
Abstract
Among the mitogen-activated protein kinase (MAPK) family members, extracellular signal–regulated kinase (ERK) promotes cell proliferation or differentiation, whereas c-jun N terminal kinase (JNK) and p38 MAPK are thought to inhibit cell growth and induce apoptosis. The MAPK family may plays some role during kidney development, when large-scale proliferation and apoptosis have been observed to occur. Also, in this period, the renin-angiotensin system is markedly activated. We have demonstrated that angiotensin-converting enzyme inhibition in the developing rat kidney increases apoptosis and decreases cell proliferation, which may account for renal growth impairment. The aim of this study, therefore, was to examine the relationship between the MAPK family and renin-angiotensin system during neonatal renal development. Newborn rat pups were treated with enalapril (30 mg · kg−1 · d−1) or normal saline for 7 d. Right kidneys of both groups were selected for immunohistochemical stains of MAPKs and activating transcription factor-2 (ATF-2), and left kidneys were selected for reverse transcriptase–PCR and immunoblot analysis of MAPKs, phospho-MAPKs, and ATF-2. To determine whether apoptosis is involved in the same tubules that highly expressed JNK and p38, we performed terminal deoxynucleotide transferase-mediated nick-end labeling stain for apoptotic cells and immunohistochemical stains for JNK-2, p38, and ATF-2 expression in the serial sections from the same kidney of the enalapril-treated group. In the enalapril-treated group, JNK-2, p38, phospho-JNK-2, phospho-p38, and ATF-2 protein expressions were significantly increased, and their immunoactivities were strongly detected in the proximal tubular epithelial cells in the cortex, compared with the control group. Especially JNK-2 and p38 expressions were highly activated and were spatially in accordance with the occurrence of apoptosis. ERK1/2 and phospho-ERK expressions were not changed by enalapril. These results suggest that the expressions of the MAPK family are modulated by angiotensin-converting enzyme inhibition in the developing kidney. JNK and p38 may be implicated to participate in angiotensin II–related intracellular signaling pathways of renal apoptosis in the developing kidney.
Similar content being viewed by others

Main
The renin-angiotensin system (RAS), which plays an important role in the complex affair of renal organogenesis, is up-regulated during renal development and in the perinatal period (1–4). Among them, angiotensin II (Ang II) has received considerable attention because of its relevant hemodynamic and growth-promoting effects in the cardiovascular system and kidney.
Especially in the developing kidney, RAS is markedly activated and is thought to play an important role in postnatal renal growth and maturation. In our previous studies, we demonstrated that Ang II modulates growth in a variety of tissues, including kidney and heart (5–7). Ang II inhibition by angiotensin-converting enzyme (ACE) inhibitor in the developing rat kidney increases apoptosis and decreases its related genes (Bcl-2 and clusterin) expression (7,8). Also, Ang II inhibition decreases cell proliferation and decreases its related growth factors (transforming growth factor-β and epidermal growth factor) expression, which may account for neonatal renal growth impairment (5,9).
The signal transduction pathways that modulate Ang II–related growth effects are not clearly defined yet. It has been suggested that the mitogenic effects of Ang II could be mediated by mitogen-activated protein kinases (MAPKs), which are rapidly activated upon stimulation of a variety of cell surface receptors (10,11). They function by transmitting signals from the cell surface to the nucleus to regulate the expression of transcription factors such as c-jun and c-Myc and modulate the process of cell growth and differentiation (12,13).
The MAPK family is serine/threonine kinases and can be divided into at least three broad families on the basis of sequence similarity; upstream activators; and, to a lesser extent, substrate specificity, namely extracellular signal–regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 MAPK (p38). The classic ERKs (ERK1 and ERK2), which were identified in the context of growth factor–related signaling and activated by various growth factors, promote cellular proliferation or differentiation, but JNK and p38 families, which were described in the setting of cell response to stress and inflammation and activated by cytokines and cellular stresses, are thought to inhibit cell growth and induce apoptosis (14–17). The MAPK family participates in a variety of metabolic pathways covering cellular growth, differentiation, survival, and cell death, so the MAPK family is suggested to play a role during normal kidney development.
All three MAPKs are activated by phosphorylation on both tyrosine and threonine residues and inactivated by the dephosphorylation of either residue. MAPKs are phosphorylated by MAPK kinases, and their phosphorylation state is determined by phosphatases as well as kinases. Therefore, the activities of MAPK families are evaluated by assessing the phosphorylated MAPKs.
MAPKs promote changes in the pattern of gene expression as they activate transcription factors. After activation, JNK phosphorylates the transcription factor c-jun and Elk-1 and activating transcription factor-2 (ATF-2), c-jun, and Elk-1 in their transactivating domains (18,19). p38 also phosphorylates and activates ATF-2 (20). Therefore, ATF-2 has been postulated as a effector of JNK as well as p38 (19–24).
The intracellular molecular mechanism in controlling renal growth and development by RAS is poorly understood. To determine the signaling molecular mechanism of Ang II–related renal growth and development, we designed the present study to investigate whether MAPKs, which are thought to be involved in proliferation and apoptosis in renal growth and development, serve as signaling intermediates in renal growth impairment by ACE inhibition. For this purpose, we first examined the changes of the expressions of MAPKs and phosphorylated MAPKs in neonatal kidney development by chronic administration of enalapril, an ACE inhibitor, to neonatal rat pups. To evaluate the functional role of JNK and p38 activation, we determined their ATF-2 expression. Finally, to determine whether apoptosis is involved in the same tubules that highly express JNK and p38, we performed terminal deoxynucleotide transferase-mediated nick-end labeling (TUNEL) stain for apoptotic cells and immunohistochemical stains for JNK-2, p38, and ATF-2 expressions in the serial sections from the same kidney of the enalapril-treated group.
METHODS
Animal preparation.
Forty-nine neonatal rat pups from five pregnant Sprague Dawley rats were breastfed by their own mother throughout the study. Body weights were measured daily from birth, and a dose of 30 mg/kg of enalapril (Sigma Chemical Co., St. Louis, MO; the enalapril-treated group, n = 23) and normal saline (the control group, n = 26) were fed via an orogastric tube. A dose of 30 mg/kg of enalapril is known to block the effects on Ang II (25). After 7 d, their kidneys were harvested and RNA analysis, protein assays, and immune histochemistry were performed. The experimental protocol was approved by the Animal Care Committee of Korea University Guro Hospital.
Immunohistochemical stain.
For assessing the expression of MAPK family, five right kidneys of the control group and the enalapril-treated group, respectively, were selected for representative immunohistochemical stains of JNK, p38, ERK, and ATF-2. Harvested kidneys were treated in 10% formalin solution (Sigma Chemical Co.) and embedded in paraffin. The samples were then cut into 4-μm sections and dried onto silicanized slides (Sigma Chemical Co.). Slides were deparaffinized and digested for 10 min with proteinase K (20 μg/mL), and an endogenous peroxidase activity was quenched in 2% hydrogen peroxide for 5 min. Sections were incubated with primary antibodies against JNK-2 (Santa Cruz Biotechnology, Santa Cruz, CA; dilution 1:50), ERK1/2 (Upstate Biotechnology, Lake Placid, NY; dilution 1:100), p38 (Santa Cruz Biotechnology; dilution 1:50), and ATF-2 (Santa Cruz Biotechnology; dilution 1:100). As negative controls, the primary antibody was substituted with PBS. The incubation time was 120 min at room temperature or overnight at 4°C. After incubating with secondary antibody at a concentration of 1:200, immunoreaction products were developed using 3,3-diaminobenzidine as the chromogen, with standardized development times. Samples were rinsed in PBS for 2 min. The sections were rinsed in water and counterstained in 0.5% methyl green solution for 5 min, and the samples were dehydrated and mounted. After dehydration, we compared them using a light microscope.
To identify the structural and functional cell type of the immunoactivated tubules in the enalapril-treated group, we performed a immunoperoxidase labeling procedure using collecting duct–specific biotinylated Dolichos biflorus agglutinin (DBA; 5 μg/mL; Vector Laboratories, Burlingame, CA). We used DBA lectin because binding sites are expressed throughout ontogeny in the ureteric bud and its derivatives (collecting ducts) but not expressed in other nonureteric bud–derived tubules (26–28).
Isolation of RNA and analysis of mRNA.
Five left kidneys of the control group and the enalapril-treated group, respectively, were selected for RNA analysis and protein assays. After being removed, the renal tissue was frozen in liquid nitrogen and stored at −70°C. Total cellular RNA was isolated using TRI-REAGENT (Molecular Research Center, Cincinnati, OH) and homogenized with tissue tearer (Model 985-370; Biospec products, Bartlesville, OK). Chloroform (37%; 200 μL/1 mL TRI reagent) was added to the homogenates and centrifuged at 12,000 rpm for 15 min in 4°C, separating into three layers: RNA, DNA, and protein material. The colorless clear upper layer was moved into another Eppendorf tube, and isopropanol was added. This was left at room temperature for 15 min and was then centrifuged at 12,000 rpm for 10 min in 4°C until the white-colored cellular RNA pellet was isolated. It was dried at room temperature for 5 min after washing in 75% ethanol and was dissolved in 25 μL of Forma zol (Molecular Research Center) at 55°C heating block for 10 min and then stored at −70°C. The RNA was quantified spectrophotometrically by absorbance at 260 nm.
cDNA synthesis by reverse transcription and PCR.
Oligo dT primed first-strand cDNA was synthesized from the template RNA 1 μg through a cDNA Synthesis Kit (Boehringer Mannheim Corp., Indianapolis, IN). AMV reverse transcriptase (RT) was used for synthesis of first-strand cDNA for use in subsequent amplification reactions. The primers used in the PCR reaction are presented in Table 1 (29–33). The PCR was carried out in a different mode of time and temperature for each reaction period using Perkin Elmer Cetus DNA Thermal Cycler (Model 2400; Foster City, CA).
For glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a housekeeping gene, PCR was performed at 30 cycles of heating for 45 s at 94°C after denaturation for 5 min at 94°C, followed by annealing for 45 s at 60°C and last a primer extension for 45 s at 70°C. For JNK-2, PCR was performed at 28 cycles of heating for 45 s at 94°C after denaturation for 5 min at 94°C, followed by annealing for 40 s at 59°C and last a primer extension for 50 s at 72°C. For ERK1, this was performed at 26 cycles of heating for 50 s at 94°C after denaturation for 5 min at 94°C, followed by annealing for 40 s at 55°C and primer extension for 50 s at 72°C. For ERK2, this was performed at 30 cycles of heating for 50 s at 94°C after denaturation for 5 min at 94°C, followed by annealing for 40 s at 57°C and primer extension for 50 s at 72°C. For p38, this was performed at 30 cycles of heating for 40 s at 94°C after denaturation for 5 min at 94°C, followed by annealing for 50 s at 57°C and primer extension for 50 s at 72°C. These amplified PCR products were visible as a fluorescent band under UV light after electrophoresis in 2% agarose gel at regular intervals and ethidium bromide staining. Polaroid photographs were scanned using Epson GT-9500 (Seiko Corp, Nagano, Japan) and quantified by densitometer (Image PC alpha 9; National Institutes of Health, Bethesda, MD), and the values were revised by GAPDH.
Protein extraction.
Alcohol (100%) was added to interface, and organic phase remained from separation of RNA. Then the DNA portion was precipitated after being centrifuged at 5000 rpm for 5 min. Isopropanol was added to the remaining ethanol upper layer, which was centrifuged and dissolved in 0.3 M of guanidine hydrochloride in 95% ethanol. Afterward, this layer was washed three times and turned into a protein pellet. The extracted protein was dissolved in 1% SDS solution and preserved at −20°C. The Bradford method was used in the quantification of protein.
Immunoblot analysis.
The extracted proteins were solubilized in 5× SDS loading buffer for 5 min at 95°C and separated by electrophoresis on 10% SDS-polyacrylamide gels under reducing conditions. Equal amounts of 5–15 μg of proteins were loaded per lane. Subsequently the proteins were transferred to nitrocellulose membranes (Amersham Life Science, Buckinghamshire, England). The nitrocellulose membranes were blocked in 5% skim milk with TBS-T [0.05% Tween 20 in 50 mM of Tris, 150 mM of NaCl, and 0.05% NaN3 (pH 7.4)] at room temperature for 1 h. The membranes were washed two times in TBS-T and incubated for 18 h at 4°C with rabbit polyclonal antiserum (JNK-2, p38, ERK1/2, and ATF-2) and mouse monoclonal antiserum (phospho-JNK-2, phospho-p38, and phospho-ERK1/2) in TBS-T. Thereafter, the membranes were washed two times with TBS-T and incubated for 40 min with an anti-rabbit IgG (Amersham Life Science) and an anti-mouse IgG (Amersham Life Science) at room temperature. Characteristics of antibodies directed against MAPKs and their phosphorylated MAPKs and ATF-2 are listed in Table 2. After washing by TBS-T four times, the secondary antibody bound to the nitrocellulose was detected by incubation for 1 min with a detection reagent (Amersham Life Science) and then exposed to medical x-ray film (Agfa, Mortsel, Belgium) for 1 min. The film was developed by a FPM-3500 Fuji x-ray Film Processor (Fuji, Otawara, Japan). After that, for stripping and reprobing, the same membranes were submerged in stripping buffer (2% SDS in 62.5 mM of Tris-HCL and 0.1 M of β-mercaptoethanol) and incubated at 50°C for 10 min with occasional shaking. To control equal loading, α-tubulin (1:1000 dilution; Oncogene, Boston, MA) and anti-mouse IgG conjugated horseradish peroxidase (1:1000 dilution; Amersham Life Science) were used as primary and secondary antibodies in the same method as described above. The developed x-rays were scanned using Epson GT-9500 (Seiko Corp, Nagano, Japan) and quantified by a densitometer (Image PC alpha 9).
Detection of apoptosis.
To determine whether apoptosis is involved in the same tubules that highly expressed JNK and p38, we performed TUNEL stains for apoptotic cells in the kidney of the enalapril-treated group. Apoptotic nuclei were labeled using the TACS TM 2 TdT In Situ Apoptosis Detection Kit (Trevigen, Gaithersburg, MD). Tissues were fixed in 4% neutral-buffered formalin for 4 h at 4°C, dehydrated in graded alcohols, and embedded in paraffin. The samples were then cut into 5-μm sections and dried onto silicanized slides (Sigma Chemical Co.). Slides were deparaffinized and digested for 10 min with proteinase K (20 μg/mL), and an endogenous peroxidase activity was quenched in 2% hydrogen peroxide for 5 min. Samples were equilibrated using a provided buffer for 2 min and labeled with biotin dNTP mixtures, TdT, CoCl2, and a labeling buffer according to the manufacturer's advice. Samples were placed inside a humidity chamber in a 37°C incubator for 60 min. Negative controls were treated with water instead of TdT. The labeling reaction was stopped using the provided stop buffer for 5 min. Samples were rinsed in PBS for 2 min and treated with the provided streptavidin-horseradish peroxidase detection solution for 10 min and finally washed two times in PBS for 2 min each wash. The color was developed using 0.05% 3,3-diaminobenzidine and 0.05% hydrogen peroxidase in PBS for 5 min. The sections were rinsed in water and counterstained in methyl green for 5 min, and the samples were dehydrated and mounted. After dehydration, we compared them using a light microscope. In addition to this, the immunohistochemical stains for JNK-2, p38, and ATF-2 expressions in the serial sections from the same kidney of the enalapril-treated group were performed and compared with the apoptotic changes spatially.
Statistical analysis.
Results are expressed as mean± SEM. Differences between groups were tested for significance by t test and Mann-Whitney rank sum test, and a value of p < 0.05 was considered statistically significant.
RESULTS
The mortality rate of the enalapril-treated group was increased significantly (30.4%, 7 of 23), compared with the control group (7.7%, 2 of 26; p < 0.05). On day 7, the body weight was 14.6 ± 0.5 g in the enalapril-treated group and 17.1 ± 0.5 g in the control group, and the kidney weight was 0.113 ± 0.004 g in the enalapril-treated group and 0.0871 ± 0.005 g in the control group. The body and kidney weights of the enalapril-treated group were significantly lower than those of the control group (p < 0.05).
Expression of JNK.
In immunohistochemical stain, JNK-2 was weakly expressed only at some glomeruli in the control group (Fig. 1B). In the enalapril-treated group, its expression was also similarly detected in some glomeruli. However, contrary to the control group, JNK-2 was highly expressed in the proximal tubular epithelial cells in the enalapril-treated group (Fig. 1C, renal cortex, and (Fig. 2B, renal corticomedullary junction). As a result of semiquantitative RT-PCR, JNK-2/GAPDH mRNA expression was 0.49 ± 0.04 in the control group and 0.48 ± 0.04 in the enalapril group (Fig. 3A). As a result of the immunoblot analysis, JNK-2/tubulin protein expression was increased significantly in the enalapril-treated group (0.99 ± 0.04) compared with the control group (0.75 ± 0.04; p < 0.05; (Fig. 3B). Also, phospho-JNK-2/tubulin protein expression was increased significantly in the enalapril-treated group (0.74 ± 0.07) compared with the control group (0.63 ± 0.08; p < 0.05; (Fig. 3C).

Immunohistochemical stains for JNK-2 (brown color, black arrows) in renal cortex of developing rat. (A) Negative control. (B) In the control group, JNK-2 was only weakly expressed in some glomeruli. In glomeruli, JNK-2 was expressed in some podocytes and mesangial cells. (C) In the enalapril-treated group, JNK-2 expression was similarly detected in some glomeruli and also mainly distributed in the proximal tubular epithelial cells compared with the control group. Especially, JNK-2 expression was highly detected at the luminal portion of the proximal tubular cells only in cytoplasm, not in nucleus. (D) In serial section from the same kidney of the enalapril-treated group, the collecting ducts (*) were detected by dark-brown staining with DBA lectin. G, glomerulus. Bar = 0.1 μm. Magnification: ×400.

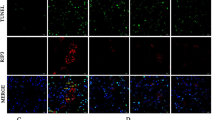
TUNEL stains for apoptotic cells and immunohistochemical stains for JNK-2, p38, and ATF-2 in the serial sections from the same kidney of the enalapril-treated group at the renal corticomedullary junction. (A) TUNEL-positive apoptotic cells were occasionally observed in the nondilated and dilated proximal tubular epithelial cells. (B) JNK expression was highly detected in the proximal tubular epithelial cells. (C) p38 expression was highly detected in the proximal tubular epithelial cells. (D) ATF-2 expression was highly detected in the proximal tubular epithelial cells. However, contrary to TUNEL-positive apoptotic cells, JNK, p38, and ATF-2 expressions were more abundant and distributed throughout the proximal tubular epithelial cells in the enalapril-treated group. G, glomerulus. Bar = 0.1 μm. Magnification: ×400.

Representative expressions of JNK-2 mRNA and protein and phospho-JNK-2 protein in the developing rat kidney. ▪, control group; □, enalapril-treated group. (A) In semiquantitative RT-PCR, JNK-2/GAPDH mRNA expression was not different between the two groups. (B) In immunoblot analysis, JNK-2/tubulin protein expression was increased significantly in the enalapril-treated group compared with the control group. (C) In immunoblot analysis, phospho-JNK-2/tubulin protein expression was increased significantly in the enalapril-treated group compared with the control group.
Expression of p38.
Immunohistochemically, p38 expression was not detectable in the kidney of the control group (Fig. 4B). However, contrary to the control group, p38 was highly expressed in the proximal tubular epithelial cells in the enalapril-treated group (Fig. 4C, renal cortex, and Fig. 2C, renal corticomedullary junction). As a result of semiquantitative RT-PCR, p38/GAPDH mRNA expression was 0.82 ± 0.03 in the control group and 0.63 ± 0.09 in the enalapril group (Fig. 5A). As a result of the immunoblot analysis, p38/tubulin protein expression was increased significantly in the enalapril-treated group (1.63 ± 0.15) compared with the control group (1.22 ± 0.21; p < 0.05; Fig. 5B). Also, phospho-p38/tubulin protein expression was increased significantly in the enalapril-treated group (0.82 ± 0.09) compared with the control group (0.58 ± 0.09; p < 0.05; Fig. 5C).

Immunohistochemical stains for p38 (brown color, black arrows) in renal cortex of the developing rat. (A) Positive control of rat kidney 1 d after birth. p38 expression was distributed in some glomeruli and tubules in varying expression. (B) In the control group, 8 d after birth, p38 was not expressed in the kidney. (C) In the enalapril-treated group, 8 d after birth, p38 was highly expressed in the proximal tubular epithelial cells compared with the control group. Especially, p38 expression was highly detected at the luminal portion of the proximal tubular cells only in cytoplasm, not in nucleus. (D) In a serial section from the same kidney of the enalapril-treated group, the collecting ducts (*) were detected by dark-brown staining with DBA lectin. G, glomerulus. Bar = 0.1 μm. Magnification: ×400.

Representative expressions of p38 mRNA and protein and phospho-p38 protein in the developing rat kidney. ▪, control group; □, enalapril-treated group. (A) In semiquantitative RT-PCR, p38/GAPDH mRNA expression was not different between the two groups. (B) In immunoblot analysis, p38/tubulin protein expression was increased significantly in the enalapril-treated group compared with the control group. (C) In immunoblot analysis, phospho-p38/tubulin protein expression was increased significantly in the enalapril-treated group compared with the control group.
Expression of ERK.
In immunohistochemical stain, ERK was weakly expressed at some glomeruli and mainly expressed in distal tubular epithelial cells in the control group (Fig. 6A). In the enalapril-treated group, ERK expression was not changed and so was detected only in distal tubular epithelial cells (Fig. 6B). As a result of semiquantitative RT-PCR, ERK-1/GAPDH mRNA expression was 0.58 ± 0.01 in the control group and 0.56 ± 0.02 in the enalapril-treated group, and ERK-2/GAPDH mRNA expression was 0.76 ± 0.02 in the control group and 0.60 ± 0.07 in the enalapril-treated group (Fig. 7A). As a result of the immunoblot analysis, ERK-1/tubulin protein was 1.45 ± 0.06 in the control group and 1.62 ± 0.11 in the enalapril-treated group, and ERK-2/tubulin protein expression was 1.43 ± 0.06 in the control group and 1.58 ± 0.09 in the enalapril-treated group (Fig. 7B). Phospho-ERK-1/tubulin protein was 0.58 ± 0.06 in the control group and 0.55 ± 0.11 in the enalapril-treated group, and phospho-ERK-2/tubulin protein expression was 0.74 ± 0.13 in the control group and 0.67 ± 0.10 in the enalapril-treated group (Fig. 7C).

Immunohistochemical stains for ERK (brown color, black arrows) in renal cortex of developing rat. (A) In the control group, ERK was mainly expressed in distal tubular epithelial cells in the control group. (B) In the enalapril-treated group, ERK expression was not changed and so was detected only in distal tubular epithelial cells. (C) In a serial section from the same kidney of the enalapril-treated group, the collecting ducts (*) were detected by dark-brown staining with DBA. G, glomerulus. Bar = 0.1 μm. Magnification: ×400.

Representative expressions of ERK-1/2 mRNAs, ERK-1/2 proteins, and phospho-ERK-1/2 proteins in the developing rat kidney. ▪, ERK-1 in the control group; □, ERK-1 in the enalapril-treated group; ▪, ERK-2 in the control group; ▪, ERK-2 in the enalapril-treated group. (A) In semiquantitative RT-PCR, ERK-1/GAPDH and ERK-2/GAPDH mRNA expressions were not different between the two groups. (B) In immunoblot analysis, ERK-1/tubulin and ERK-2/tubulin protein expressions were not different between the two groups. (C) In immunoblot analysis, phospho-ERK-1/tubulin and phospho-ERK-2/tubulin protein expressions were not different between the two groups.
Expression of ATF-2.
In immunohistochemical stain, ATF-2 was expressed only in the renal vessels of the control group (Fig. 8A). In the enalapril-treated group, its expression was also similarly detected in the renal vessels. However, contrary to the control group, ATF-2 was highly expressed in the proximal tubular epithelial cells in the enalapril-treated group (Fig. 8B, renal cortex, and Fig. 2D, renal corticomedullary junction). As a result of the immunoblot analysis, ATF-2/tubulin protein expression was increased significantly in the enalapril-treated group (0.67 ± 0.07) compared with the control group (0.56 ± 0.08; p < 0.05; (Fig. 9).

Immunohistochemical stains for ATF-2 (brown color, black arrows) in renal cortex of the developing rat. (A) In the control group, ATF-2 was expressed only in the renal vessels. (B) In the enalapril-treated group, ATF-2 expression was also similarly detected in the renal vessels and highly detected in the proximal tubular epithelial cells compared with the control group. (C) In serial section from the same kidney of the enalapril-treated group, the collecting ducts (*) were detected by dark-brown staining with DBA lectin. G, glomerulus. Bar = 0.1 μm. Magnification: ×400.

Representative expressions of ATF-2 protein in the developing rat kidney. ▪, control group; □, enalapril-treated group. In immunoblot analysis, ATF-2/tubulin protein expression was increased significantly in the enalapril-treated group compared with the control group.
Spatial relationship of TUNEL-positive apoptotic cells and expression of JNK-2, p38, and ATF-2. In the enalapril-treated group, TUNEL-positive apoptotic cells were occasionally observed in the nondilated and dilated proximal tubular epithelial cells in renal cortex (Fig. 2A). JNK, p38, and ATF-2 expressions were highly detected in the proximal tubular epithelial cells also. However, contrary to TUNEL-positive apoptotic cells, JNK, p38, and ATF-2 expressions were more abundant and distributed throughout all proximal tubular epithelial cells in the renal cortex and were not restricted to TUNEL-positive apoptotic cells (Fig. 2B–D).
DISCUSSION
The octapeptide Ang II, the central product of the RAS, is well known to cause potent increases in systemic and local blood pressure via its vasoconstrictive effect, to influence renal tubules to retain sodium and water, and to stimulate aldosterone release from the adrenal gland. Ang II also acts as a renal growth factor that modulates cell growth and extracellular matrix synthesis and degradation (34–37) and is thought to be important in renal development, growth, and maturation. Determining the mechanism by which Ang II modulates cell growth in developing kidney is fundamental to our understanding of physiologic renal growth and development. One of the major signal transduction pathways that link activation of the Ang II receptor to the nuclear events necessary for cell growth is the MAPK cascade (38).
The present study using in vivo animal study demonstrates that MAPK families may play a role in renal growth impairment by ACE inhibition. Especially, JNK-2 protein expression was increased significantly in the enalapril-treated group. The JNK family, which is also known as stress-activated protein kinase, is generally responsive to cell stressors such as hypertonicity, UV light, heat shock, and proinflammatory cytokines. Members of this family include the widely expressed JNK-1 (p46) and JNK-2 (p54), as well as the brain-specific JNK-3 (p49). Activation of the JNK cascade, which is dependent in part on renal growth impairment, is considered as an important intermediate of cell apoptosis. Recent study in rat renal development by Omori et al. (39) demonstrated that JNK was not detectable in the fetal kidney, became detectable at 7 d after birth, and was most abundant in the adult kidney. Furthermore, JNK was confined to tubular cells, collecting duct cells, and glomerular epithelial cells in adult kidney. These observations suggest that JNK may participate in the renal growth and differentiation at a late stage of kidney development. In addition, it is tempting to speculate that JNK may be exerting a growth inhibitory action.
We previously described that Ang II inhibition by ACE inhibitor in the developing rat kidney increases apoptosis, which may account for neonatal renal growth impairment (7,8). We show here that increased renal apoptosis as a result of ACE inhibition may be associated with strong JNK activation. JNK-2 protein and phospho-JNK-2 protein expressions were significantly increased by ACE inhibition, and especially in immunohistochemical stain, JNK-2 expression was strongly increased in the cytoplasm of the proximal tubular epithelial cells. The immunolocalization of JNK in the proximal tubular epithelial cells in the enalapril-treated group correlated with the distribution of increased apoptotic cells by ACE inhibition in our previous developing rat kidney study. These observations show that JNK may be implicated to participate in Ang II–related intracellular signaling pathways of renal apoptosis in the developing kidney.
Another member of the MAPK family, p38 is also activated by cytokines and cellular stresses and is thought to inhibit cell growth and induce apoptosis. Although p38 has been implicated in apoptosis, some reports suggested that its expression and activation correlated with proliferation rather than apoptosis in rat embryonic and developing kidney (39,40). They also suggested that p38 in the developing kidney may have a role in differentiation of renal cells. Contrary to JNK, p38 was predominantly expressed in the developing kidney with the highest levels in the embryo and became undetectable after birth and was not detectable in the adult kidney. In the present study, we found that p38 protein and phospho-p38 protein expressions were significantly increased by enalapril. Also in immunohistochemical stain, by ACE inhibition, p38 expression appeared strongly in the proximal tubular epithelial cells that were correlated with the distribution of increased apoptotic cells by ACE inhibition. Thus, p38 may participate in Ang II–related intracellular signaling pathways of renal apoptosis in the developing kidney.
In our previous studies (7,8), TUNEL-positive apoptotic cells were occasionally observed in the dilated cortical tubules, and their actual numbers are counted 1.27 ± 0.07 on 250 × 250-μm area. To determine whether apoptosis is involved in the same tubules that highly express JNK and p38, we performed TUNEL stains for apoptotic cells and immunohistochemical stains for JNK-2, p38, and ATF-2 expressions in serial sections from the same kidney of the enalapril-treated group. In the enalapril-treated group, TUNEL-positive apoptotic cells were occasionally observed in some proximal tubular epithelial cells in renal cortex, but JNK and p38 expressions were even highly detected in the proximal tubular epithelial cells. Contrary to TUNEL-positive apoptotic cells, JNK and p38 expressions were more abundant and distributed throughout the proximal tubular epithelial cells in the renal cortex and were not restricted only to TUNEL-positive apoptotic cells.
ATF-2 has been postulated as an effector of JNK as well as p38. Therefore, in the present study, to evaluate the functional role of JNK and p38 activation, we determined the effects of ACE inhibition on their phosphorylated transcription factor ATF-2 in the developing kidney. In the enalapril-treated group, similar to JNK and p38 expressions, the immunoreactivity of ATF-2 was highly expressed in the proximal tubular epithelial cells, and ATF-2 protein expression was increased significantly compared with the control group. These results suggest that ATF-2 participates in MAPK signaling processes that mediate the renal growth impairment as a result of ACE inhibition.
Among the MAPK family members, ERK has been implicated in protection against cell injury, and its activation has been considered a sign of renal regeneration and protection (41,42). The ERK1/2 pathway is generally but not exclusively responsive to activators of both receptor tyrosine kinases and G protein–coupled receptors. From a physiologic perspective, ERK signaling has been implicated in mitogenesis and promotes cell proliferation and differentiation. In rat renal development, ERK was present in the kidney throughout the stage, from the fetal kidney to adult kidney, and the location of ERK expression was changed markedly during kidney development. Also in the neonatal kidney, the location of ERK correlated with the distribution of proliferating cells during kidney development (39). In our present study, we found that ERK protein and mRNA expressions are not changed in the enalapril-treated group compared with the control group. Also, immunohistochemically, the ERK expression was not changed and so was weakly detected in some glomeruli and mainly detected in distal tubular epithelial cells in both groups. In our previous study (7,8), we also reported that ACE inhibition in the developing kidney decreased the renal cell proliferation, which may account for neonatal renal growth impairment. In the present study, although the change of ERK and phospho-ERK expression was not found in either group, ERK expression was confined to the distal tubular epithelial cells, where proliferating cell nuclear antigen–positive cells were most abundant in both groups of the previous study. These observations suggest that ERK is thought to be involved in signaling processes in the renal growth and proliferation but not involved in RAS-related regulation of renal growth and development.
MAPK families, especially JNK, are usually activated in response to various cellular stresses. The long-term ACE inhibition may have an effect on the renal hemodynamics, as a result of hypotension, microcirculation, or local ischemia in an in vivo model. In previous reports, there is no direct evidence that enalapril-induced hemodynamic changes affect cellular responses in the developing rat kidney (6,43). The effect of antihypertensive drugs (e.g. enalapril, verapamil, hydralazine) on growth during the neonatal period is limited to enalapril alone, causing growth retardation in maturing kidney and heart. Hence, a compilation of these results suggest that the cellular actions of Ang II in the developing kidney are independent of hemodynamic changes.
CONCLUSION
In conclusion, our data suggest that the expression of the MAPK family is modulated by ACE inhibition in the developing kidney. Inhibition of endogenous Ang II production by chronic administration of an ACE inhibitor to neonatal rat seems to impair the renal growth, and, in this process, the MAPK family may play a role in renal growth impairment. Especially, JNK-2 and p38 expressions were activated in the kidney of the enalapril-treated group, and their expressions were strongly detected in the proximal tubular epithelial cells in the enalapril-treated group. With regard to the correlation between JNK and p38 expressions and the occurrence of apoptosis, JNK and p38 may be implicated to participate in the Ang II–related intracellular signaling pathways of renal apoptosis in the developing kidney. However, the expression of JNK and p38 was distributed throughout all proximal tubular epithelial cells in the enalapril-treated group and not restricted to apoptotic cells after chronic administration of enalapril. These results suggest that the MAPK family plays multiple roles in signaling pathways that participate in a variety of cellular events beyond apoptosis during renal development. Further studies are needed to look into the intracellular signaling mechanisms involved in a variety of metabolic pathways underlying cellular growth, differentiation, survival, and cell death, which in turn control renal growth and development by RAS.
Abbreviations
- ACE:
-
angiotensin-converting enzyme
- Ang II:
-
angiotensin II
- ATF-2:
-
activating transcription factor-2
- DBA:
-
Dolichos biflorus agglutinin
- ERK:
-
extracellular signal–regulated kinase
- GAPDH:
-
glyceraldehyde-3-phosphate dehydrogenase
- JNK:
-
c-Jun NH2-terminal kinase
- MAPK:
-
mitogen-activated protein kinase
- RAS:
-
renin-angiotensin system
- TUNEL:
-
terminal deoxynucleotide transferase-mediated nick-end labeling
References
Gomez RA, Norwood VF 1995 Developmental consequences of the renin-angiotensin system. Am J Kidney Dis 26: 409–431
Guron G, Friberg P 2000 An intact renin-angiotensin system is a prerequisite for normal renal development. J Hypertens 18: 123–137
Niimura F, Okubo S, Fogo A, Ichikawa I 1997 Temporal and spatial expression pattern of the angiotensinogen gene in mice and rats. Am J Physiol 272: R142–R147
Alcorn D, McCausland JE, Maric C 1996 Angiotensin receptors and development: the kidney. Clin Exp Pharmacol Physiol Suppl 3: S88–S92
Yoo KH, Wolstenholme JT, Chevalier RL 1997 Angiotensin-converting enzyme inhibition decreases growth factor expression in the neonatal rat kidney. Pediatr Res 42: 588–592
Choi JH, Yoo KH, Cheon HW, Kim KB, Hong YS, Lee JW, Kim SK, Kim CH 2002 Angiotensin converting enzyme inhibition decreases cell turnover in the neonatal rat heart. Pediatr Res 52: 325–332
Yoo KH, Cheon HW, Choi BM, Hong YS, Lee JW, Kim SK 1998 Angiotensin converting enzyme inhibition increases apoptosis and BCL-2 expression in the neonatal rat kidney. [abstract]. J Am Soc Nephrol 9: 512A
Yoo KH, Cheon H, Choi BM, Hong YS, Lee JW, Kim SK 1999 Effects of angiotensin converting enzyme inhibition into apoptosis and related genes in the neonatal rat kidney. J Korean Pediatr Soc 42: 1086–1095
Kang NS, Yim HE, Bae IS, Choi JH, Choi BM, Yoo KH, Hong YS, Lee JW, Kim SK 2003 ACE inhibition modulates transforming growth factor-β receptors in the young rat. Pediatr Nephrol 18: 865–871
Griendling KK, Ushio-Fukai M, Lassegue B, Alexander RW 1997 Angiotensin II signaling in vascular smooth muscle. New concepts. Hypertension 29: 366–373
Huwiler A, van Rossum G, Wartmann M, Pfeilschifter J 1998 Angiotensin II stimulation of the stress-activated protein kinases in renal mesangial cells is mediated by the angiotensin AT1 receptor subtype. Eur J Pharmacol 343: 297–302
Marshall CJ 1995 Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80: 179–185
Bonventre JV, Force T 1998 Mitogen-activated protein kinases and transcriptional responses in renal injury and repair. Curr Opin Nephrol Hypertens 7: 425–433
Schaeffer HJ, Weber MJ 1999 Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19: 2435–2444
Bokemeyer D, Sorokin A, Dunn MJ 1996 Multiple intracellular MAP kinase signaling cascades. Kidney Int 49: 1187–1198
Tian W, Zhang Z, Cohen DM 2000 MAPK signaling and the kidney. Am J Physiol 279: F593–F604
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME 1995 Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270: 1326–1331
Hibi M, Lin A, Smeal T, Minden A, Karin M 1993 Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 7: 2135–2148
van Dam H, Wilhelm D, Herr I, Steffen A, Herrlich P, Angel P 1995 ATF-2 is preferentially activated by stress-activated protein kinases to mediate c-jun induction in response to genotoxic agents. EMBO J 14: 1798–1811
Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, Ulevitch RJ, Davis RJ 1995 Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267: 682–685
Livingstone C, Patel G, Jones N 1995 ATF-2 contains a phosphorylation-dependent transcriptional activation domain. EMBO J 14: 1785–1797
Hazzalin CA, Cano E, Cuenda A, Barratt MJ, Cohen P, Mahadevan LC 1996 p38/RK is essential for stress-induced nuclear responses: JNK/SAPKs and c-Jun/ATF-2 phosphorylation are insufficient. Curr Biol 6: 1028–1031
Waas WF, Lo HH, Dalby KN 2001 The kinetic mechanism of the dual phosphorylation of the ATF2 transcription factor by p38 mitogen-activated protein (MAP) kinase alpha. Implications for signal/response profiles of MAP kinase pathways. J Biol Chem 276: 5676–5684
Gupta S, Campbell D, Derijard B, Davis RJ 1995 Transcription factor ATF2 regulation by the JNK signal transduction pathway. Science 267: 389–393
Gomez RA, Lynch KR, Chevalier RL, Everett AD, Johns DW, Wilfong N, Peach MJ, Carey RM 1988 Renin and angiotensinogen gene expression and intrarenal renin distribution during ACE inhibition. Am J Physiol 254: F900–F906
Holthofer H 1988 Cell type-specific glycoconjugates of collecting duct cells during maturation of the rat kidney. Cell Tissue Res 253: 305–309
Holthofer H, Schulte BA, Spicer SS 1987 Expression of binding sites for Dolichos bifloris agglutinin at the apical aspect of collecting duct cells in rat kidney. Cell Tissue Res 249: 481–485
Laitinen L, Virtanen I, Saxen L 1987 Changes in the glycosylation pattern during embryonic development of mouse kidney as revealed with lectin conjugates. J Histochem Cytochem 35: 55–65
Tisdale EJ 2001 Glyceraldehyde-3-phosphate dehydrogenase is required for vesicular transport in the early secretory pathway. J Biol Chem 276: 2480–2486
Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, Hibi M, Nakabeppu Y, Shiba T, Yamamoto KI 1999 JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol Cell Biol 19: 7539–7548
Maisonpierre PC, Le Beau MM, Espinosa R 3rd Ip NY, Belluscio L, de la Monte SM, Squintos Furth ME, Yancopoulos GD 1991 Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics 10: 558–568
Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD 1991 ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 65: 663–675
Han J, Lee JD, Bibbs L, Ulevitch RJ 1994 A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 265: 808–811
Timmermans PB, Wong PC, Chiu AT, Herblin WF, Benfield P, Carini DJ, Lee RJ, Wexler R, Saye JA, Smith RO 1993 Angiotensin II receptors and angiotensin II receptor antagonists. Pharmacol Rev 45: 205–251
Wolf G, Neilson EG 1993 Angiotensin II as a renal growth factor. J Am Soc Nephrol 3: 1531–1540
Ruiz-Ortega M, Lorenzo O, Suzuki Y, Ruperez M, Egido J 2001 Proinflammatory actions of angiotensins. Curr Opin Nephrol Hypertens 10: 321–329
Ruiz-Ortega M, Egido J 1997 Angiotensin II modulates cell growth-related events and synthesis of matrix proteins in renal interstitial fibroblasts. Kidney Int 52: 1497–1510
Robinson MJ, Cobb MH 1997 Mitogen-activated protein kinase pathways. Curr Opin Cell Biol 9: 180–186
Omori S, Hida M, Ishikura K, Kuramochi S, Awazu M 2000 Expression of mitogen-activated protein kinase family in rat renal development. Kidney Int 58: 27–37
Hida M, Omori S, Awazu M 2002 ERK and p38 MAP kinase are required for rat renal development. Kidney Int 61: 1252–1262
di Mari JF, Davis R, Safirstein RL 1999 MAPK activation determines renal epithelial cell survival during oxidative injury. Am J Physiol 277: F195–F203
Masaki T, Foti R, Hill PA, Ikezumi Y, Atkins RC, Nikolic-Paterson DJ 2003 Activation of the ERK pathway precedes tubular proliferation in the obstructed rat kidney. Kidney Int 63: 1256–1264
Fogo A, Yoshida Y, Yared A, Ichikawa I 1990 Importance of angiogenic action of angiotensin II in the glomerular growth of maturing kidneys. Kidney Int 38: 1068–1074
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was supported by Korea Research Foundation Grant (K.H.Y., KRF-2001-0410-F00160).
Rights and permissions
About this article
Cite this article
Choi, B., Yoo, K., Bae, I. et al. Angiotensin-Converting Enzyme Inhibition Modulates Mitogen-Activated Protein Kinase Family Expressions in the Neonatal Rat Kidney. Pediatr Res 57, 115–123 (2005). https://doi.org/10.1203/01.PDR.0000148064.27632.1D
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000148064.27632.1D
This article is cited by
-
Angiotensin inhibition in the developing kidney; tubulointerstitial effect
Pediatric Research (2019)
-
Postnatal early overnutrition causes long-term renal decline in aging male rats
Pediatric Research (2014)
-
MAPK and angiotensin II receptor in kidney of newborn rats from losartan-treated dams
Pediatric Nephrology (2008)
-
Angiotensin receptor blockers in the treatment of NASH/NAFLD: Could they be a first-class option?
Advances in Therapy (2008)
-
AT1 antagonist modulates activin-like kinase 5 and TGF-β receptor II in the developing kidney
Pediatric Nephrology (2006)
