
Abstract
Menkes disease (MD) is a rare and severe X-linked recessive disorder of copper metabolism. The MD gene, ATP7A (ATPase Cu++ transporting alpha polypeptide), encodes an ATP-dependent copper-binding membrane protein. In this report, we describe a girl with typical clinical features of MD, carrying a balanced translocation between the chromosomes X and 16 producing the disruption of one copy of ATP7A gene and the silencing of the other copy because of the chromosome X inactivation. Fluorescence in situ hybridization experiments with bacterial derived artificial chromosome probes revealed that the breakpoints were located within Xq13.3 and 16p11.2. Replication pattern analysis demonstrated that the normal X chromosome was late replicating and consequently inactivated, whereas the der(X)t(X;16), bearing the disrupted ATP7A gene, was active. An innovative approach, based on FMR1 (fragile X mental retardation 1) gene polymorphism, has been used to disclose the paternal origin of the rearrangement providing a new diagnostic tool for determining the parental origin of defects involving the X chromosome and clarifying the mechanism leading to the cytogenetic rearrangement that occurred in our patient.
Similar content being viewed by others

Main
Menkes disease (MD) is a recessive X-linked syndrome described by Menkes (1) more than 40 y ago. It is characterized by progressive cerebral and cerebellar degeneration, growth retardation, and peculiar scalp hair (2). The incidence is 1/250,000 (ORPHA565, http://www.orpha.net) and, in most of the cases, it is responsible for death before 3 y of age. Patients with MD have been found to have a defect in copper metabolism and low levels of serum copper and ceruloplasmin (3). MD phenotype is due to mutations occurring in the ATP7A (ATPase Cu++ transporting alpha polypeptide) gene, which encodes an ATP-dependent membrane protein (4) that regulates the release of Cu ions at the outer cell membrane, affecting the amount of copper-loaded enzymes, as ceruloplasmin and metallothionein (5).
Diagnosis can be established by detecting low levels of copper and ceruloplasmin in the serum, and high levels in cutaneous fibroblasts. The diagnosis can be confirmed by identification of the gene mutations. Genetic analysis also allows screening of carrier females, who may have patches with cutaneous and hair anomalies, and prenatal diagnosis through chorionic villus sampling.
Because MD is X-linked recessive, affected females are very rare. So far, only eight cases have been described in literature and cytogenetic studies have been reported only in five cases (Table 1): one displayed a mosaicism 45,X/46,XX and the remaining four showed a de novo balanced translocation between the chromosome X and an autosome. The breakpoint on the X chromosome is Xq13 where the gene ATP7A has been mapped.
In this article, we describe a female with MD carrying a balanced translocation between the chromosomes X and 16 producing the disruption of one copy of ATP7A gene and silencing of the other copy. Moreover, the late-replicating chromosome X has been investigated and an easy and reliable innovative approach has been used to disclose the parental origin of the rearrangement.
Clinical report.
A 6.5-mo-old girl presenting failure to thrive, facial dysmorphisms, and psychomotor delay was admitted for evaluation. She was the only daughter of a remotely consanguineous couple (cousins in fourth degree); the father had two other twin daughters who died in the newborn period with neonatal hepatitis. Family history was negative for neurologic diseases.
Her gestation was the result of in vitro fertilization, and she was born at 37 wk of gestation by emergency caesarean section because of meconium staining of amniotic fluid. Apgar score was 5 and 9 at first and fifth minute after birth, needing resuscitation with cardiac massage and positive pressure ventilation. Birth weight was 2270 g (below the 10th percentile).
At 5 mo, during hospitalization for respiratory syncytial virus (RSV), bronchiolitis, psychomotor delay, failure to thrive, and dysmorphic features facies (i.e. frontal bossing, epicanthus, broad-based nose, low set ears) were noticed. Laboratory investigations showed reduced circulating levels of ceruloplasmin.
At 6.5 mo, she was referred to our hospital for a metabolic evaluation. At admission weight, height, and head circumference were all below the third percentile; the skin was pale and dry, hair was scarce, distorted and harsh, with alopecia in the occipital region. Physical examination showed axial hypotonia, motor delay with no head control, with absence of abdominal organomegaly. Ophthalmogical examination was normal.
As shown in Table 2, laboratory studies displayed marked reduction of serum copper and ceruloplasmin levels. Blood lactate was elevated (49 mg/dL; normal <20) and urine organic acid analysis showed increased excretion of lactate, succinate, malate, and 2-ketoglutarate.
The 64Cu-uptake analysis in cultured fibroblasts showed a significant increase in copper uptake, confirming the clinical suspect of MD.
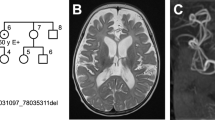
Cerebral imaging by angio-MRI disclosed increased tortuosity of all intracranial arteries (Fig. 1).

Angio-RMI showing a typical winding feature of the cerebral arteries of the patients with Menkes disease. Scale bar = 1 cm.
The karyotype analysis showed a balanced translocation between the long arm of X chromosome and the short arm of chromosome 16 described as 46,X,t(X;16)(q13.3;p11.2).
At 11 mo, she developed partial epileptic seizures that were controlled with carbamazepine. At 17 mo, right ureteral ectasia was detected.
In the last evaluation, at the age of 26 mo, the child maintained an inadequate growth (below third percentile), with severe psychomotor delay, just being able to control the head and visually following an object. She developed no language, had bilateral convergent strabismus, severe axial hypotonia with limb hypertonia, and right ureteral ectasia with no evidence of urinary tract infections. Occasionally, she had brief seizures treatment consisted in carbamzepine, oral baclophen, cefixime as prophylaxis for urinary tract infections, and rehabilitation physiotherapy.
The study was approved by the Bambino Gesù Children's Hospital Ethics Committee, and informed consent was obtained from the patient's parents.
MATERIALS AND METHODS
Culturing, harvesting of peripheral blood cells, and chromosome banding were performed according to standard methods. GTG-banded chromosomes were studied and the karyotype was described consistent with the International System for Human Cytogenetic Nomenclature (6).
Fluorescence in situ hybridization (FISH) was carried out as previously described (7) using whole chromosome painting probes for the chromosomes X and 16 (Appligene Oncor, Downers Grove, IL). To characterize the rearrangement, bacterial artificial chromosome (BAC) clones RP11-666O2 (AC020765; 16p11.2), and RP11-101E7 (AC009019; 16p12.1) and P1 artificial chromosome clone RP3-465G10 (AL645821; Xq21.1, containing ATP7A gene) were used as probes in FISH analysis. They were selected according to the NCBI database (www.ncbi.nlm.nih.gov, Build 35.1). The clones were obtained from the Roswell Park Cancer Institute library (http://www.chori.org/bacpac/). BAC and P1 artificial chromosome probes were directly labeled with Texas-Red-dUTP and fluorescein-dUTP (PerkinElmer Lifesciences, Boston, MA). Further FISH experiments were carried out using X inactive-specific transcript (XIST) site probe (Appligene Oncor) Digital images were obtained using a Nikon Eclipse E1000 epifluorescence microscope equipped with a cooled CCD camera Photometrics CoolSNAP FX. Pseudo coloring and merging of images were performed with Genikon software v3.6.16.
To evaluate the chromosome X late-replicating further experiments were performed using BrdU-Hoechst-Giemsa method. The thymidine analog, 2′deoxycytidine hydrochloride and 5-Bromo-2′-deoxyuridine (BrdU), is incorporated into chromosomes during the S phase (DNA synthesis) of the cell cycle. If the BrdU have added when the S phase is ending and all DNA regions has been synthesized except late replicating regions, only those that late replicate (e.g. inactive X) will incorporate BrdU. The procedure, having thymidine present for the beginning of the cell cycle and employing a terminal pulse of BrdU, produces pale late-replicating regions and well-colored early replicating regions when stained with Hoechst followed by Giemsa and observed at bright-field microscope. Blood cells were cultured after standard techniques, and 5 h before harvesting, BrdU was added to the culture medium. BrdU was incorporated in late-replicating DNA and was revealed in fixed chromosomes on slide with Hoechst after ultraviolet rays exposure and Giemsa staining (8). A total of 50 nuclei were recorded.
The fragile X mental retardation 1 gene (FMR1) methylation status analysis of the proband and her parents was carried out using the kit specific for Fragile X syndrome with primers for FMR1 gene (Abbott, Des Plaines, IL) following the manufacturer's instruction and with minor modifications. Shortly, DNA was extracted from peripheral blood according to phenol-chloroform standard method and digested with restriction enzymes sensitive to methylation (HpaII and HhaI).
Digested and undigested DNA were used as template for polymerase chain reaction (PCR) with Fragile-X kit by means of GeneAmp PCR System 2700 (Applied Biosystems, Foster City, CA). PCR products were run on ABI Prism 310 Genetic Analyzer (Applied Biosystems), using GeneMapper v3.0 as software.
RESULTS
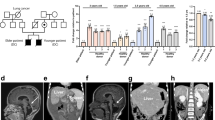
G-banding analysis of metaphase spreads from peripheral blood cultures revealed the following karyotype: 46,X,t(X;16)(q13.3;p11.2) (Fig. 2A). Parental chromosomes were normal, indicating a de novo origin of the rearrangement.

Karyotype of the patient showing chromosomes stained with (A) G-banding and (B) BrdU-Hoechst-Giemsa method.
FISH experiments using the whole chromosome painting probes for the chromosomes X and 16 confirmed the translocation between these chromosomes (Fig. 3A and B). The clone RP3-465G10 showed one signal on the normal chromosome X and one splitted signal on der(X) and der(16) as expected because it contains ATP7A gene, that is responsible for MD (Fig. 3C). Regarding the BACs RP11-101E7 and RP11-666O2 localized on the chromosome 16, the former displayed two signals on the chromosomes 16 and der(X) whereas the latter showed three spots on the normal chromosome 16, der(16) and der(X), indicating exactly the breakpoint location in 16p11.2 (Fig. 3C). Hybridization with XIST probe showed signal on both the normal X chromosome and the derivative X, excluding the involvement of XIST gene in nonrandom inactivation of X chromosome.

Results of some FISH experiments. (A) WCP probe (red) specific for chromosome X painted entirely the chromosomes X and partially the der(X) and der(16). (B) WCP probe (green) specific for chromosome 16 displayed the chromosome 16 fully painted and the der(16) and der(X) partially covered. (C) The clone RP3-465G10 (Xq21.1, red), containing the gene ATP7A, was cohybridized with the clone RP11-666O2 (16p11.2, green) both displaying splitting signals, respectively, on the chromosomes X, der(X) and der(16) and on the chromosomes 16, der(16) and der(X) revealing where the breakpoints of the reciprocal translocation were located.
Further investigations carried out by means of BrdU-Hoechst-Giemsa method revealed that the normal chromosome X was inactive in all the 50 examined cells, whereas der(X)t(X;16) was always active (Fig. 2B).
Molecular analysis was performed to evaluate the methylation status of the polymorphic CGG repeat of the FMR1 gene both in the patient and in her parents to disclose the parental origin of the rearrangement. It is well known that in females the chromosome X maintains the inactivation status through methylation, thus, by use of a restriction enzyme cutting not-methylated CpG islands, only active chromosome X will be digested. Because FMR1 gene contains CpG islands, after digestion, only the methylated chromosome X, which it is not digested, will be amplified. All the cells of the patient showed an electropherogram pattern corresponding only to the inactivated X chromosome. After scoring the number of CGG triplet in the patient and comparing it with those of her parents, it was possible to assign the parental origin of the rearrangement to the paternal allele (Table 3).
DISCUSSION
Females who are carrier of a recessive X-linked mutation may exhibit a disease phenotype if, during embryogenesis, X chromosome bearing a normal allele is inactivated in the majority of the cell clones. In the case of balanced X-autosome translocations arising in females, selective inactivation of the entire X chromosome often occurs. This phenomenon of lyonization can be the consequence of cell death of the clones inactivating the derivative X-autosome chromosome. In fact, cells carrying derivative inactive X-autosome chromosome would have partial monosomy of the autosome involved and partial disomy of the chromosome X, with gene unbalance. The efficiency of this process depends on the position of the X breakpoint and the size of the noninactivated region and of the autosome genomic segment (9). In the case of breakage of a recessive gene located on the chromosome X, due to a translocation X-autosome, the prevalence of the clones carrying the normal inactive X does not allow the expression of the normal allele of that gene, producing the disease phenotype in a female who is usually not affected by X-linked diseases (10).
This is exactly what happened in our patient, who is, to the best of our knowledge, the fifth female case of MD carrying a balanced X-autosome translocation disclosed at the chromosome level. The gene ATP7A, responsible for MD, is interrupted in the active chromosome der(X)t(X;16) where it is located.
In the genomic region, where the breakpoint of the chromosome 16 was mapped, seven known genes (CLN3, APOB48R, IL27, NUPR1, CCDC101, SULT1A2, SULT1A1) and two hypothetical proteins (LOC440350, LOC440350) were located. CLN3 (ceroid-lipofuscinosis neuronal 3) encodes a protein that is involved in lysosomal function and mutations in this gene cause neurodegenerative diseases; the protein codified by APOB48R (apolipoprotein B48 receptor) is a macrophage receptor binding to the apolipoprotein B48; IL27 (IL-27) produces one of the subunits of a heterodimeric cytokine complex; NUPR1 (nuclear p8 protein isoform a) encodes a protein that promotes cellular growth; CCDC101 (coiled-coil domain containing 101) produces a regulator of transcription; SULT1A2 and SULT1A1 (sulfotransferase family, cytosolic, 2A and 1A) encode sulfotransferase enzymes. It seems that these genes are not involved in generating the phenotype of our patient.
Interestingly, blood lactate was elevated and urine organic acid analysis showed increased excretion of lactate, and Krebs cycle intermediates in our patient. We have already observed this pattern in other male patients with MD (11), together with a deficiency in the muscle tissue of the enzyme activity of cytochrome c oxidase, a copper-dependent enzyme. These abnormalities can be explained with a mitochondrial dysfunction due to reduce mitochondrial copper concentrations in mitochondria, as has been observed in fibroblasts of patients (12).
After the literature, eight cases of MD-affected females were described (13), but only in five cases abnormal karyotypes were reported: one case displayed a mosaic karyotype and four cases showed a balanced de novo translocation X-autosome (3,13–16) (Table 1).
Studies of late-replicating X were performed only in two cases (3,13) and revealed that normal chromosome X was inactive, respectively, in 16/20 and in 50/50 metaphases.
Females affected by MD are very rare and they often present with milder clinical manifestation than males probably because XIST gene does not produce a complete inactivation of the X chromosome where it is expressed generating a cellular mosaicism. On the contrary, our patient showed a severe phenotype because of inactivation of the normal X chromosome in all the cells, and this was unequivocally shown by X late-replicating experiments and molecular investigations of the methylation status of the polymorphic CGG triplet in the FMR1 gene.
The majority of abnormalities are likely to occur during meiosis, when the chromosomes are breaking and rejoining in the process of recombination. The amount and pattern of recombination differ markedly in the male and female gametogenesis and it is not unreasonable to assume that this might influence the parental origin of structural rearrangements. In fact, spermatozoa display a higher frequency of structural rearrangements than oocytes, mainly because of the large sex difference in the number of premeiotic germ cell divisions (17). Meiotic recombination between mispaired chromosomes is a common cause of translocations, especially in spermatogenesis (18). Conversely, oocytes in women over 35-y, show a reduced number of crossing-over between sister chromatids delivering an abnormal segregation and consequent numerical abnormalities. Thus, structural aberrations may have a preferential paternal origin and numerical abnormalities a maternal origin (19,20).
Recently, some authors have addressed the paternal origin of de novo structural chromosome rearrangements (20–25); along these lines, paternal meiosis seems to be more prone to chromosomal exchange, as happened in our case.
MD, being an X-linked recessive syndrome, is rare in females and, in this context, X chromosome inactivation plays a critical role in the expression of the clinical phenotype. In our case, we reported an extensive cyto-molecular characterization of the X-autosome translocation in association with a thorough study of the X-inactivation. Moreover, we provide a simple and reliable method to study the parental origin of the causative cytogenetic rearrangement, which is useful to better clarify the molecular mechanisms underlying X-linked disease.
Abbreviations
- ATP7A:
-
ATPase Cu++ transporting alpha polypeptide
- BAC:
-
bacterial artificial chromosome
- BrdU:
-
5-Bromo-2′-deoxyuridine
- FMR1:
-
fragile X mental retardation 1
- MD:
-
Menkes disease
- WCP:
-
whole chromosome painting probe
- XIST:
-
X inactive-specific transcript
References
Menkes JH, Alter M, Steigleder GK, Weakley DR, Sung JH 1962 A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration. Pediatrics 29: 764–779
Tønnesen T, Kleijer WJ, Horn N 1991 Incidence of Menkes disease. Hum Genet 86: 408–410
Sugio Y, Sugio Y, Kuwano A, Miyoshi O, Yamada K, Niikawa N, Tsukahara M 1998 Translocation t(X;21)(q13.3;p11.1) in a girl with Menkes disease. Am J Med Genet 79: 191–194
Kaler SG 1998 Diagnosis and therapy of Menkes syndrome, a genetic form of copper deficiency. Am J Clin Nutr 67: 1029S–1034S
Harris ED 2000 Cellular copper transport and metabolism. Annu Rev Nutr 20: 291–310
Mitelman F 1995 An International System for Human Cytogenetic Nomenclature. Karger, Basel
Lichter P, Tang Chang CJ, Call K, Hermanson G, Evans GA, Housman D, Ward DC 1990 High resolution mapping of human chromosomes 11 by in situ hybridization with cosmid clones. Science 247: 64–69
Tsukahara M, Kajii T 1985 Replication of X chromosome in complete moles. Hum Genet 71: 7–10
Schmidt M, Du Sart D 1992 Functional disomies of the X chromosome influence the cell selection and hence the X inactivation pattern in females with balanced X-autosome translocations: a review of 122 cases. Am J Med Genet 42: 161–169
Migeon BR 1994 X-chromosome inactivation: molecular mechanisms and genetic consequences. Trends Genet 10: 230–235
Rizzo C, Bertini E, Piemonte F, Leuzzi V, Sabetta G, Federici G, Luchetti A, Dionisi-Vici C 2000 Oxidative abnormalities in Menkes disease. J Inherit Metab Dis 23: 349–351
Kodama H, Okabe I, Yanagisawa M, Kodama Y 1989 Copper deficiency in the mitochondria of cultured skin fibroblasts from patients with Menkes syndrome. J Inherit Metab Dis 12: 386–389
Abusaad I, Mohammed SN, Ogilvie CM, Ritchie J, Pohl KR, Docherty Z 1999 Clinical expression of Menkes disease in a girl with X;13 translocation. Am J Med Genet 87: 354–359
Kapur S, Higgins JV, Delp K, Rogers B 1987 Menkes syndrome in a girl with X-autosome translocation. Am J Med Genet 26: 503–510
Beck J, Enders H, Schliephacke M, Buchwald-Saal M, Tümer Z 1994 X;1 translocation in a female Menkes patient: characterization by fluorescence in situ hybridization. Clin Genet 46: 295–298
Gerdes AM, Tønnesen T, Horn N, Grisar T, Marg W, Müller A, Reinsch R, Barton NW, Guiraud P, Joannard A, Richard MJ, Guttler F 1990 Clinical expression of Menkes syndrome in females. Clin Genet 38: 452–459
Chandley AC 1991 On the parental origin of the novo mutation in man. J Med Genet 28: 217–223
Strachan T, Read AP 1999 Genetica Umana Molecolare 2. Wiley, New York, pp 52–55
Hunt PA, Hassold TJ 2002 Sex matters in meiosis. Science 296: 2181–2183
Thomas NS, Durkie M, Van Zyl B, Sanford R, Potts G, Youings S, Dennis N, Jacobs P 2006 Parental and chromosomal origin of unbalanced de novo structural chromosome abnormalities in man. Hum Genet 119: 444–450
De Gregori M, Ciccone R, Magini P, Pramparo T, Gimelli S, Messa J, Novara F, Vetro A, Rossi E, Maraschio P, Bonaglia MC, Anichini C, Ferrero GB, Silengo M, Fazzi E, Zatterale A, Fischetto R, Previdere C, Belli S, Turci A, Calabrese G, Bernardi F, Meneghelli E, Riegel M, Rocchi M, Guerneri S, Lalatta F, Zelante L, Romano C, Fichera M, Mattina T, Arrigo G, Zollino M, Giglio S, Lonardo F, Bonfante A, Ferlini A, Cifuentes F, Van Esch H, Backx L, Schinzel A, Vermeesch JR, Zuffardi O 2007 Cryptic deletions are a common finding in ‘‘balanced” reciprocal and complex chromosome rearrangements: a study of 59 patients. J Med Genet 44: 750–762
Ashley T, Gaeth AP, Inagaki H, Seftel A, Cohen MM, Anderson LK, Kurahashi H, Emanuel BS 2006 Meiotic recombination and spatial proximity in the etiology of the recurrent t(11;22). Am J Hum Genet 79: 524–538
Gribble SM, Prigmore E, Burford DC, Porter KM, Ng BL, Douglas EJ, Fiegler H, Carr P, Kalaitzopoulos D, Clegg S, Sandstrom R, Temple IK, Youings SA, Thomas NS, Dennis NR, Jacobs PA, Crolla JA, Carter NP 2005 The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Genet 42: 8–16
Ciccone R, Giorda R, Gregato G, Guerrini R, Giglio S, Carrozzo R, Bonaglia MC, Priolo E, Lagana C, Tenconi R, Rocchi M, Pramparo T, Zuffardi O, Rossi E 2005 Reciprocal translocations: a trap for cytogenetists?. Hum Genet 117: 571–582
Miyake N, Kurotaki N, Sugawara H, Shimokawa O, Harada N, Kondoh T, Tsukahara M, Ishikiriyama S, Sonoda T, Miyoshi Y, Sakazume S, Fukushima Y, Ohashi H, Nagai T, Kawame H, Kurosawa K, Touyama M, Shiihara T, Okamoto N, Nishimoto J, Yoshiura K, Ohta T, Kishino T, Niikawa N, Matsumoto N 2003 Preferential paternal origin of microdeletions caused by prezygotic chromosome or chromatid rearrangements in Sotos syndrome. Am J Hum Genet 72: 1331–1337
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sirleto, P., Surace, C., Santos, H. et al. Lyonization Effects of the t(X;16) Translocation on the Phenotypic Expression in a Rare Female With Menkes Disease. Pediatr Res 65, 347–351 (2009). https://doi.org/10.1203/PDR.0b013e3181973b4e
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/PDR.0b013e3181973b4e
