
Abstract
X-linked hypohidrotic ectodermal dysplasia (XLHED), which is characterized by hypodontia, hypotrichosis, and hypohidrosis, is caused by mutations in ED1, the gene encoding ectodysplasin-A (EDA). This protein belongs to the tumor necrosis factor ligand superfamily. We analyzed ED1 in two Japanese patients with XLHED. In patient 1, we identified a 4-nucleotide insertion, c.119-120insTGTG, in exon 1, which led to a frameshift mutation starting from that point (p.L40fsX100). The patient's mother was heterozygous for this mutation. In patient 2, we identified a novel missense mutation, c.1141G>C, in exon 9, which led to a substitution of glycine with arginine in the TNFL domain of EDA (p.G381R). This patient's mother and siblings showed neither symptoms nor ED1 mutations, so this mutation was believed to be a de novo mutation in maternal germline cells. According to molecular simulation analysis of protein structure and electrostatic surface, p.G381R increases the distance between K375 in monomer A and K327 in monomer B, which suggests an alteration of overall structure of EDA. Thus, we identified two novel mutations, p.L40fsX100 and p.G381R, in ED1 of two XLHED patients. Simulation analysis suggested that the p.G381R mutation hampers binding of EDA to its receptor via alteration of overall EDA structure.
Similar content being viewed by others


Main
Hypohidrotic ectodermal dysplasia (HED) is a congenital disorder characterized by the impaired development of teeth (hypodontia), hair (hypotrichosis), and eccrine sweat glands (hypohidrosis) (1). Most HED patients have shown an X-linked inheritance pattern, although a minority of patients had an autosomal dominant or recessive trait (2,3).
The ED1 gene responsible for XLHED was mapped to chromosome Xq12-q13 and was identified by positional cloning (4). ED1 encodes the protein ectodysplasin-A (EDA) that belongs to the tumor necrosis factor ligand (TNFL) superfamily (5,6). EDA consists of a small N-terminal intracellular domain, a transmembrane domain, and a larger C-terminal extracellular domain containing a furin-cleavage site, a collagen-like domain, and a TNFL domain. The collagen-like domain has been believed to be necessary for trimerization of EDA proteins (7). However, it is not known whether the collagen-like domain is solely responsible for the trimerization: constructs of EDA-A1 and EDA-A2 lacking the collagen- like region can pack in the crystals as dimers of trimers (8). The TNFL domain has the ability to interact with the EDA receptor (EDAR) and X-linked ectodysplasin-A2 receptor (XEDAR) (9).
EDA-A1 and EDA-A2 are the longest EDA isoforms, but they differ by only an insertion/deletion of two amino acids (E308, V309) as determined by alternative splicing of ED1 pre-mRNA. This insertion/deletion functions to regulate receptor-binding specificity, such that EDA-A1 binds EDAR, whereas EDA-A2 binds XEDAR (10). EDAR interacts with its adapter EDAR-associated death domain (EDARADD) to build an intracellular complex and activate the nuclear factor-κB pathway, which is essential for the proper development of ectodermal derivatives (3,11,12). EDAR and EDARADD are the genes responsible for the autosomal forms of HED (2,3,13).
Crystal structure analysis has provided a new understanding of EDA-A1 and EDA-A2. For example, higher-order assembly of the EDA trimers has been clarified. The crystallographic asymmetric unit of both EDA-A1 and EDA-A2 crystals contains more than one copy of the biologically relevant trimer. The EDA-A1 asymmetric unit comprises four trimers, and the EDA-A2 asymmetric unit comprises two trimers. Ligation of trimers into higher-order assemblies may increase receptor affinity by avidity or may change the geometry of the ligand-receptor complex (8).
Many mutations in ED1 in various countries have been reported (14–17). In Japan, several mutations have been identified in XLHED patients (18–20). Affected males showed most or all of the typical phenotypes of XLHED. However, only a few studies have reported the relationship between nucleotide substitution and protein structure (8,21,22).
In this study, we analyzed the ED1 gene in two unrelated Japanese patients with XLHED, and we identified two novel mutations: p.L40fsX100 and p.G381R. The former was inherited from the mother, and the latter was a de novo mutation occurred in maternal germline cells. Our molecular simulation analysis of EDA suggested that the p.G381R mutation hampers the binding of EDA to its receptor via alteration of the overall structure of EDA.
PATIENTS AND METHODS
Patients.
Two Japanese patients with XLHED were enrolled in this study. Informed consent was obtained from the parents before DNA sampling. This study was approved by the Kobe University ethical committee. One hundred healthy Japanese adults volunteered to participate in the study as control subjects.
Patient 1 was a 1-y-5-mo-old boy of unrelated parents in family A (Fig. 2A). He had no siblings. The mother was healthy from her appearance; she had no abnormal features and no missing permanent teeth. The patient was initially diagnosed with HED on the basis of his sparse, thin hair; lack of eyebrows and eyelashes; and characteristic facial appearance, including a prominent forehead and saddle nose. The qualitative sweating test with starch and iodine produced almost no response in the patient. The mother showed a slight decrease in sweat response, but she did not undergo further quantitative analysis. Histologic examination of the skin biopsy specimen showed complete lack of hair follicles, eccrine sweat glands, and sebaceous glands, which corresponded to the characteristic findings of HED (Fig. 1A). XLHED was diagnosed on the basis of molecular analysis of ED1.

Family A with XLHED. (A) Pedigree. (B) DHPLC screening and direct DNA sequencing of ED1 exon 1. The patient carried a c.119-120insTGTG in ED1 exon 1, which made a premature stop codon at amino acid 100 (p.L40fsX100). His mother was heterozygous for the mutation. Arrows indicate the TGTG insertion. (C) X-chromosome inactivation assay. The 310-bp band was from the normal allele, and the 280-bp band was from the affected allele. In patient 1, no amplification of the 280-bp band was obtained, because the patient X-chromosome is not methylated. In his mother, with HpaII treatment produced a higher intensity 310-bp band compared with the 280-bp band, which suggests that the normal allele was mainly inactivated by methylation. The degree of skewed X-chromosome inactivation toward the normal allele was calculated as 80%.

Histologic examination of the skin biopsy specimen. (A) Patient 1. H&E staining. Scale bar, 0.5 mm. (B) Patient 2. H&E staining. Scale bar, 0.5 mm. Both patients showed complete lack of hair follicles, eccrine sweat glands, and sebaceous glands.
Patient 2 was an 8-mo-old twin boy of unrelated parents in family B (Fig. 3A). His elder sister, twin brother, and parents were healthy and showed no dysmorphologic features. He was referred to a city hospital because of recurrent episodes of high temperature. The diagnosis of HED was based on his thin skin, sparse hair; lack of eyebrows; no teeth; and characteristic facial features, including a full forehead and saddle nose. The acetylcholine test demonstrated no sweat response in the patient. Histologic examination of the skin biopsy specimen revealed complete lack of hair follicles, eccrine sweat glands, and sebaceous glands (Fig. 1B). XLHED was diagnosed on the basis of molecular analysis of ED1.

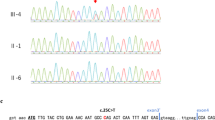
Family B with XLHED. (A) Pedigree. (B) DHPLC screening of ED1 exon 9. DHPLC demonstrated a heteroduplex peak for patient 2 but not other family members. (C) Direct DNA sequencing of ED1 exon 9. The patient carried a c.1141G>C in ED1 exon 9, which led to substitution of glycine with arginine at amino acid 381 (p.G381R). The arrow indicates the c.1141G>C. (D) Amino acid sequence alignments of EDA and its closest relatives in the H-strand of the TNFL domain. Bold letters mean conserved amino acids. The glycine at amino acid 381 in EDA is highly conserved among the TNFL superfamily.
DNA extraction.
Genomic DNA was extracted from 2–5 mL of whole blood from each individual by using a DNA extraction kit (SepaGene; Sanko Junyaku, Tokyo, Japan), according to the manufacturer's instructions. The extracted DNA samples were stored at –20°C until analysis.
Polymerase chain reaction.
PCR was carried out by using a PC 701 thermal cycler (Astec, Tokyo, Japan). Table 1 shows the primer sequences for ED1 analysis.
Denaturing HPLC (DHPLC).
To screen for a mutation in ED1, DHPLC analysis was performed according to our previous reports (23,24). Hybridization of the PCR product mixture of the test DNA with nucleotide substitution and reference DNA without nucleotide substitution produced a heteroduplex peak in the DHPLC chart. The appearance of this heteroduplex peak suggested the presence of a nucleotide substitution in the PCR-amplified fragment. The DHPLC conditions, including column temperatures (Table 1), were determined empirically to maximize resolution of the heteroduplex and homoduplex peaks.
DNA sequencing.
To identify and confirm the mutation, direct sequencing analysis was performed with a BigDye Terminator V3.0 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and a genetic analyzer (ABI Prism 310; Applied Biosystems), with DNA Sequencing Analysis Software (Applied Biosystems).
Skewed X-chromosome inactivation assay.
To determine the X-chromosome inactivation status of the mother of family A, a manifested carrier of XLHED, we quantified the activated and inactivated CpG dinucleotides in exon 1 of the androgen receptor gene (AR) by the method described in our previous report (25). To explain briefly, unmethylated (activated) and methylated (inactivated) alleles were measured via a combination of DNA digestion by HpaII and PCR amplification of digested and nondigested products. HpaII digested unmethylated CpG but not methylated CpG. PCR amplified only nondigested products, i.e. the inactivated allele. The degree of inactivation skewing to one allele was calculated according to the formula presented by Lau et al. (26).
Parentage testing.
To confirm that all siblings in family B were born to the same parents, parentage testing was performed. Fifteen additional loci on 13 chromosomes were genotyped in all family members by using AmpFLSTR PCR Amplification Kits (Applied Biosystems). The loci tested were D3S1358, vWA, D16S539, D2S1338, D8S1179, D21S11, D18S51, D19S433, THo1, FGA, D5S818, D13S317, D7S820, TPOX, and CSF1PO. Genomic DNA was amplified in a standard PCR reaction, and alleles were analyzed via the ABI PRISM 310 Genetic Analyzer with GeneScan and Genotyper software programs (Applied Biosystems).
Molecular simulation analysis of the protein structure and the electrostatic surface.
The three-dimensional structures of EDA-A1 and EDA-A2 were derived from Protein Data Bank (accession numbers: EDA-A1, 1RJ7; EDA-A2, 1RJ8). The hydrogen atoms in these model structures were added by means of PyMOL software (27). Following preparation of p.G381R mutants of EDA-A1 and EDA-A2 by means of SWISS-MODEL and Swiss-PDB Viewer (28), the mutant coordinates were optimized by using the MINIMIZE program of the TINKER software package (29) with the AMBER99 force field parameter. The minimizations were executed for root mean square (RMS) gradient values of 0.01 kcal/mol/Å. Then, the molecular structure and electrostatic surface were analyzed by using PyMOL software with the APBS (Adaptive Poisson-Boltzmann Solver) plugin (30).
RESULTS
Patient 1 and Family A
Identification of a novel insertion mutation.
A heteroduplex peak in the DHPLC chart for ED1 exon 1 suggested the presence of a mutation in the patient and his mother (Fig. 2B). Direct sequencing showed that the patient had a 4-bp insertion between nucleotides 119 and 120 of ED1 exon 1 (c.119-120insTGTG) (Fig. 2B). The mother was heterozygous for the mutation. The insertion c.119-120insTGTG led to a frameshift mutation starting from that point and would result in truncation of the protein at amino acid 100 (p.L40fsX100).
Determination of skewed X-chromosome inactivation.
Figure 2C shows the result of the skewed X-chromosome inactivation assay. The affected allele of patient 1 was digested completely by HpaII treatment, and no amplification of the 280-bp band of the affected allele was observed. For his mother, two bands, the 310-bp band from the normal allele and the 280-bp band from the affected allele, were obtained with and without HpaII treatment. Without HpaII treatment, the 310-bp and 280-bp bands manifested the same intensity. However, with HpaII treatment, the 310-bp band had a much higher intensity compared with the 280-bp band, which suggests that the normal allele was mainly inactivated by methylation. The degree of skewed X-chromosome inactivation toward the normal allele was calculated to be 80%, i.e. only 20% of the activated X-chromosome was normal.
Patient 2 and Family B
Identification of a novel missense mutation.
A heteroduplex peak in the DHPLC chart for ED1 exon 9 suggested the presence of a mutation in the patient (Fig. 3B). Direct DNA sequencing showed that the patient had a G-to-C transversion at nucleotide 1141 (Fig. 3C). This c.1141G>C transversion led to substitution of glycine with arginine at amino acid 381 (p.G381R) in the TNFL domain. p.G381R likely affects the binding of EDA to its receptor; the glycine residue at amino acid 381 is highly conserved among TNFL domains in TNFL superfamily members (Fig. 3D).
DHPLC did not detect the index transversion in family members. The transversion in the patient may be a maternal germline mutation, because parentage testing verified the relationships among family members with a cumulative likelihood ratio of more than 99.99% (data not shown).
To test the possibility that c.1141G>C in ED1 was a polymorphism, we screened 100 Japanese control subjects for the transversion. None carried this transversion, which suggests that it is not a polymorphism but is instead a disease-causing mutation.
Molecular structure and electrostatic surface of G381R mutants.
To investigate whether p.G381R could alter the overall structure of EDA, the electrostatic surface and the molecular structure were analyzed by using wild type and simulated mutant molecules. Figure 4A and B show the molecular surfaces of wild-type and p.G381R-mutated EDA colored according to the calculated electrostatic surface potential. Blue indicates positive (maximum +10 kT/e), white indicates neutral, and red indicates negative (minimum –10 kT/e). The EDA surface contains the putative receptor-binding site along the monomer–monomer interface. The relatively large receptor interaction areas (upper and lower yellow boxes in Fig. 4A and B) include dispersed binding determinant parts. The mutation alters the positively charged area in the receptor binding sites (upper yellow boxes in Fig. 4A and B) of EDA. In wild-type isoforms, K375 in monomer A and K327 in monomer B are adjacent, whereas in p.G381R-mutant isoforms, the two amino acids are separated by a considerable distance.

Effect of the p.G381R mutation on the electrostatic surface of EDA-A1 and EDA-A2. (A) Electrostatic surfaces of wild-type EDA-A1 (left) and p.G381R-mutant EDA-A1 (right), with predicted receptor binding sites (yellow boxes). As shown in the enlargements of the upper yellow boxed areas, the p.G381R mutation increased the distance between K375 in monomer A and K327 in monomer B. (B) Electrostatic surfaces of wild-type EDA-A2 (left) and p.G381R-mutant EDA-A2 (right), with predicted receptor binding sites (yellow boxes). As shown in the enlargements of the upper boxed areas, similar to EDA-A1, the p.G381R mutation increased the distance between K375 in monomer A and K327 in monomer B.
DISCUSSION
Mutations.
We identified two novel mutations in ED1 in two Japanese patients with XLHED. Patient 1 carried a new 4-bp insertion, c.119-120insTGTG, which led to a frameshift mutation starting from amino acid 40 and made a stop codon at amino acid 100 in EDA (p.L40fsX100). The truncated EDA would lack part of the collagen-like domain and the whole TNFL domain. The collagen-like domain is crucial for trimerization of EDA, and the TNFL domain is required for binding of EDA to its receptor (7). However, it is not known whether the collagen-like domain is solely responsible for the trimerization, as described in the Introduction section. Schneider et al. (9) also reported that in-frame deletions in collagen-like domain do not affect the ability of EDA to trimerize nor to further multimerize, suggesting that the collagen-like domain may serve additional functions. Thus, the 4-bp insertion may disrupt the functions of the EDA protein because of the loss two important functional domains. Patient 2 carried a novel missense mutation, c.1141G>C, which led to the substitution of glycine with arginine at amino acid 381 in EDA (p.G381R). The mutation is located in the TNFL domain of the EDA protein. Several mutations in the TNFL domain have previously been reported (8,9,22). Schneider et al. (9) showed that all missense mutations in the TNFL domain resulted in abolished or impaired binding of EDA to its receptors.
Inheritance traits.
The mother of patient 1 had no symptoms except for slightly decreased sweating, with X-chromosome inactivation being skewed mainly toward the normal allele in blood cells. Carriers who are heterozygous for an XLHED mutation may have variable clinical features (14,17,31,32). Lexner et al. (17) reported that in two female carriers with pronounced clinical symptoms, the normal allele in their blood cells was mainly inactivated. Martínez et al. (31) suggested that skewed X-chromosome inactivation in blood cells can explain symptomatic differences among female carriers. However, the finding for our family A were not consistent with such reports. Our result suggests that X-chromosome inactivation is different in skin from blood cells: ectodermal and mesodermal tissues may differ with regard to factors related to X-chromosome inactivation. It also leads us to the idea that unlike the inference in previous reports, skewed X-chromosome inactivation in blood cells does not predict the clinical situation in the carrier status of XLHED.
The p.G381R mutation in patient 2 was a germline mutation. The recurrence risk of the germline mutation may depend on the proportions of germ cells with and without the mutation (24). The possibility of mutation recurrence in the next male sibling of a patient should be explained to parents during genetic counseling, even though the disease-causing mutation was identified as a de novo mutation and the risk of recurrence of the disease cannot be exactly determined.
Protein structure.
The p.G381R mutation in patient 2 replaced the small amino acid, glycine, with a larger one, arginine, that could not be accommodated structurally. Hymowitz et al. (8) divided the disease-causing mutations that affect the TNFL domain of EDA into three groups: 1) mutations that probably affect the overall structure of EDA, 2) mutations that affect the receptor binding site, and 3) mutations whose effect is uncertain but that may define a novel interaction site. It is most reasonable to expect that p.G381R belongs to the first group and may affect EDA function via alteration of overall EDA structure. Our simulation data offered some evidence for this hypothesis. The mutation increased the distance between K375 in monomer A and K327 in monomer B, which resulted in a drastic change in the electrostatic surface and surface conformation of the receptor binding site (upper boxes in Fig. 4A and B).
The question then arises: how does this p.G381R-induced alteration in structure affect EDA function? One possible explanation is that the electrostatic and structural changes in the surface caused by the mutation may hamper receptor binding. It should be noted that the mutation produced a large alteration in the “upper box” part of the receptor-binding site and a small alteration in the “lower box” part. This finding suggests that these parts of the receptor-binding site play different roles, that is, maintaining binding affinity and providing the receptor specificity, respectively. Another possible explanation is that unexpected physiochemical changes related to p.G381R may have a negative effect on receptor binding or assembly of the three monomers. An additional possibility is that the mutation alters the solubility or folding properties of EDA, which are related to its secretion by cells.
In conclusion, we found two novel mutations, one a frameshift (p.L40fsX100) and one a missense (p.G381R), in ED1 and discussed their inheritance trait in two unrelated families with XLHED. Molecular simulation analysis of the structure of the EDA protein suggested that the missense mutation hampers the binding of EDA into its receptor via alteration of the overall structure of EDA.
Abbreviations
- DHPLC:
-
denaturing high-performance liquid chromatography
- EDA:
-
ectodysplasin-A
- HED:
-
hypohidrotic ectodermal dysplasia
- TNFL:
-
tumor necrosis factor ligand
- XLHED:
-
X-linked HED
References
McKusick VA 1998 Mendelian Inheritance in Man, 12th Ed. Johns Hopkins University Press, Baltimore, pp 3307–3309
Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J 1999 Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet 22: 366–369
Headon DJ, Emmal SA, Ferguson BM, Tucker AS, Justice MJ, Sharpe PT, Zonana J, Overbeek PA 2001 Gene defect in ectodermal dysplasia implicates a death domain adapter in development. Nature 414: 913–916
Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson B, Munoz F, Morgan D, Clarke A, Baybayan P, Chen EY, Ezer S, Saarialho-Kere U, de la Chapelle A, Schlessinger D 1996 X-linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 13: 409–416
Bayés M, Hartung AJ, Ezer S, Pispa J, Thesleff I, Srivastava AK, Kere J 1998 The anhidrotic ectodermal dysplasia gene (EDA) undergoes alternative splicing and encodes ectodysplasin-A with deletion mutations in collagenous repeats. Hum Mol Genet 7: 1661–1669
Monreal AW, Zonana J, Ferguson B 1998 Identification of a new splice form of the EDA1 gene permits detection of nearly all X-linked hypohidrotic ectodermal dysplasia mutations. Am J Hum Genet 63: 380–389
Ezer S, Bayés M, Elomaa O, Schlessinger D, Kere J 1999 Ectodysplasin is a collagenous trimeric type II membrane protein with a tumor necrosis factor-like domain and co-localizes with cytoskeletal structures at lateral and apical surfaces of cells. Hum Mol Genet 8: 2079–2086
Hymowitz SG, Compaan DM, Yan M, Wallweber HJ, Dixit VM, Starovasnik MA, de Vos AM 2003 The crystal structures of EDA-A1 and EDA-A2: splice variants with distinct receptor specificity. Structure 11: 1513–1520
Schneider P, Street SL, Gaide O, Hertig S, Tardivel A, Tschopp J, Runkel L, Alevizopoulos K, Ferguson BM, Zonana J 2001 Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J Biol Chem 276: 18819–18827
Yan M, Wang LC, Hymowitz SG, Schilbach S, Lee J, Goddard A, de Vos AM, Gao WQ, Dixit VM 2000 Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science 290: 523–527
Yan M, Zhang Z, Brady JR, Schilbach S, Fairbrother WJ, Dixit VM 2002 Identification of a novel death domain-containing adaptor molecule for ectodysplasin-A receptor that is mutated in crinkled mice. Curr Biol 12: 409–413
Mikkola ML, Thesleff I 2003 Ectodysplasin signaling in development. Cytokine Growth Factor Rev 14: 211–224
Bal E, Baala L, Cluzeau C, El Kerch F, Ouldim K, Hadj-Rabia S, Bodemer C, Munnich A, Courtois G, Sefiani A, Smahi A 2007 Autosomal dominant anhidrotic ectodermal dysplasias at the EDARADD locus. Hum Mutat 28: 703–709
Vincent MC, Biancalana V, Ginisty D, Mandel JL, Calvas P 2001 Mutational spectrum of the ED1 gene in X-linked hypohidrotic ectodermal dysplasia. Eur J Hum Genet 9: 355–363
Visinoni AF, de Souza RL, Freire-Maia N, Gollop TR, Chautard-Freire-Maia EA 2003 X-linked hypohidrotic ectodermal dysplasia mutations in Brazilian families. Am J Med Genet A 122: 51–55
Zhao J, Hua R, Zhao X, Meng Y, Ao Y, Liu Q, Shang D, Sun M, Lo WH, Zhang X 2008 Three novel mutations of the EDA gene in Chinese patients with X-linked hypohidrotic ectodermal dysplasia. Br J Dermatol 158: 614–617
Lexner MO, Bardow A, Juncker I, Jensen LG, Almer L, Kreiborg S, Hertz JM 2008 X-linked hypohidrotic ectodermal dysplasia. Genetic and dental findings in 67 Danish patients from 19 families. Clin Genet 74: 252–259
Yotsumoto S, Fukumaru S, Matsushita S, Oku T, Kobayashi K, Saheki T, Kanzaki T 1998 A novel point mutation of the EDA gene in a Japanese family with anhidrotic ectodermal dysplasia. J Invest Dermatol 111: 1246–1247
Aoki N, Ito K, Tachibana T, Ito M 2000 A novel arginine → serine mutation in EDA1 in a Japanese family with X-linked anhidrotic ectodermal dysplasia. J Invest Dermatol 115: 329–330
Hashiguchi T, Yotsumoto S, Kanzaki T 2003 Mutations in the ED1 gene in Japanese families with X-linked hypohidrotic ectodermal dysplasia. Exp Dermatol 12: 518–522
RamaDevi AR, Reddy EC, Ranjan S, Bashyam MD 2008 Molecular genetic analysis of patients from India with hypohidrotic ectodermal dysplasia reveals novel mutations in the EDA and EDAR genes. Br J Dermatol 158: 163–167
Li S, Li J, Cheng J, Zhou B, Tong X, Dong X, Wang Z, Hu Q, Chen M, Hua ZC 2008 Non-syndromic tooth agenesis in two Chinese families associated with novel missense mutations in the TNF domain of EDA (ectodysplasin A). PLoS One 3: e2396
Sutomo R, Akutsu T, Takeshima Y, Nishio H, Sadewa AH, Harada Y, Matsuo M 2002 Rapid SMN1 deletion test using DHPLC to screen patients with spinal muscular atrophy. Am J Med Genet 113: 225–226
Sadewa AH, Sasongko TH, Gunadi Lee MJ, Daikoku K, Yamamoto A, Yamasaki T, Tanaka S, Matsuo M, Nishio H 2008 Germ-line mutation of KCNQ2, p.R213W, in a Japanese family with benign familial neonatal convulsion. Pediatr Int 50: 167–171
Ishihara H, Kanda F, Nishio H, Sumino K, Chihara K 2001 Clinical features and skewed X-chromosome inactivation in female carriers of X-linked recessive spinal and bulbar muscular atrophy. J Neurol 248: 856–860
Lau AW, Brown CJ, Peñaherrera M, Langlois S, Kalousek DK, Robinson WP 1997 Skewed X-chromosome inactivation is common in fetuses or newborns associated with confined placental mosaicism. Am J Hum Genet 61: 1353–1361
DeLano WL 2002 The PyMOL Molecular Graphics System. Available at: http://www.pymol.org Accessed August 30, 2008
Guex N, Peitsch MC 1997 SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18: 2714–2723
Ren P, Ponder JW 2003 Polarizable atomic multipole water model for molecular mechanics simulation. J Phys Chem B 107: 5933–5947
Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA 2001 Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA 98: 10037–10041
Martínez F, Millán JM, Orellana C, Prieto F 1999 X-linked anhidrotic (hypohidrotic) ectodermal dysplasia caused by a novel mutation in EDA1 gene: 406T>G (Leu55Arg). J Invest Dermatol 113: 285–286
Vincent MC, Cossée M, Vabres P, Stewart F, Bonneau D, Calvas P 2002 Pitfalls in clinical diagnosis of female carriers of X-linked hypohidrotic ectodermal dysplasia. Arch Dermatol 138: 1256–1258
Acknowledgements
We thank Dr. Ahmad Hamim Sadewa and Dr. Teguh Haryo Sasongko (Kobe University Graduate School of Medicine) for their technical assistance and Drs. Surini Yusoff and Indra Sari Kusuma Harahap (Kobe University Graduate School of Medicine) for their helpful comments on interpretation of the data.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by a Grant-Aid from the Ministry of Education, Science, Sports and Culture of Japan.
Rights and permissions
About this article
Cite this article
Gunadi, Miura, K., Ohta, M. et al. Two Novel Mutations in the ED1 Gene in Japanese Families With X-Linked Hypohidrotic Ectodermal Dysplasia. Pediatr Res 65, 453–457 (2009). https://doi.org/10.1203/PDR.0b013e3181991229
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181991229
This article is cited by
-
NRG1 variant effects in patients with Hirschsprung disease
BMC Pediatrics (2018)
-
Mutational spectrum in 101 patients with hypohidrotic ectodermal dysplasia and breakpoint mapping in independent cases of rare genomic rearrangements
Journal of Human Genetics (2016)
-
Orofacial Features of Hypohidrotic Ectodermal Dysplasia
Head and Neck Pathology (2012)
