
Abstract
Beta cells are partially replaced in neonatal rodents after deletion with streptozotocin (STZ). Exposure of pregnant rats to a low protein (LP) diet impairs endocrine pancreas development in the offspring, leading to glucose intolerance in adulthood. Our objective was to determine whether protein restriction has a similar effect on the offspring in mice, and if this alters the capacity for beta cell regeneration after STZ. Pregnant Balb/c mice were fed a control (C) (20% protein) or an isocaloric LP (8% protein) diet during gestation. Pups were given 35 mg/kg STZ (or vehicle) from d 1 to 5 for each dietary treatment. Histologic analysis showed that C-fed offspring had largely replaced beta cell mass (BCM) after STZ by d 30, but this was not sustained over time. Female LP-fed offspring showed an initial increase in BCM by d 14 but developed glucose intolerance by d 130. In contrast, male LP offspring showed no changes in BCM or glucose tolerance. However, LP exposure limited the capacity for recovery of BCM in both genders after STZ treatment.
Similar content being viewed by others


Main
Poor fetal growth and low birth weight are associated with increased risk of impaired glucose tolerance (1), type 2 diabetes (2), the metabolic syndrome (3), and cardiovascular disease (4) in adult life. Animal models that have been developed to understand the role of the intrauterine environment in susceptibility to postnatal disease have included maternal dietary calorie restriction (5), nutritional imbalance (6), and pharmacological (7) or surgical intervention (8). Administration of a low protein (LP) diet to rats during pregnancy results in reduced birth weight (6), impaired islet cell development, deficient insulin release (9), an increased islet apoptotic rate (10,11), and decreased beta cell mass (BCM) in the offspring (9–11). Offspring of LP-fed dams were glucose intolerant at 130 d of age (12). Our first objective was to characterize an LP model in mice, to facilitate future exploration into the mechanisms, whereby nutritional insult can predispose to adult disease by using genetic manipulation.
Pancreatic beta cells have a significant capacity to regenerate after injury in early life. Subtotal deletion of beta cells with streptozotocin (STZ) is followed by their partial regeneration and remission of hyperglycemia in young rodents (13,14). However, this regenerative ability decreases with age (15). New beta cells have been postulated to be generated through three possible mechanisms: 1) beta cell replication (16), 2) pancreatic duct progenitor cell differentiation (17), and 3) acinar cell transdifferentiation (18). It remains to be determined, which mechanism is most important for beta cell regeneration and could be exploited for beta cell therapy. It is not known if administration of an LP diet to the pregnant rodent will alter beta cell renewal, or by which mechanism, in the offspring after an STZ insult, which constitutes our second objective in this study. Understanding how the intrauterine environment alters plasticity of BCM in the offspring could lead to strategies to intervene before the onset of type 2 diabetes.
MATERIALS AND METHODS
Animals.
Balb/c mice (Charles River Ltd, Montreal, QC, Canada) were housed in a temperature controlled room with 12-h light:dark cycle. Water and food (Bio-Serv, Frenchtown, NJ) were given ad libitum. The LP diet [similar to Snoeck et al. (6)] was isocaloric with 8% protein versus 20% in the control (C) diet. Pregnancy was confirmed by a vaginal plug and mothers were randomly assigned to C or LP diet for the duration of gestation. A total of 61 C litters and 52 LP litters were used, with 15 litters/diet followed for maternal weight and food intake measurements every 5th d until weaning. All dams were maintained on the C diet after parturition. On d 1 through 5, the offspring received injections of either 35 mg/kg dose of STZ (Sigma Chemical Co., St.Louis, MO) in sodium citrate buffer (0.1 mol/L, pH 4.5) or equal volume of the vehicle (sham). The offspring were divided into four groups: C = control, sham injected; C + STZ = control, STZ injected; LP = low protein, sham injected; LP + STZ = low protein, STZ injected. On d 1, 7, 14, 30, and 130, the weights of the offspring were recorded and blood glucose levels were measured after a 2- to 4-h fast using an Ascencia Breeze glucometer (Bayer Inc., Toronto, ON, Canada). Intraperitoneal glucose tolerance test (IPGTT) was performed at d 42 and 130 for 90 min after a 4-h fast, as described previously (13). Mice were killed, and the pancreata were removed and fixed in 10% formalin for morphologic analysis. Three mice from independent litters were included per group for each gender. At d 130 the total abdominal adipose tissue was removed and weighed. All animal procedures were approved by the Animal Care Committee of the University of Western Ontario in accordance with the guidelines of the Canadian Council for Animal Care.
Immunohistochemistry.
Simultaneous immunohistochemistry was performed on sections for glucagon and insulin as described previously (19). Sections were mounted under glass cover slips with Permount (Fisher Scientific, Toronto, ON, Canada). A triple stain was performed to determine colocalization of proliferating cell nuclear antigen (PCNA) with insulin or glucagon. Glucagon was localized as above, using a rabbit anti-glucagon antibody (1:300; Santa Cruz Biotechnology, Santa Cruz, CA) with a goat anti-rabbit secondary (1:50; Vector Laboratories, Burlingame, CA). PCNA was localized using a mouse anti-PCNA antibody (1:50; Dako Cytomation, Mississauga, ON, Canada) with a horse anti-mouse secondary (1:50; Vector) followed by Vectastain ABC-AP kit (Vector) and visualized using Vector Blue Alkaline Phosphatase substrate kit III (Vector). Tween-20 1× Tris buffered saline was used as a wash, with PBS used for the final insulin staining portion. Insulin was localized as earlier with a guinea pig anti-insulin antibody (1:300; Abcam, Cambridge, MA) with a goat anti-guinea pig secondary (1:50; Vector).
Pdx-1 and insulin dual immunofluorescent staining was performed using a rabbit anti-Pdx-1 primary antibody (1:200; generous gift from Dr. Chris Wright, Vanderbilt University, Nashville, TN) with donkey Cy3 anti-rabbit secondary (1:50; Jackson ImmunoResearch Laboratories Inc., West Grove, PA), mouse anti-insulin primary antibody (1:200; Sigma Chemical Co.), and Alexafluor 488 goat anti-mouse secondary (1:50; Invitrogen Canada Inc., Burlington, ON, Canada) with DAPI nuclear counterstain (Invitrogen). Sections were mounted under glass cover slips with Fluroscent mounting medium (Dako). Pdx-1+ cells were counted for every islet/section, and the Pdx1+/insulinlow cell population was expressed as percentage of total Pdx1+ cells.
Morphometric analysis.
Three representative sections (coefficient of variation = 10.45%) from 3 animals/group were imaged and analyzed using Northern Eclipse software (version 7.0; Empix Imaging, Mississauga, ON, Canada). Total pancreas area for each section and the area of insulin and glucagon immunoreactivity for each islet were measured. Islets with an area <300 μm2 (≤3 cells) were excluded from analysis. Beta (and alpha) cell mass was calculated as previously described (11). Individual alpha and beta cell size were determined by averaging the individual cell area measured from 10 to 15 random islets for each of three sections per pancreas and three pancreata per group.
Serum and pancreatic insulin.
On d 7, 14, and 30 at least 20 μL of serum was collected and stored at −80°C. Insulin was measured using an Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem Inc., Downers Grove, IL) according to the manufacturer's instructions with sensitivity 0.1 ng/mL (intra- and inter-assay coefficient of variance ≤8% and ≤10%, respectively). Insulin was extracted from d 14 whole pancreata (∼50 mg) by mechanical homogenization in 500 μL acid ethanol (165 mM HCL in 75% ethanol). After overnight incubation at 4°C, samples were centrifuged (2000 × g) for 5 min at 4°C. Insulin was quantified in the supernatant by rat insulin RIA (Linco Research Inc, St. Charles, MO; 100% reactivity to mouse insulin) and normalized to pancreatic DNA content. Assay sensitivity was 0.1 ng/mL, with an intra- and inter-assay coefficient of variance of 4.6% and 9.4%, respectively.
Statistical analysis.
Three-way ANOVA was performed revealing significant differences with respect to gender. The genders were separated to examine the effects of diet and treatment using a two-way ANOVA with a Bonferroni posttest or an unpaired t test. All values are means ± SEM and significance level p < 0.05.
RESULTS
Body, organ weights, and diabetes onset.
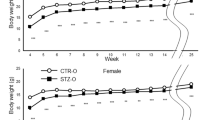
No difference in weight gain or food intake (g consumed/d) was observed between the C and LP fed dams at any time during pregnancy or lactation (n = 15 dams per group). The mean litter size for the C (5.1 ± 0.3 pups; n = 61) and the LP (5.8 ± 0.3 pups; n = 52) groups was not significantly different. On d 1, male and female LP offspring had significantly reduced weights (p < 0.05), and at d 7, the LP diet significantly (p < 0.01) reduced body weight in both genders, although all mice recovered weight by d 14 (Table 1). Pancreas weight (% body weight) was significantly decreased (p < 0.05) in both male C + STZ and LP mice on d 7 compared with controls, whereas the effect of LP treatment continued to significantly (p < 0.05) reduce pancreas weight (%) in male offspring at d 14 (Table 1).
All female mice survived to d 130 and showed no difference in weight between groups (Table 1). Male C + STZ and LP + STZ mice became overtly diabetic between 60 and 80 d, at which point they were killed. The survival rates for male C + STZ and LP + STZ mice were 67% and 33%, respectively (n = 6). Therefore, only male C and LP mice were examined at 130 d, where no difference in weight was observed (Table 1). At d 130, previous STZ treatment caused a decrease in mean abdominal adipose weight (% body weight), but the LP diet had no effect in either gender (Female: C = 2.5 ± 0.3, C + STZ = 1.8 ± 0.4, LP = 2.9 ± 0.4, LP + STZ = 1.6 ± 0.4; Male: C = 2.0 ± 0.4, LP = 1.9 ± 0.3; n = 4–5).
Beta cell mass.
BCM was unaffected in male LP offspring throughout the period studied. However, female LP mice showed a significant increase (p < 0.001) in BCM by d 14 compared with C animals (Fig. 1B). At d 30 and 130, the BCM in LP females were comparable to C values (Fig. 1B). Therefore, LP exposure alone had no long term effects on BCM of either gender.

Changes in beta cell mass with diet and STZ treatment. BCM for C (black bars), C + STZ (white bars), LP (gray bars), and LP + STZ (hatched bars) offspring over time for male (A) and female (B) mice. Means ± SEM; n = 3 animals per group and three sections per pancreas. (A) **p < 0.05 vs sham; §p < 0.001 vs sham; ∥p < 0.05 vs C + sham; (B) **p < 0.05 vs sham; §p < 0.001 vs sham; ¶p < 0.001 vs C + sham; *p < 0.01 effect of LP diet; †p < 0.01 effect of STZ; ‡p < 0.001 effect of STZ.
Beta cell loss occurred following STZ treatment for both control and LP-treated mice as shown on d 7, 2 d after the final STZ injection (Figs. 1 and 2A and B; p < 0.05). No histologic evidence of insulitis was observed in animals receiving STZ. STZ administration decreased BCM by 51 and 45% in female C + STZ and LP + STZ mice compared with the respective sham-treated groups on d 7. In the male C + STZ and LP + STZ groups there was a 64% and 52% decrease respectively. On d 14 in males given STZ, with or without LP exposure, there was a partial recovery of BCM (Fig. 1A). By d 30, male C + STZ mice fully recovered BCM versus controls. In contrast LP + STZ mice showed a significant deficit (p < 0.05) compared with LP sham-treated mice (Fig. 1A). This indicates that the initial increase in BCM in male LP + STZ was not sustained. Male STZ-treated mice became overtly diabetic before d 130 as mentioned above. In female mice, LP + STZ animals had a significantly lower BCM (p < 0.01) compared with LP sham injected mice on d 7 (Fig. 1B). Subsequently, female LP mice showed a significant increase (p < 0.001) in BCM by d 14 compared with C animals, which was not seen following STZ treatment (Fig. 1B). BCM was completely recovered by d 30 in C + STZ but not LP + STZ mice. However, BCM was not maintained over the long term as measured at d 130 in the C + STZ group, which was significantly lower (p < 0.001) than in C mice, whereas LP + STZ mice continued to show a reduced (p < 0.001) BCM compared with LP animals (Fig. 1B).

Representative Immuno-histochemical and fluorescent images. Localization of insulin (red) and glucagon (brown) in an islet from (A) C and (B) C + STZ mice showing a decrease in insulin immunoreactive positive beta cells on d 7. Identification of Pdx-1+/Inslow progenitor cells (arrows) from an LP + STZ islet. (C) Insulin; (D) Pdx-1; (E) insulin and Pdx-1; (F) merged image with nuclear DAPI stain (blue). Magnification bars represent 100 μm.
Alpha cell mass.
To determine whether the effects of LP diet were specific to beta cells, the ACM was also examined (Fig. 3). ACM in male LP mice was significantly reduced (p < 0.01) at d 7, but was comparable with C mice for the remaining period of study (Fig. 3A). In female mice, initially there was a significant effect (p < 0.05) of the LP diet reducing ACM at d 7, however, LP mice significantly increased (p < 0.01) ACM at d 14 and 30 when compared with C mice (Fig. 3A). In male C + STZ mice, there was a transient decrease (p < 0.001) in ACM on d 7 compared with C (Fig. 3B). In female mice, STZ treatment under LP conditions significantly reduced ACM at d 14 and 30 (p < 0.05; Fig. 3B).

Changes in alpha cell mass with diet and STZ treatment. ACM for C (black bars), C + STZ (white bars), LP (gray bars), and LP + STZ (hatched bars) offspring over time for male (A) and female (B) mice. Means ± SEM; n = 3 animals per group and three sections per pancreas. (A) §p < 0.001 vs sham; ∥p < 0.05 vs C + sham; (B) **p < 0.05 vs sham; §p < 0.001 vs sham; †p < 0.01 vs C + sham; ‡p < 0.001 vs C + sham; *p < 0.05 effect of LP diet.
Islet size distribution.
Islets were grouped into large (>10,000 μm2), medium (5,000–10,000 μm2) and small (300–5000 μm2) sizes by measuring mean islet area and normalized to total pancreas area (mm2). The LP diet did not alter the islet distribution (Table 2), and therefore the change in BCM in LP female mice was not attributed to an increase in new small islets or small islets becoming large. In addition, STZ treatment did not alter the distribution of islet sizes on d 7, demonstrating that the reduced BCM associated with STZ affected all sizes of islets equally (Table 2). By d 14, C + STZ male, but not female mice, showed a significant reduction in the number of large islets compared with sham controls. By d 30, there were no longer any differences in islet size distribution for control-fed animals receiving STZ, but LP female mice showed reduced numbers of islets of all sizes following STZ (Table 2). This shows that the relative deficit in BCM in these animals shown in Fig. 1 was due to an overall reduction in the number of islets, rather than a failure of smaller islets to become large.
Islet cell proliferation.
The percentage of PCNA positive alpha and beta cells was measured to determine the contribution of replication to the observed changes in islet mass. On d 14, there was a significant increase (p < 0.01) in alpha and beta cell replication in male C + STZ mice compared with C mice (Fig. 4A). This may contribute to the 4.5-fold increase in BCM from d 7 to 14 and continued expansion to d 30, as well as the increased ACM. However, in female mice there were no significant changes in alpha or beta cell proliferation (Fig. 4B).

Alpha and beta cell proliferation. Percentage of PCNA positive proliferating beta (A and C) and alpha (B and D) cells at d 14 for male (A and B) and female (C and D) mice. White bars = sham; black bars = STZ. Means ± SEM; n = 3 animals per group, and three sections per pancreas. **p < 0.01 vs C.
Individual cell size.
We examined cell size to determine the relative contribution of cell hypertrophy to the increased ACM and BCM in d 14 female LP mice. Individual beta cell area was significantly (p < 0.01) increased in the LP group (71.77 ± 3.18 μm2) compared with the C group (61.01 ± 1.41 μm2). This change in the LP group is associated with an approximate 5.8-fold increase in BCM compared with the C group. No difference was found for individual alpha cell area.
Pdx-1+/insulinlow islet cells.
Female mice were further analyzed to identify a possible precursor cell population that might contribute to regeneration of BCM. Between 4 and 8% of islet cells were found to be Pdx-1+/insulinlow in C animals, and this did not change after STZ (Table 4; Fig. 2C–F). However, the abundance of these cells was significantly (p < 0.01) increased in LP mice after STZ.
Glucose homeostasis.
Both male STZ-treated groups were relatively hyperglycemic compared with sham-injected mice at d 7, 14, and 30 (Table 1), whereas female STZ-treated mice were able to maintain euglycemia, despite changes in BCM. At d 42, STZ treatment in male and female mice led to fasting hyperglycemia (data not shown). At this age, an IPGTT was performed. Blood glucose levels in the C + STZ and LP + STZ groups were elevated over the time course compared with non-STZ treatment and did not reach baseline within 90 min. The area under the curve (AUC) for STZ treated groups was significantly increased (p < 0.05; Fig. 5A and B insets), indicating a relative impairment of glucose tolerance. Exposure to LP diet did not impair glucose tolerance at this age. At d 130, fasting hyperglycemia was observed in female STZ treated mice, whereas no difference was found between C and LP groups for either gender (Table 1). Following an IPGTT at d 130, female STZ, LP and LP + STZ mice displayed glucose intolerance determined by an elevated AUC (p < 0.05; Fig. 5D inset). However, the LP male mice had a comparable glucose response with the C mice during the IPGTT (Fig. 5C).

Intraperitoneal glucose tolerance test. IPGTTs were performed on d 42 (A and B) and d 130 (C and D) for male (A and C) and female (B and D) offspring of C (solid lines) or LP dams (dashed lines), with (open squares) or without (closed squares) subsequent STZ treatment. Insets represent the corresponding AUC, where white bars = sham; black bars = STZ. Means ± SEM for n = 10–13 (A and B); n = 3–5 (C and D). *p < 0.05 effect of STZ treatment (A, B, and D insets); §p < 0.05 effect of LP diet (D, inset).
Serum and pancreatic insulin.
Measurements of serum insulin concentration were made on d 7, 14, and 30. Despite changes in BCM, serum insulin concentrations were similar between groups and genders, with d 14 mean values listed in Table 3. On d 14, pancreatic insulin content was significantly reduced (p < 0.05) in male mice exposed to LP, but not in females (Table 3). STZ treatment significantly reduced (p < 0.05) insulin content in females, but the extent of the reduction was greater in animals receiving LP diet. No change in pancreatic insulin content was observed in males treated with STZ; however, this may have been more evident after the initial beta cell loss on d 7.
DISCUSSION
The pancreas has the ability to regenerate new beta cells after injury (14,20,21), however, regeneration after subtotal deletion with a single, high dose of STZ may be incomplete (14) and this ability decreases with age (15). We used multiple low doses of STZ to create partial beta cell deletion in young mice that was not accompanied by lymphocyte infiltration, with the expectation that BCM would largely regenerate. No significant change in blood glucose levels, or circulating insulin was observed in female mice following STZ, although there was a decrease in pancreatic insulin content consistent with BCM loss. Male STZ-treated mice showed elevated blood glucose levels throughout the study, which may have been due initially to a marginally greater loss of BCM following STZ in males than in females.
Glucose tolerance was impaired in C + STZ females on d 42 and BCM was reduced by d 130. This suggests that while beta cell replacement is possible, it is functionally incomplete, consistent with previous findings in rat (14). Recovery of BCM by d 30 was not due to significant increases in beta cell proliferation, or beta cell hypertrophy, but may be the result of stem/precursor cell differentiation. A Pdx1+/insulinlow islet cell population was described (22), which were capable of maturing into insulin-expressing cells. Our findings show that 4%to 8% of islet cells in neonatal mice were Pdx1+/insulinlow.
As in females, BCM was fully replaced by d 30 but in males this was attributed to an increase in beta cell proliferation when sampled at d 14. Male C + STZ mice remained hyperglycemic, which led to overt diabetes in 33% of mice. The gender difference in blood glucose after STZ treatment is consistent with previous findings (23), however, here we report a novel finding that male and female mice seem to replace BCM by different mechanisms.
This is the first study to describe the effects of LP during gestation on the development of the endocrine pancreas in the mouse offspring. Protein restriction did not affect maternal weight gain or food intake, contrary to the findings in rats where maternal weight gain decreased, even with increased food intake (6,9). Birth weight was decreased, as observed previously (6,24,25), however, a relative overgrowth was reported by 3 wk (24) or 8 mo (25), which was not observed here. BCM was significantly increased in the female offspring on d 14, unlike the findings in rat (6,9,11), indicating important species and gender differences. This could represent a compensatory response to meet the demands of increased nutrient availability, leading to beta cell hypertrophy, but no increase in pancreatic insulin content was found. On d 14, the BCM in female LP mice was approximately 3-fold greater than in C. Hypertrophy of individual beta cells could account for this, whereas proliferation was unchanged. Male LP offspring did not show increased BCM on d 14 and had comparable values with control-fed animals on d 30. These gender differences could be related to changes in endocrine hormones. Studies have shown in the LP-fed rat that maternal plasma levels of prolactin, leptin, estradiol, and progesterone are all altered during gestation (26). These changes may differentially affect fetal trajectory of beta cell growth and glucose homeostasis later in life.
A study in C57Bl/6J mice found that male, but not female mice, showed glucose intolerance at 8 mo of age after exposure to LP in early life (25). In contrast, glucose tolerance at d 130 was impaired predominantly in female LP mice in this study, although the mouse strains and time course used differed. Our results agree with the corresponding rat model where LP-fed female rats became glucose intolerant at 130 d of age (12). Research has also shown a strong link between poor human fetal development during times of famine and glucose intolerance later in life (27). Despite the compensatory beta cell response shown in LP mice early in life, the long-term effects are consistent with rat and human studies.
We found that LP exposure affected pancreatic endocrine plasticity after STZ. LP + STZ mice failed to reach an equivalent BCM to that of LP mice, unlike control-fed animals, indicating a reduced capacity for beta cell regeneration. In female animals at d 30, this was associated with a reduced number of islets relative to STZ treatment alone, but this was not seen in males. Consistent with our observations, other studies have shown that nutrient deficiency early in life effects tissue plasticity. LP diet during gestation significantly impaired recovery of male adult rats after ischemia reperfusion (28), whereas maternal calorie restriction during gestation and lactation impaired beta cell replacement after STZ (29). Changes in islet morphometry resulting from previous exposure to LP were not specific to beta cells, as the ACM was also increased at d 14 and 30 in females. Thus, the increased BCM in LP-fed females at d 14 is likely to represent a more general increase in islet mass. In the LP + STZ group, ACM was significantly reduced versus LP alone by d 30. This suggests that the inability of LP-fed animals to recover BCM after STZ also resulted in a delayed ability to increase ACM as would normally occur with age, implying that the mechanisms involved might be common to multiple endocrine cell types.
Pdx-1+/insulinlow cells secrete less insulin then Pdx-1+/insulin+ cells and over time, this population has the ability to become Pdx-1+/insulin+ without cell replication (22), perhaps functioning as a strategic reserve. In our model, the proportion of the Pdx-1+/insulinlow islet cells was not altered by LP diet but was increased after STZ. This may indicate that normal maturational pathways that allow such cells to differentiate into functional beta cells are impaired by LP exposure, resulting in a reduced BCM. In addition to a risk for later diabetes, the loss of plasticity may compromise the female offspring during pregnancy, where a substantial expansion of maternal BCM normally occurs.
In conclusion, we describe a mouse model in which exposure to a LP diet during gestation has critical effects on the development of the endocrine pancreas and beta cell recovery after STZ. We have shown that mouse pancreatic development in a protein-deficient environment was altered to produce morphologic changes in islet mass in early life and glucose intolerance in females later in life. In addition, recovery from STZ-induced injury was impaired by previous LP exposure. Our results therefore suggest that the previous dietary environment is a determinant of subsequent beta cell plasticity in a mouse model.
Abbreviations
- ACM:
-
alpha cell mass
- AUC:
-
area under the curve
- BCM:
-
beta cell mass
- C:
-
control diet
- IPGTT:
-
intraperitoneal glucose tolerance test
- LP:
-
low protein diet
- PCNA:
-
proliferating cell nuclear antigen
- STZ:
-
streptozotocin
References
Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD 1991 Fetal and infant growth and impaired glucose tolerance at age 64. BMJ 303: 1019–1022
Hales CN, Barker DJ 1992 Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35: 595–601
Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM 1993 Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36: 62–67
Barker DJ, Winter PD, Osmond C, Margetts B, Simmonds SJ 1989 Weight in infancy and death from ischaemic heart disease. Lancet 2: 577–580
Holemans K, Van Bree R, Verhaeghe J, Meurrens K, Van Assche FA 1997 Maternal semistarvation and streptozotocin-diabetes in rats have different effects on the in vivo glucose uptake by peripheral tissues in their female adult offspring. J Nutr 127: 1371–1376
Snoeck A, Remacle C, Reusens B, Hoet JJ 1990 Effect of a low protein diet during pregnancy on the fetal rat endocrine pancreas. Biol Neonate 57: 107–118
Oh W, Gelardi NL, Cha CJ 1988 Maternal hyperglycemia in pregnant rats: its effect on growth and carbohydrate metabolism in the offspring. Metabolism 37: 1146–1151
De Prins FA, Van Assche FA 1982 Intrauterine growth retardation and development of endocrine pancreas in the experimental rat. Biol Neonate 41: 16–21
Heywood WE, Mian N, Milla PJ, Lindley KJ 2004 Programming of defective rat pancreatic beta-cell function in offspring from mothers fed a low-protein diet during gestation and the suckling periods. Clin Sci (Lond) 107: 37–45
Holemans K, Aerts L, Van Assche FA 2003 Lifetime consequences of abnormal fetal pancreatic development. J Physiol 547: 11–20
Petrik J, Reusens B, Arany E, Remacle C, Coelho C, Hoet JJ, Hill DJ 1999 A low protein diet alters the balance of islet cell replication and apoptosis in the fetal and neonatal rat and is associated with a reduced pancreatic expression of insulin-like growth factor-II. Endocrinology 140: 4861–4873
Chamson-Reig A, Thyssen SM, Arany E, Hill DJ 2006 Altered pancreatic morphology in the offspring of pregnant rats given reduced dietary protein is time and gender specific. J Endocrinol 191: 83–92
Thyssen S, Arany E, Hill DJ 2006 Ontogeny of Regeneration of {beta}-Cells in the Neonatal Rat after Treatment with Streptozotocin. Endocrinology 147: 2346–2356
Wang RN, Bouwens L, Kloppel G 1994 Beta-cell proliferation in normal and streptozotocin-treated newborn rats: site, dynamics and capacity. Diabetologia 37: 1088–1096
Wang RN, Bouwens L, Kloppel G 1996 Beta-cell growth in adolescent and adult rats treated with streptozotocin during the neonatal period. Diabetologia 39: 548–557
Dor Y, Brown J, Martinez OI, Melton DA 2004 Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429: 41–46
Xu X, D'Hoker J, Stangé G, Bonné S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, Bouwens L, Scharfmann R, Gradwohl G, Heimberg H 2008 Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132: 197–207
Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA 2008 In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455: 627–632
Chamson-Reig A, Arany EJ, Summers K, Hill DJ 2009 A low protein diet in early life delays the onset of diabetes in the non-obese diabetic mouse. J Endocrinol 201: 231–239
Cantenys D, Portha B, Dutrillaux MC, Hollande E, Roze C, Picon L 1981 Histogenesis of the endocrine pancreas in newborn rats after destruction by streptozotocin. An immunocytochemical study. Virchows Arch B Cell Pathol Incl Mol Pathol 35: 109–122
Dutrillaux MC, Portha B, Roze C, Hollande E 1982 Ultrastructural study of pancreatic B cell regeneration in newborn rats after destruction by streptozotocin. Virchows Arch B Cell Pathol Incl Mol Pathol 39: 173–185
Szabat M, Luciani DS, Piret JM, Johnson JD 2009 Maturation of adult beta-cells revealed using a Pdx1/insulin dual-reporter lentivirus. Endocrinology 150: 1627–1635
Rossini AA, Williams RM, Appel MC, Like AA 1978 Sex differences in the multiple-dose streptozotocin model of diabetes. Endocrinology 103: 1518–1520
Chen JH, Martin-Gronert MS, Tarry-Adkins J, Ozanne SE 2009 Maternal protein restriction affects postnatal growth and the expression of key proteins involved in lifespan regulation in mice. PLoS One 4: e4950
Bhasin KK, van Nas A, Martin LJ, Davis RC, Devaskar SU, Lusis AJ 2009 Maternal low-protein diet or hypercholesterolemia reduces circulating essential amino acids and leads to intrauterine growth restriction. Diabetes 58: 559–566
Fernandez-Twinn DS, Ozanne SE, Ekizoglou S, Doherty C, James L, Gusterson B, Hales CN 2003 The maternal endocrine environment in the low-protein model of intra-uterine growth restriction. Br J Nutr 90: 815–822
Ravelli AC, Van Der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, Bleker OP 1998 Glucose tolerance in adults after prenatal exposure to famine. Lancet 351: 173–177
Elmes MJ, Gardner DS, Langley-Evans SC 2007 Fetal exposure to a maternal low-protein diet is associated with altered left ventricular pressure response to ischaemia-reperfusion injury. Br J Nutr 98: 93–100
Garofano A, Czernichow P, Breant B 2000 Impaired beta-cell regeneration in perinatally malnourished rats: a study with STZ. FASEB J 14: 2611–2617
Acknowledgements
We thank Christine Beamish, Astrid Chamson-Reig, Tom Chrones, Kelly Marchand, Mike Nicholson, Jake Pace, Brenda Strutt, Angela Taylor, and the animal care staff for their expert assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported through grants from the Canadian Diabetes Association and the Canadian Institutes of Health Research.
Rights and permissions
About this article
Cite this article
Cox, A., Gottheil, S., Arany, E. et al. The Effects of Low Protein During Gestation on Mouse Pancreatic Development and Beta Cell Regeneration. Pediatr Res 68, 16–22 (2010). https://doi.org/10.1203/PDR.0b013e3181e17c90
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181e17c90
