–49.
Hirokawa M, Kuma S, Miyauchi A, et al. Morules in cribriform-morular variant of papillary thyroid carcinoma: Immunohistochemical characteristics and distinction from squamous metaplasia. APMIS. 2004;112:275–82.
Kameyama K, Mukai M, Takami H, et al. Cribriform-morular variant of papillary thyroid carcinoma: ultrastructural study and somatic/germline mutation analysis of the APC gene. Ultrastruct Pathol. 2004;28:97–102.
Lee S, Hong SW, Shin SJ, et al. Papillary thyroid carcinoma associated with familial adenomatous polyposis: molecular analysis of pathogenesis in a family and review of the literature. Endocr J. 2004;51:317–23.
Nakatani Y, Masudo K, Nozawa A, et al. Biotin-rich, optically clear nuclei express estrogen receptor-beta: tumors with morules may develop under the influence of estrogen and aberrant beta-catenin expression. Hum Pathol. 2004;35:869–74.
Tomoda C, Miyauchi A, Uruno T, et al. Cribriform-morular variant of papillary thyroid carcinoma: clue to early detection of familial adenomatous polyposis-associated colon cancer. World J Surg. 2004;28:886–9.
Chuah KL, Hwang JS, Ng SB, et al. Cytologic features of cribriform-morular variant of papillary carcinoma of the thyroid: a case report. Acta Cytol. 2005;49:75–80.
Shim YR, Bae YK, Choi JH, et al. Cribriform-morular variant of papillary thyroid carcinoma. A report of two cases. Korean J Pathol. 2005;39:48–53.
Chung DC, Maher MM, Faquin WC. Case records of the Massachusetts General Hospital. Case 37-2006. A 19-year-old woman with thyroid cancer and lower gastrointestinal bleeding. N Engl J Med. 2006;355:2349–57.
Dalal KM, Moraitis D, Iwamoto C, et al. Clinical curiosity: cribriform-morular variant of papillary thyroid carcinoma. Head Neck. 2006;28:471–6.
Uchino S, Noguchi S, Yamashita H, et al. Mutational analysis of the APC gene in cribriform-morular variant of papillary thyroid carcinoma. World J Surg. 2006;30:775–9.
Herraiz M, Barbesino G, Faquin W, et al. Prevalence of thyroid cancer in familial adenomatous polyposis syndrome and the role of screening ultrasound examinations. Clin Gastroenterol Hepatol. 2007;5:367–73.
Cameselle-Teijeiro J, Menasce LP, Yap BK, et al. Cribriform-morular variant of papillary thyroid carcinoma: molecular characterization of a case with neuroendocrine differentiation and aggressive behavior. Am J Clin Pathol. 2009;131:134–42.
Dong Y, Zhou X, Yin H, et al. Cribriform-morular variant of papillary thyroid carcinoma: report of three cases and review of the literature. Pathology. 2009;41:509–12.
Donnellan KA, Bigler SA, Wein RO. Papillary thyroid carcinoma and familial adenomatous polyposis of the colon. Am J Otolaryngol. 2009;30:58–60.
Jung CK, Choi YJ, Lee KY, et al. The cytological, clinical, and pathological features of the cribriform-morular variant of papillary thyroid carcinoma and mutation analysis of CTNNB1 and BRAF genes. Thyroid. 2009;19:905–13.
Nikiel B, Chekan M, Jarzab M, et al. Endogenous avidin biotin activity (EABA) in thyroid pathology: immunohistochemical study. Thyroid Res. 2009;2:5.
Hirokawa M, Maekawa M, Kuma S, et al. Cribriform-morular variant of papillary thyroid carcinoma--cytological and immunocytochemical findings of 18 cases. Diagn Cytopathol. 2010;38:890–6.
Baloch ZW, Segal JP, Livolsi VA. Unique growth pattern in papillary carcinoma of the thyroid gland mimicking adenoid cystic carcinoma. Endocr Pathol. 2011;22:200–5.
Ito Y, Miyauchi A, Ishikawa H, et al. Our experience of treatment of cribriform morular variant of papillary thyroid carcinoma; difference in clinicopathological features of FAP-associated and sporadic patients. Endocr J. 2011;58:685–9.
Schaeffer DF, Yoshida EM, Owen DA, et al. Familial adenomatous polyposis-rendering a diagnosis based on recognition of an unusual primary thyroid neoplasm. Case Rep Med. 2011;2011:767610.
Crippa S, Saletti P, Barizzi J, et al. The clinical management in familial adenomatous polyposis deserves continuous monitoring for thyroid carcinoma. BMJ Case Rep. 2012;2012:bcr2012007046.
Gronnier C, Donatini G, Leteurtre E, et al. Cribriform-morular variant of papillary thyroid carcinoma: characteristic histologic feature of adenomatous polyposis. A case report. Ann Endocrinol. 2012;73:213–5.
Rossi ED, Revelli L, Martini M, et al. Cribriform-morular variant of papillary thyroid carcinoma in an 8-year-old girl: a case report with immunohistochemical and molecular testing. Int J Surg Pathol. 2012;;20:629–32.
Boonyaarunnate T, Olson MT, Bishop JA, et al. Cribriform morular variant of papillary thyroid carcinoma: clinical and cytomorphological features on fine-needle aspiration. Acta Cytol. 2013;57:127–33.
Chong Y, Shin JH, Oh YL, et al. Cribriform-morular variant of papillary thyroid carcinoma: ultrasonographic and clinical characteristics. Thyroid. 2013;23:45–9.
Nakazawa T, Celestino R, Machado JC, et al. Cribriform-morular variant of papillary thyroid carcinoma displaying poorly differentiated features. Int J Surg Pathol. 2013;21:379–89.
Giannelli SM, McPhaul L, Nakamoto J, et al. Familial adenomatous polyposis-associated, cribriform morular variant of papillary thyroid carcinoma harboring a K-RAS mutation: case presentation and review of molecular mechanisms. Thyroid. 2014;24:1184–9.
Levy RA, Hui VW, Sood R, et al. Cribriform-morular variant of papillary thyroid carcinoma: an indication to screen for occult FAP. Fam Cancer. 2014;13:547–51.
Yeoh EC, Lim LC, Ng SB, et al. Cribriform morular variant of papillary thyroid carcinoma in a patient with an incidental neck lump: a case report and review of the literature. Endocr Pathol. 2014;25:302–6.
Abdullah Suhaimi SN, Nazri N, Nani Harlina ML, et al. Familial adenomatous polyposis-associated papillary thyroid cancer. Malays J Med Sci. 2015;22:69–72.
Alikhan M, Koshy A, Hyjek E, et al. Discrepant serum and urineβ-hCG results due to production of β-hCG by a cribriform-morular variant of thyroid papillary carcinoma. Clin Chim Acta. 2015;438:181–5. PMID: 25181612
Kumamoto K, Ishida H, Ohsawa T, et al. Germline and somatic mutations of the APC gene in papillary thyroid carcinoma associated with familial adenomatous polyposis: analysis of three cases and a review of the literature. Oncol Lett. 2015;10:2239–43.
Kwon MJ, Rho YS, Jeong JC, et al. Cribriform-morular variant of papillary thyroid carcinoma: a study of 3 cases featuring the PIK3CA mutation. Hum Pathol. 2015;46:1180–8.
Liyanapathirana N, Seneviratne SA, Samarasekera DN. A distinct variant of papillary thyroid carcinoma indicating familial adenomatous polyposis (FAP): a case report and brief review. BMC Res Notes. 2015;8:795.
Oh EJ, Lee S, Bae JS, et al. TERT promoter mutation in an aggressive cribriform morular variant of papillary thyroid carcinoma. Endocr Pathol. 2017;28:49–53.
Uchino S, Ishikawa H, Miyauchi A, et al. Age- and gender-specific risk of thyroid cancer in patients with familial adenomatous polyposis. J Clin Endocrinol Metab. 2016;101:4611–7.
Giardiello FM, Offerhaus GJ, Lee DH, et al. Increased risk of thyroid and pancreatic carcinoma in familial adenomatous polyposis. Gut. 1993;34:1394–6.
Iwama T, Mishima Y, Utsunomiya J. The impact of familial adenomatous polyposis on the tumorigenesis and mortality at the several organs. Its rational treatment. Ann Surg. 1993;217:101–8.
Cetta F, Toti P, Petracci M, et al. Thyroid carcinoma associated with familial adenomatous polyposis. Histopathology. 1997;31:231–6.
Truta B, Allen BA, Conrad PG, et al. Genotype and phenotype of patients with both familial adenomatous polyposis and thyroid carcinoma. Fam Cancer. 2003;2:95–9.
Jarrar AM, Milas M, Mitchell J, et al. Screening for thyroid cancer in patients with familial adenomatous polyposis. Ann Surg. 2011;253:515–21.
Plail RO, Bussey HJ, Glazer G, et al. Adenomatous polyposis: an association with carcinoma of the thyroid. Br J Surg. 1987;74:377–80.
Casellas-Cabrera N, Díaz-Algorri Y, Carlo-Chévere VJ, et al. Risk of thyroid cancer among Caribbean Hispanic patients with familial adenomatous polyposis. Fam Cancer. 2016;15:267–74.
Cetta F, Montalto G, Gori M, et al. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: results from a European cooperative study. J Clin Endocrinol Metab. 2000;85:286–92.
Chen CS, Phillips KD, Grist S, et al. Congenital hypertrophy of the retinal pigment epithelium (CHRPE) in familial colorectal cancer. Fam Cancer. 2006;5:397–404.
Fujimoto T, Hirokawa M, Ota H, et al. Characteristic sonographic features of cribriform papillary thyroid carcinoma for differentiation from other thyroid nodules. J Med Ultrason. 2015;42:83–7.
Lee JH, Shin JH, Lee HW, et al. Sonographic and cytopathologic correlation of papillary thyroid carcinoma variants. J Ultrasound Med. 2015;34:1–15.
Perea del Pozo E, Ramirez Plaza C, Padillo Ruiz J, et al. Cribiform-variant of papillary thyroid cancer and familial adenomatous polyposis. Int J Surg Case Rep. 2015;16:192–4.
Lam AK, Fridman M. Characteristics of cribriform morular variant of papillary thyroid carcinoma in post-Chernobyl affected region. Hum Pathol. 2018. https://doi.org/10.1016/j.humpath.2018.01.006. [Epub ahead of print] PubMed PMID: 29320754.
Rosen EJ, Newlands SD, Rampy BA. Pathology quiz case: cribriform-morular variant of papillary thyroid carcinoma. Arch Otolaryngol Head Neck Surg. 2003;129:679–81.
Mogoş V, Mogoş S, Sfarti C, et al. Familial syndromic papillary thyroid carcinoma report of two cases. Rev Med Chir Soc Med Nat Iasi. 2012;116:1048–54.
Cetta F, Moltoni L, Barellini L, et al. Familial adenomatous polyposis-associated papillary thyroid carcinoma shows an indolent course and usually, but not always, belongs to the cribriform-morular variant of papillary thyroid carcinoma. Acta Cytol. 2012;56:107–8.
Mohindra S, Sakr H, Sturgis C, et al. LEF-1 is a sensitive marker of cribriform morular variant of papillary thyroid carcinoma. Head Neck Pathol 2017. https://doi.org/10.1007/s12105-017-0873-3. [Epub ahead of print] PMID: 29243023.
Kouzmenko AP, Takeyama K, Ito S, et al. Wnt/beta-catenin and estrogen signaling converge in vivo. J Biol Chem. 2004;279:40255–8.
Gamachi A, Kashima K, Daa T, et al. Aberrant intranuclear localization of biotin, biotin-binding enzymes, and beta-catenin in pregnancy-related endometrium and morule-associated neoplastic lesions. Mod Pathol. 2003;16:1124–31.
Nesland JM. Ultrastructural and molecular analysis of cribriform-morular variant of papillary thyroid carcinoma. Ultrastruct Pathol. 2004;28:53.
Cameselle-Teijeiro J, Alberte-Lista L, Chiarelli S, et al. CD10 is a characteristic marker of tumours forming morules with biotin-rich, optically clear nuclei that occur in different organs. Histopathology. 2008;52:389–92.
Cameselle-Teijeiro J, Alberte-Lista L, Peteiro-González D, et al. CDX2 expression in some variants of papillary thyroid carcinoma. Am J Clin Pathol. 2012;138:907–9.
Cameselle-Teijeiro J, Febles-Pérez C, Sobrinho-Simões M. Papillary and mucoepidermoid carcinoma of the thyroid with anaplastic transformation: a case report with histologic and immunohistochemical findings that support a provocative histogenetic hypothesis. Pathol Res Pract. 1995;191:1214–21.
Cameselle-Teijeiro JM, Eloy C, Amendoeira I, et al. Rare familial tumours. In: Cameselle-Teijeiro JM, Eloy C, Sobrinho-Simões M, editors. Rare tumors of the thyroid gland: diagnosis and WHO classification. 1st ed. Berlin: Springer; 2018. p. 57–77.
Kuma S, Hirokawa M, Xu B, et al. Cribriform-morular variant of papillary thyroid carcinoma. Report of a case showing morules with peculiar nuclear clearing. Acta Cytol. 2004;48:431–6.
Priyani AA, Opatha ST, Gunathilake NW, et al. Cribriform morular variant of papillary thyroid carcinoma: cytomorphology, differential diagnosis and diagnostic implications in patients with adenomatous polyposis coli. J Cytol. 2016;33:235–8.
Koo JS, Jung W, Hong SW. Cytologic characteristics and β-catenin immunocytochemistry on smear slide of cribriform-morular variant of papillary thyroid carcinoma. Acta Cytol. 2011;55:13–8.
Monappa V, Kudva R. Cytomorphologic diversity of papillary thyroid carcinoma. J Cytol. 2017;34:183–7.
Colletta G, Sciacchitano S, Palmirotta R, et al. Analysis of adenomatous polyposis coli gene in thyroid tumours. Br J Cancer. 1994;70:1085–8.
Curtis L, Wyllie AH, Shaw JJ, et al. Evidence against involvement of APC mutation in papillary thyroid carcinoma. Eur J Cancer. 1994;30A:984–7.
Yang K, Wang X, Zhang H, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest. 2016;96:116–36.
Cameselle-Teijeiro J, Peteiro-González D, Carreira M, et al. Molecular alterations in the cribriform-morular variant of papillary thyroid carcinoma. Virchows Arch. 2016;469:S72.
Peteiro González D. Clinical and molecular study of the cribiform-morular variant of papillary thyroid carcinoma. Doctoral Thesis. University of Santiago de Compostela. Spain. 2016 (in Spanish).
Subramaniam MM, Putti TC, Anuar D, et al. Clonal characterization of sporadic cribriform-morular variant of papillary thyroid carcinoma by laser microdissection-based APC mutation analysis. Am J Clin Pathol. 2007;128:994–1001.
Schuetze D, Hoschar AP, Seethala RR, et al. The T1799A BRAF mutation is absent in cribriform-morular variant of papillary carcinoma. Arch Pathol Lab Med. 2009;133:803–5.
Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676–90.
Onder S, Ozturk Sari S, Yegen G, et al. Classic architecture with multicentricity and local recurrence, and absence of TERT promoter mutations are correlates of BRAF(V600E) harboring pediatric papillary thyroid carcinomas. Endocr Pathol. 2016;27:153–61.
Melo M, da Rocha AG, Vinagre J, et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J Clin Endocrinol Metab. 2014;99:E754–65.
Abbosh PH, Nephew KP. Multiple signaling pathways converge on beta-catenin in thyroid cancer. Thyroid. 2005;15:551–61.
Fang D, Hawke D, Zheng Y, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–9.
Wang Y, van der Zee M, Fodde R, et al. Wnt/Β-catenin and sex hormone signaling in endometrial homeostasis and cancer. Oncotarget. 2010;1:674–84.
Acknowledgements
This work was supported by Grant PI15/01501-FEDER from the Instituto de Salud Carlos III, Ministry of Economy and Competitiveness, Spain. This work was also financed by FEDER - Fundo Europeu de Desenvolvimento Regional funds through the COMPETE 2020—Operacional Program for Competitiveness and Internationalization (POCI), Portugal 2020, and by Portuguese funds through FCT—Fundação para a Ciência e a Tecnologia/Ministério da Ciência, Tecnologia e Inovação in the framework of the project “Institute for Research and Innovation in Health Sciences” (POCI-01-0145-FEDER-007274). Further funding was obtained from the project “Advancing cancer research: from basic knowledgment to application”; NORTE-01-0145-FEDER-000029; “Projetos Estruturados de I&D&I”, funded by Norte 2020—Programa Operacional Regional do Norte. Further funded by the European Regional Development Fund (ERDF) through the Operational Program for Competitiveness and Internationalization—COMPETE 2020, and Portuguese national funds via FCT—Fundação para a Ciência e a Tecnologia, under project POCI-01-0145-FEDER-016390:CANCEL STEM”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Cameselle-Teijeiro, J.M., Peteiro-González, D., Caneiro-Gómez, J. et al. Cribriform-morular variant of thyroid carcinoma: a neoplasm with distinctive phenotype associated with the activation of the WNT/β-catenin pathway. Mod Pathol 31, 1168–1179 (2018). https://doi.org/10.1038/s41379-018-0070-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-018-0070-2
This article is cited by
-
Molecular pathology of endocrine gland tumors: genetic alterations and clinicopathologic relevance
Virchows Archiv (2024)
-
Cribriform Morular Thyroid Carcinoma – Ultimobranchial Pouch-Related? Deep Molecular Insights of a Unique Case
Endocrine Pathology (2023)
-
Cribriform morular thyroid carcinoma: a case report with pathological, immunohistochemical, and molecular findings suggesting an origin from follicular cells (or their endodermal precursors)
Virchows Archiv (2023)
-
Cribriform-morular variant of papillary thyroid carcinoma with poorly differentiated features: report of a case and review of the literature
Surgical and Experimental Pathology (2022)
-
Overview of the 2022 WHO Classification of Familial Endocrine Tumor Syndromes
Endocrine Pathology (2022)
