
Abstract
The “hard wiring” encoded within the genome that determines the emergence of the laryngotracheal groove and subsequently early lung branching morphogenesis is mediated by finely regulated, interactive growth factor signaling mechanisms that determine the automaticity of branching, interbranch length, stereotypy of branching, left-right asymmetry, and finally gas diffusion surface area. The extracellular matrix is an important regulator as well as a target for growth factor signaling in lung branching morphogenesis and alveolarization. Coordination not only of epithelial but also endothelial branching morphogenesis determines bronchial branching and the eventual alveolar-capillary interface. Improved prospects for lung protection, repair, regeneration, and engineering will depend on more detailed understanding of these processes. Herein, we concisely review the functionally integrated morphogenetic signaling network comprising the critical bone morphogenetic protein, fibroblast growth factor, Sonic hedgehog, transforming growth factor-β, vascular endothelial growth factor, and Wnt signaling pathways that specify and drive early embryonic lung morphogenesis.
Similar content being viewed by others


THE 70 M2 ALVEOLAR SURFACE ORIGINATES FROM THE LARYNGOTRACHEAL GROOVE
Lung development begins with the appearance of the laryngotracheal groove, which is a small diverticulum that arises from the floor of the primitive pharynx at E9 in mouse and 4 wk in human. The proximal portion of the laryngotracheal groove gives rise to the larynx and trachea, whereas the distal portion gives rise to the left and right mainstem bronchial buds, which in turn give rise to the left and right lobar branches of the bronchial tree. In mice, four lobes form in the right lung, whereas the left lung forms a single lobe. In humans, the left lung is trilobed, whereas the right lung is bilobed. Lobation of the lungs, as well as the first 16 of 23 airway generations in humans is stereotypic, which implies the presence of a hard-wired, genetic program that controls early embryonic lung branching morphogenesis. The latter seven generations of lower airway branching and onward into the folding and expansion phase of the alveolar surface is nonstereotypic, but nevertheless follows a recognizable, proximal-distal fractal pattern that is repeated automatically at least 50 million times. The lung morphogenetic program thus drives the formation of an alveolar gas diffusion surface 0.1 μm thick by 70 m2 in surface area that is perfectly matched to the alveolar capillary and lymphatic vasculature. This huge gas exchange surface area is packed into the chest in a highly spatially efficient manner (1,2).
SOLUBLE FACTORS ARISING FROM THE MESENCHYME DETERMINE LUNG EPITHELIAL MORPHOGENESIS
Soluble factors arising from the mesenchyme have long been implicated in lung branching morphogenesis distal to the trachea, since it was discovered that transposition of early stage distal lung mesenchyme onto the primitve tracheal epithelium can induce supernumary branching (3–7) as well as distal epithelial marker gene expression (8–10). Null mutation in mice of Fgf10, or its cognate receptor Fgfr2, as well as misexpression of dominant negative kinase dead FGFR, or of soluble FGFR, all display a spectrum of closely related tracheobronchial truncation phenotypes, in which epithelial, endothelial, and lymphatic branching of the bronchi distal to the trachea is abrogated (11–15). This strongly suggests that the genetic controls of laryngotracheal groove formation are separate from those that induce the peripheral lung. Herein, we review the little that is known about the molecular signals that induce the formation of the laryngotracheal groove. Then, we consider the rather more extensive information on some of the key molecular signaling mechanisms that mediate branching of the airways during early lung development.
THE LARYNX AND TRACHEA ARE GENETICALLY DISTINGUISHABLE FROM THE DISTAL AIRWAYS
Null mutation of Hnf3β results in failure of closure of the primitive foregut, thus no laryngotracheal bud can form (16). Hoxa3 and 5 also play key roles in specifying the larynx and trachea (17–20). Retinoic acid deficiency in mice has long been associated with total agenesis of the lungs, in association with other complex anomalies (21,22). Certain RAR null mutations are also among the few known causes of complete laryngo-tracheo-pulmonary agenesis in mice (22). The other known null mutations associated with complete absence of the larynx, trachea, and lung being the double Gli2/3 null mutation (23). Recently, it has been shown that retinoic acid induces Fgf10 expression within the mesoderm subjacent to the site of origin of the laryngotracheal groove (24). Blockade of RAR signaling in E9 foregut cultures not only blocks the local expression of Fgf10 at this restricted site but also disrupts expression of Nkx2.1 and Sp-C in the floor of the primitive pharynx. Thus, retinoic acid signaling may connect formation of the larnygotracheal groove with activation of Fgf10-dependent bronchial morphogenesis. Further evidence for distinctly separate embryonic cell lineages that form the trachea and proximal bronchi versus the peripheral lung was provided by tetracycline driven Sp-C-Cre recombination, which activated lineage marker expression within the peripheral lung but not the trachea, upper bronchi, or gastrointestinal tract (25).
LEFTY-1, LEFTY-2, AND NODAL ARE IMPLICATED IN DETERMINATION OF LEFT-RIGHT LATERALITY OF THE LUNG
Lefty-1 null mutant mice show a variety of left-right isomerisms in visceral organs, but the most common feature is thoracic left isomerism. The lack of Lefty-1 expression results in abnormal bilateral expression of Nodal, Lefty-2, and Pitx2 (a homeobox gene normally expressed on the left side). This suggests that Lefty-1 normally restricts Lefty-2 and Nodal expression to the left side, and that Lefty-2, Pitx2, and/or Nodal encode a signal for “leftness” in the lung (26,27). We speculate that proximal-distal, anteroposterior, and left-right stereotypy must be superimposed on the automatic morphogenetic branching mechanisms proposed below.
TRACHEOESOPHAGEAL SEPTATION
Septation of the trachea from the esophagus is normal in the Fgf10 null mutation, indicating that this process can also be distinguished genetically from peripheral lung branching. However, null mutations of the transcriptional factor Nkx2.1 and of Shh display severe anomalies of larngyo-tracheal-esophageal septation, together with abrogation of distal lung morphogenesis (28–31). Haploinsufficency of Foxf1, which is itself a SHH target, causes esophageal atresia and tracheoesophageal fistula (32). SHH also plays a key role in determining correct patterning of the tracheal cartilages, inasmuch as peripheral expression of Shh under the control of the Sp-C promoter fails to rescue tracheal cartilage morphogenesis on the Shh null background (33). SHH also regulates Gli3 gene expression (34). Laryngeal clefts, tracheal atresias, and esophageal atresias, with or without tracheoesophageal fistula, occur quite frequently in humans. Clinical associations with multiple other anomalies, including the VACTERL association, duodenal atresia and pancreatic malformations, have been noted. In the Pallister-Hall syndrome, among other anomalies, bifid epiglottis and cleft larynx have been described as an effect of Gli3 mutation (34). Defects in the Shh-Gli pathway as well as the FGF pathway have also been noted in the adriamycin-mediated teratogenic model of tracheoesophageal anomalies (35–38). Hox family genes are also likely to play a key role in tracheobronchial differentiation, since in Hoxa-5−/− mice, tracheal occlusion and respiratory distress are associated with marked decrease in surfactant protein production, together with altered gene expression in the pulmonary epithelium (19). Because Hoxa-5 expression is restricted to the lung mesenchyme, the null mutant phenotype strongly supports the inference that Hoxa-5 expression is necessary for induction of epithelial gene expression by the underlying mesenchyme (39,40).
BMP, EGF, FGF, SHH, TGF-β, VEGF, AND WNT SIGNALING PATHWAYS ALL PLAY CRITICAL ROLES TO DRIVE EARLY EMBRYONIC DISTAL LUNG BRANCHING MORPHOGENESIS
EGF, FGF, and PDGF signal through cognate transmembrane tyrosine kinase receptors to exert a positive effect on lung morphogenesis. In contrast, TGF-β and BMP family peptides signal through transmembrane serine-threonine kinase receptors. In general, TGF-β family peptides exert an overriding inhibitory effect on lung epithelial cell proliferation and hence negatively regulate lung morphogenesis. However, BMP4 appears to exert a complex negative or positive regulatory influence, depending on whether mesenchymal signaling is intact. SHH family peptide signaling represents another key mechanism. The SHH cognate receptor, patched (PTC), exerts a negative effect on SHH signaling both through the release of the transcriptional repressor Smoothened (SMO) and the induction of the Hedgehog interacting protein (Hip). Recently Wnt signaling has also emerged as exerting a critical influence on matrix fibronectin deposition and hence the specification of branch points. Also, the VEGF pathway has emerged as playing a critical role in early lung morphogenesis. These peptide growth factors and their cognate receptors are expressed in repeating patterns within morphogenetic centers that surround and direct each new branch tip in the early embryonic lung. Important mesenchymally expressed morphogenetic genes include Fgf10, Sprouty4 (Spry4), patched, smoothened, Wnt, and Hox family members, whereas Bmp4, Shh, mSpry2, Smads 2, 3, and 4, and VEGF are expressed in the adjacent epithelium. The interactions of subsets of these ligand signals, particularly SHH, BMP4, and FGF10, have been extensively reviewed recently and several models have been proposed to explain how they may interact to induce and then regulate epithelial branching morphogenesis (1–3,5,41).
BMP PLAY MULTIPLE ROLES IN LUNG DEVELOPMENT
BMP4 is expressed predominantly in the epithelium and is increased at branch tips. Until recently, BMP4 was postulated to mediate localized suppression of epithelial proliferation, thus providing a negative modulatory influence on FGF signaling to mediate arrest of branch extension and hence to set up branch points. This hypothesis was based upon the hypoplastic phenotype of the epithelium in transgenic misexpression studies of Bmp4 in the epithelium, as well as upon addition of BMP4 ligand to naked epithelial explants in culture. However, two groups have now shown that BMP4 is actually a potent stimulator of branching in the presence of mesenchyme and at physiologic concentrations in lung explants. Moreover, the effects of BMP4 are in turn negatively modulated by the BMP binding proteins Gremlin and Noggin. Abrogation of Gremlin function results in abnormal lung formation in neonatal Gremlin null mutant mice (42). Therefore, it seems unlikely that BMP4 signaling merely serves to inhibit epithelial proliferation, particularly since BMP4-specific Smads are also expressed in the mesenchyme away from the epithelium at certain developmental stages. BMP have also been reported to control differentiation of the endoderm along the proximal-distal axis (43). Inhibition of BMP signaling at the tip of the lung bud by overexpression in the distal epithelium of Noggin (a secreted inhibitor of BMP) or of a dominant negative form of the Bmp type I receptor, activin receptor-like kinase 6 (Alk6), results in a distal epithelium exhibiting differentiation characteristics, at the molecular and cellular level, of the proximal epithelium. Several additional BMP, including BMP3, 5, and 7, are expressed during embryonic lung development (44–46). The expression of Bmp5 and Bmp7 has been detected in the mesenchyme and the endoderm of the developing embryonic lung respectively, whereas Bmp4 expression is restricted to the distal epithelial cells and the adjacent mesenchyme (44,46). Recently, parallels have been drawn between genetic hard wiring of tracheal morphogenesis in Drosophila melanogaster and mammals (1). Dpp, the Drosophila BMP4 orthologue, has been reported to be essential for the formation of the dorsal and ventral branches of the tracheal system, controlling tracheal branching and outgrowth possibly through induction of the zinc finger proteins Kni and Knrl (47,48). Because conventional murine knockouts for BMP4 and BMP-specific Smads cause early embryonic lethality, their functions in lung development in vivo still need to be further defined. Interestingly, germ line mutations in BMP type II receptors were recently found in familial primary pulmonary hypertension (49).
EGFR NULL MUTATION REDUCES BRANCHING BY 50%
EGF is expressed in the distal epithelium and mesenchyme. Null mutation of Egfr results in a 50% reduction in branching and a neonatal lethal failure of lung maturation (50,51). EGF was the first growth factor to be shown to exert an inductive role on chick trachea to induce ectopic branching (52), as well as to accelerate branching through activation of its cognate EGFR tyrosine kinase in mouse embryonic lung in culture (53,54). In addition, null mutation of TNF-α converting enzyme (TACE), a cell surface protein sheddase that regulates release of active from latent forms of TGF-α, amphiregulin, neuregulin-1, HB-EGF, and HerbB4, also causes a hypoplastic phenotype of lung in the perinatal stage and lethality in new born mice (55).
FGF LIGANDS AND RECEPTORS DIRECT EPITHELIAL MIGRATION AND DIFFERENTIATION
FGF10 promotes directed growth of the lung epithelium and induces both proliferation and chemotaxis of isolated endoderm (56,57). The chemotaxis response of the lung endoderm to FGF10 involves the coordinated movement of an entire epithelial tip, containing hundreds of cells, toward an FGF10 source. How this population of cells monitors the FGF gradient and which receptors trigger this effect remains unknown. FGF10 also controls the differentiation of the epithelium by inducing SP-C expression and by up-regulating the expression of BMP4, a known regulator of lung epithelial differentiation (44,57–59). In vitro binding assays have shown that FGF10 acts mostly through FGFR1b and FGFR2b (60). Although there is good evidence that FGF10 acts through FGFR2b in vivo, there are as yet no conclusive data involving FGFR1b (or any other receptor) in vivo. The biologic activities mediated through these two epithelial receptors are likely to be different, as FGF7 (acting mostly through FGFR2b) exhibits a different activity compared with FGF10 (56). This hypothesis is also supported by our recent unpublished findings showing that Fgf10−/− lungs exhibit a more severe phenotype than Fgfr2b−/− lungs.
THE MAMMALIAN FGF RECEPTOR FAMILY COMPRISES FOUR GENES (FGFR1 TO FGFR4), WHICH ENCODE AT LEAST SEVEN PROTOTYPE RECEPTORS
Fgfr1, 2, and 3 encode two receptor isoforms (termed IIIb or IIIc) that are generated by alternative splicing, and each binds a specific repertoire of FGF ligands (60). FGFR2-IIIb (FGFR2b) is found mainly in epithelia and binds four known ligands (FGF1, FGF3, FGF7, and FGF10), which are primarily expressed in mesenchymal cells. Peters et al. (61) reported the first evidence of a key role for Fgfr2 during lung development. They showed that misexpression of a dominant negative form of Fgfr2 in the embryonic lung under the SP-C promoter led to a severe reduction in branching morphogenesis. Further evidence came from Fgfr2 inactivation in the embryo. Whereas mice null for the Fgfr2 gene die early during embryogenesis, those that are null for the Fgfr2b isoform, but retain Fgfr2c, survive to birth (62–65). Mice deficient for Fgfr2b show agenesis and dysgenesis of multiple organs, including the lungs, indicating that signaling through this receptor is critical for mesenchymal-epithelial interactions during early organogenesis. This idea is supported by the recent finding that prenatally induced misexpression of a dominant negative FGFR, to abrogate FGF signaling, results in a hypoplastic, emphysematous lung phenotype (66). In contrast, induced abrogation of FGF signaling postnatally did not produce any recognizable phenotype.
THE FGF LIGAND FAMILY IS COMPRISED OF AT LEAST 23 MEMBERS
Many FGF have been implicated in multiple aspects of vertebrate development (for review, see Ref. 67). FGF 1, 2, 7, 9, 10, and 18 play overlapping, yet distinct roles in the lung. In particular, FGF10 has been associated with instructive mesenchymal-epithelial interactions, such as those that occur during branching morphogenesis, whereas FGF9 and 18 appear to play a role in the mesenchyme and FGF1 and 7 appear to play roles in mediating postnatal lung repair (68). In the developing lung, Fgf10 is expressed in the distal mesenchyme at sites where prospective epithelial buds will appear. Moreover, its dynamic pattern of expression and its ability to induce epithelial expansion and budding in organ cultures have led to the hypothesis that FGF10 governs the directional outgrowth of lung buds during branching morphogenesis (56). Furthermore, FGF10 was shown to induce chemotaxis of the distal lung epithelium (57,69). Consistent with these observations, mice deficient for Fgf10 show multiple organ defects including lung agenesis (70–73). FGF10 is the main ligand for FGFR2b during the embryonic phase of development as evidenced by the remarkable similarity of phenotypes exhibited by embryos where these genes have been inactivated (64,73,74). Thus, the paradigm proposed so far is that FGF10 expressed by the mesenchyme acts on the epithelium (which expresses FGFR1b and 2b).
CONSERVED FUNCTIONS OF FGF SIGNALING IN DROSOPHILA TRACHEA MORPHOGENESIS
In D. melanogaster, Branchless (bnl), the Drosophila counterpart of FGF10, has been involved in the directional growth of the ectoderm-derived cells from the tracheal placode (75). Bnl expressed by the cells surrounding the placode acts on the ectoderm expressing the Fgfr2b ortholog, breathless (btl). An additional unsuspected function of bnl in the development of the male genital imaginal disc has been recently reported (76). Here, FGF signal expressed by ectoderm-derived cells of the male genital disc induces the FGFR-expressing mesodermal cells to migrate into the male disc. These mesodermal cells also undergo a mesenchymal to epithelial transition. The authors suggest that bnl, the FGF10 ortholog, is likely to be involved in this process. Thus, FGF10 is a multifunctional growth factor and additional roles for FGF10 in lung development likely remain to be identified.
SPROUTY FAMILY MEMBERS FUNCTION AS INDUCIBLE NEGATIVE REGULATORS OF FGF SIGNALING IN LUNG DEVELOPMENT
The first example of an FGF-inducible signaling antagonist arose from the discovery of the sprouty mutant during Drosophila trachea development, in which supernumerary tracheal sprouts arise. In the Drosophila tracheae, bnl binds to btl, inducing primary, secondary, and terminal branching. The function of bnl is inhibited by Sprouty (Spry), a downstream effector in the bnl pathway (77). Spry feeds back negatively on bnl, thereby limiting the number of sites at which new secondary tracheal buds form. Spry is not only found downstream in the FGFR pathway, but also appears to be an inhibitor of other tyrosine kinase signaling pathways such as EGF and Torso (78). mSpry2 is the gene that is most closely related to Drosophila Spry and is 97% homologous to hSpry2. mSpry2 is localized to the distal tips of the embryonic lung epithelial branches and is down-regulated at sites of new bud formation (74). On the other hand, mSpry4 is predominantly expressed throughout the distal mesenchyme of the embryonic lung. Abrogation of mSpry2 expression stimulates murine lung branching morphogenesis and increased expression of specific lung epithelial maturation/differentiation markers (58). Conversely, overexpression of mSpry2 under the control of a SP-C promoter or by intratracheal microinjection of an adenovirus containing the mSpry2 cDNA, results in smaller lungs with a particular “moth-eaten” dysplastic appearance along the edges of the lobes, with decreased epithelial cell proliferation (74). Thus, not only is the function of Spry conserved during respiratory organogenesis, but also as seen by loss of function and gain of function studies, Spry plays a vital role in regulating lung branching morphogenesis. In Drosophila, in vitro co-precipitation studies show that Spry binds to Gap1 and Drk (a Grb2 orthologue), resulting in inhibition of the Ras-MAPK pathway (77). Upon further investigation of the mechanism by which mSPRY2 negatively regulates FGF10 in mouse lung epithelial cells (MLE15), we recently determined that mSPRY2 differentially binds to FGF downstream effector complexes (78).
FGFR ARE DIFFERENT FROM OTHER TYROSINE KINASE RECEPTORS IN THAT THEY REQUIRE ADAPTER OR DOCKING PROTEINS
These adapter or docking proteins include phospholipase C γ, Shc, FRS2, and various others to recruit the Grb2/Sos complex upon stimulation. Stimulation of FGFR not only results in formation of the FRS2/Grb2/Sos complex, but the binding of a positive tyrosine phosphatase regulator, Shp2, to FRS2, which is required for full potentiation of MAP-kinase activation (79). Complex formation leads to catalysis of GDP to GTP on Ras, which is required for Raf (serine/threonine kinase) activation. Raf causes direct activation of ERK, leading to phosphorylation of cytoplasmic proteins followed by cell growth and differentiation (80). We found in unpublished studies that in the native state mSPRY2 associates with Shp2 and Gap, which is a GTPase-activating protein that hydrolyzes GTP to GDP. It is possible that in this state the binding of Shp2 to mSPRY2 regulates mSPRY2 activity. Upon FGFR activation, mSPRY2 disassociates from Shp2 and Gap and translocates to the plasma membrane, where it binds to both FRS2 and Grb2, thus blocking the formation of the FRS2/Grb2/Sos complex, resulting in a net reduction of MAP-kinase activation. Thus, Sprouty would inhibit the formation of specific signaling complexes downstream tyrosine-kinase receptors resulting in modulation and co-ordination of cell growth and development during organogenesis.
DYNAMIC RELATIONSHIP BETWEEN FGF SIGNALING AND SPRY DURING DEVELOPMENT
It is interesting to note that overexpression of Spry in chick limb buds results in a reduction in limb bud outgrowth that is consistent with a decrease in FGF signaling (81). This suggests a possible co-regulatory relationship between FGF signaling and Spry during development. In further support of this model, Spry4 inhibits branching of endothelial cells as well as sprouting of small vessels in cultured mouse embryos. Endothelial cell proliferation and differentiation in response to FGF and VEGF are also inhibited by mSpry4, which acts by repressing ERK activation. Thus, Spry4 may negatively regulate angiogenesis (82).
SPRY2, SPRY4, AND SPRED SHARE COMMON INHIBITORY MECHANISMS
It has been suggested that both Spry2 and Spry4 share a common inhibitory mechanism. Both Sprouty translocate to membrane ruffles upon EGF stimulation. However, only SPRY2 was shown to associate with microtubules (83). The C-terminus of hSPRY2 has been shown to be important for modulation of cellular migration, proliferation, and membrane co-localization (83,84). Interestingly, the C-terminus is the region that is most conserved throughout the Spry family and contains potential regulatory sites that would modulate Spry activity. Spry has also been shown to interact with c-Cbl, resulting in increased EGFR internalization (85). Although Spry is not a specific inhibitor to the FGFR signaling pathway nor to respiratory organogenesis, it appears that Spry plays a vital role in modulating several signaling pathways to limit the effects of excessive growth factor receptor tyrosine kinase signaling. The Sprouty-related Spred family of genes are also expressed in the developing lung, however, their function is less clear (86). Nevertheless, SPRED can inhibit cell motility and Rho-mediated actin reorganization as well as growth factor–induced ERK activation (86–89) and therefore may function in a similar fashion to Sprouty to negatively modulate key aspects of lung morphogenesis.
PROTEOGLYCANS
Proteoglycans are deposited within the extracellular matrix during early embryonic lung branching morphogenesis and inhibition of proteoglycan synthesis or treatment with heparitinase severely affects branching (90–92). More recently, it has been shown that both heparan sulfate and chondroitin sulfate proteoglycans are required for lung branching and in fact mediate the inductive effects of FGF10 binding to the epithelium (93–95).
SHH, PATCHED, AND HIP
The role of SHH signaling in lung morphogenesis has recently been reviewed (96). Hedgehog signaling is essential for lung morphogenesis because Shh null produces profound hypoplasia of the lungs and failure of tracheoesophageal septation (30,97). However, proximodistal differentiation of the endoderm is preserved in the Shh null mutant, at least in so far as expression of surfactant protein-C, (SP-C) and Clara cell protein 10, (CC10) genes are concerned. The expression of the SHH receptor, Patched, is also decreased in the absence of Shh as are the Gli1 and Gli3 transcriptional factors. On the other hand, lung-specific misexpression of Shh results in severe alveolar hypoplasia and a significant increase in interstitial tissue (98). Fgf10 expression, which is highly spatially restricted in wild type, is not spatially restricted and is widespread in the mesenchyme in contact with the epithelium of the Shh null mutant mouse lung. Conversely, local suppression of SHH signaling by the induction of Ptc and Hip at branch tips may serve to facilitate FGF signaling locally, where branch outgrowth is stereotypically programmed to take place (99). It is interesting to note that the cecum, which forms as a single bud from the mouse midgut and does not branch, also expresses Fgf10 throughout its mesenchyme (Burns and Bellusci, unpublished results). Thus, temporospatial restriction of Fgf10 expression by SHH appears to be essential to initiate and maintain branching of lung.
TGF-β FAMILY PEPTIDE EXPRESSION AND ACTIVATION IS ESSENTIAL FOR NORMAL LUNG DEVELOPMENT
The TGF-β superfamily can be divided into three subfamilies: activin, TGF-β, and BMP (100). There are three TGF-β isoforms in mammals: TGF-β1, 2, and 3. All of them have been detected in murine embryonic lungs (101–104). In early mouse embryonic lungs (E11.5), TGF-β1 is expressed in the mesenchyme, particularly in the mesenchyme underlying distal epithelial branching points, whereas TGF-β2 is localized in distal epithelium, and TGF-β3 is expressed in proximal mesenchyme and mesothelium (105). Mice lacking Tgfβ1 develop normally but die within the first month or two of life of aggressive pulmonary inflammation. When raised under pulmonary pathogen–free conditions, these mice live somewhat longer but die of other forms of inflammation (106). Thus, physiologic concentrations of TGF-β1 appear to suppress the pulmonary inflammation that occurs in response to exogenous factors such as infection and endotoxin. On the other hand, Tgfβ2 null mutants die in utero of severe cardiac malformations, whereas Tgfβ3 mutants die neonatally of lung dysplasia and cleft palate (107,108). Embryonic lung organ and cell cultures reveal that TGF-β2 plays a key role in branching morphogenesis, whereas TGF-β3 plays a key role in regulating alveolar epithelial cell proliferation during the injury repair response (109,110). Thus, finely regulated and correct physiologic concentrations and temporospatial distribution of TGF-β1, 2, and 3 are essential for normal lung morphogenesis and defense against lung inflammation. Overexpression of Tgfβ1, driven by the SP-C promoter, in lung epithelium of transgenic mice causes hypoplastic phenotypes (111). Similarly, addition of exogenous TGF-β to early embryonic mouse lungs in culture resulted in inhibition of lung branching morphogenesis although each TGF-β isoform has a different IC50 (TGF-β2>1>3) (105,111). In contrast, abrogation of TGF-β type II receptor stimulated embryonic lung branching through releasing cell cycle G1 arrest (112). Moreover, overexpression of constitutively active TGF-β1, but not latent TGF-β1, in airway epithelium is sufficient to have significant inhibitory effects on lung branching morphogenesis (109). However, no inhibitory effect on lung branching was observed when TGF-β1 was overexpressed in the pleura and subjacent mesenchymal cells. Furthermore, adenoviral overexpression of a TGF-β inhibitor, Decorin, in airway epithelium, completely abrogated exogenous TGF-β1-induced inhibition of embryonic lung growth in culture (113). On the other hand, reduction of decorin expression by DNA antisense oligonucleotides was able to restore TGF-β1-mediated lung growth inhibition (113). Therefore, TGF-β signaling in distal airway epithelium seems to be sufficient for its inhibitory function for embryonic lung growth. Interestingly, TGF-β specific signaling elements, such as Smad2/3/7, are exclusively expressed in distal airway epithelium (114–116). Attenuation of Smad2/3 expression by a specific antisense oligonucleotide approach blocked the exogenous TGF-β1-induced inhibitory effects on lung growth. Moreover, expression of Smad7 in airway epithelium, which was induced by TGF-β, had negative regulatory functions for the TGF-β–Smad pathway in cultured cells, specifically blocking exogenous TGF-β–induced inhibitory effects on lung branching morphogenesis as well as on Smad2 phosphorylation in cultured lung explants. Inasmuch as blockade of TGF-β signaling not only stimulates lung morphogenesis in culture per se, but also potentiates the stimulatory effects of EGF and PDGF-A, it follows that TGF-β signaling functions downstream of or can override tyrosine kinase receptor signaling.
DEVELOPMENTAL SPECIFICITY OF TGF-β1 OVEREXPRESSION PHENOTYPES
During embryonic and fetal life, epithelial misexpression of TGF-β1 results in hypoplastic branching and decreased epithelial cell proliferation (109). In contrast, neonatal misexpression of TGF-β1 using an adenoviral vector approach phenocopies bronchopulmonary dysplasia (BPD) with alveolar hypoplasia, some interstitial fibrosis, and emphysema (117). Adult misexpression of TGF-β1, on the other hand, results in a chronic, progressive interstitial pulmonary fibrosis, resulting mainly from increased proliferation and matrix secretion by the mesenchyme; a process that depends on transduction through Smad3 (118,119). Thus, the phenotype caused by excessive TGF-β1 production and signaling is always adverse, but the precise effect depends on the developmental stage of the lung: hypoplasia in embryonic, fetal, and neonatal lung; fibrosis in premature and adult lung.
VEGF ISOFORMS INDUCE VASCULOGENESIS, ANGIOGENESIS, AND LYMPHOANGIOGENESIS DURING LUNG DEVELOPMENT
Vascularization must perfectly match epithelial morphogenesis to ensure optimal gas exchange. Several VEGF isoforms are expressed in the developing epithelium, whereas their cognate receptors are expressed in and direct the emergence of developing vascular and lymphatic capillary networks within the mesenchyme. It is possible that VEGF signaling may lie downstream of FGF signaling, inasmuch as in vivo abrogation of FGF signaling severely affects both epithelial and endothelial morphogenesis. Vasculogenesis is initiated as soon as the lung evaginates from the foregut (121). A critical growth factor during embryonic lung development is VEGF. The loss of even a single allele of Vegf leads to embryonic lethality between days E9.5 and E10.5 in the mouse (121). VEGF is diffusely distributed in pulmonary epithelial and mesenchymal cells and is involved in controlling endothelial proliferation and the maintenance of vascular structure. VEGF is localized in the basement membrane of epithelial cells (122).
BOTH HUMANS AND MICE HAVE THREE DIFFERENT VEGF ISOFORMS
VEGF-120, VEGF-164, and VEGF-188 are all expressed in mice during development, but VEGF-164 isoform is the most highly expressed and active during embryogenesis. VEGF signals through the cognate receptors FLK-1 (fetal liver kinase-1, or VEGFR2) and FLT-1 (fetal liver tyrosinase-1, or VEGFR1). VEGF signaling is responsible for the differentiation of embryonic mesenchymal cells into endothelial cells. Interactions between the epithelium and mesenchyme contribute to lung neovascularisation, which is crucial in normal lung formation (123). In fact, epithelial cells of the airways are positive for VEGF, particularly at the budding regions of the distal airway (124). Also, lung mesenchyme cultured alone in the absence of epithelium degenerates significantly and only a few Flk-1 positives cells are maintained (120).
Vegf KNOCKOUT MICE HAVE A LETHAL PHENOTYPE WITHIN THE EARLY STAGES OF EMBRYONIC DEVELOPMENT (E8.5–E9)
Vegf misexpressing transgenic mice, where the Vegf transgene is under the control of the SP-C promoter, show gross abnormalities in lung morphogenesis that are associated with a decrease in acinar tubules and mesenchyme (121). VEGF-treated human lung explants show an increase of cellular proliferation in the distal airway epithelial cells with an up-regulation of the mRNA expression of Surfactant Protein-A (Sp-A) and C (Sp-C) but not Sp-B (124).
VEGF PLAYS A ROLE IN MAINTAINING ALVEOLAR STRUCTURE
VEGF has also been demonstrated to play a role in maintaining alveolar structure (125,126). Lungs from newborn mice treated with antibodies to FLT-1 are reduced in size and display significant immaturity with a less complex alveolar pattern (127). In contrast, the accumulation of VEGF in the alveoli appears to make transgenic VEGF mice more resistant to injury by hyperoxia (128,129). VEGF is a target of hypoxia-inducible transcription factor-2α (HIF-2α). Hif-2α–deficient newborn mice die from respiratory distress syndrome (129). In Hif-2α null mice the expression of VEGF is dramatically reduced in alveolar epithelial type 2 cells. Additionally, we have recently observed that addition of VEGF to early mouse embryonic lung explants markedly stimulates epithelial as well as vascular morphogenesis (unpublished results). Thus, we speculate that VEGF signaling plays an important role in matching the epithelial-capillary interface during lung morphogenesis.
VEGF-C AND VEGF-D ARE TWO ADDITIONAL MEMBERS OF THE VEGF FAMILY
These factors have a restricted expression pattern, with high levels mainly in lung tissues (130). VEGF-C and -D stimulate lymphoangiogenesis through their cognate receptor VEGFR-3 (131). Signaling via VEGFR-3 has been shown to be sufficient for lymphoangiogenesis through null mutation (132). Finally, VEGF-C also interacts with VEGFR-2 and is therefore able to induce angiogenesis in vivo (133).
WNT SIGNALING CONTROLS EPITHELIAL AND MESENCHYMAL DIFFERENTIATION AND PLAYS AN IMPORTANT ROLE IN LUNG DEVELOPMENT
The Wnt growth factor family in the mouse is comprised of 19 different secreted ligands that interact with 10 known seven-span transmembrane receptors of the Frizzled (Fz) gene family and either one of two single-span transmembrane proteins, low-density-lipoprotein-receptor-related proteins (LRP-5 and LRP-6) (134–136). Historically, Wnt proteins have been grouped into two classes, canonical and noncanonical. Canonical Wnt bind to frizzled receptors, inhibiting glycogen-synthase kinase-3β (GSK-3β)-mediated phosphorylation of β-catenin. Hypophosphorylated β-catenin accumulates in the cytoplasm, after which it translocates to the nucleus, where it heterodimerizes with members of the TCF/LEF transcription factor family to activate the transcription of TCF/LEF target genes. Noncanonical Wnt activate other Wnt signaling pathways, such as the planar-cell-polarity (PCP)-like pathway that guides cell movements during gastrulation (137). Activation of the PCP pathway can antagonize the canonical pathway intracellularly (138–140). Secreted Wnt antagonists can be divided into two functional classes, the secreted Frizzled-related protein (sFRP) class and the Dickkopf class. Members of the sFRP class bind directly to Wnts, thereby altering their ability to bind to the Wnt receptor complex, whereas members of the Dickkopf class inhibit Wnt signaling by binding to the LRP5/LRP6 component of the Wnt receptor complex.
In addition to LRP5/6, DKK1 interacts with the high affinity DKK1 co-receptors Kremen1 (Krm1) or Kremen2 (Krm2), which functionally cooperate with Dkk1 to block Wnt/beta-catenin signaling (141,142). DKK1, DKK3, and DKK4 act as inhibitors of canonical Wnt signaling, whereas DKK2 can function both as an inhibitor in the presence of Kremen2 or as an activator in its absence (143,144).
Between E10.5 and 17.5, β-catenin is localized in the cytoplasm and often also in the nucleus of the pulmonary epithelium and adjacent mesenchyme (146). Wnt ligands, Fz receptors, and the Tcf/Lef1 transcription factors are expressed during early lung development. In the early mouse lung (at E12.5), Wnt7b is expressed in the lung epithelium with a more intense expression distally, Wnt2a is highly expressed in the distal mesenchyme, and Wnt5a shows a low expression in the mesenchyme and epithelium and is highest around the trachea and pharynx. Tcf1 (T cell factor1), Lef1 (Lymphoid enhancer factor 1), sFrp1 (secreted Frizzled-related protein 1), and sFRP2 are highly expressed in the mesenchyme adjacent to the pulmonary epithelium. Tcf3 is highly expressed at the apical side of the pulmonary epithelium whereas Tcf4 and sFrp4 are detected in both the epithelium and adjacent mesenchyme (145). Fz8 is highly expressed throughout the epithelium whereas Fz2, 3, 6 and 7 are expressed both in the epithelium and mesenchyme (145). These expression patterns suggest that Wnt signaling can originate from the epithelium and mesenchyme and can target both tissues in an autocrine and/or paracrine fashion. TOPGAL or BATGAL mice, which harbor a β-galactosidase gene under the control of a LEF/TCF and β-catenin inducible promoter (146), reveal that from E10.5 until E12.5, canonical Wnt signaling occurs throughout the epithelium and in the mesenchyme adjacent to the proximal airways where the bronchial smooth muscle cells (SMC) arise (147–149). From E13.5, TOPGAL activity is no longer present in the mesenchyme and the activity in the epithelium is reduced distally concomitant with the onset of expression of Dkk1 in the distal epithelium (149).
CONDITIONAL INACTIVATION OF THE β-CATENIN GENE IN THE EPITHELIUM OF THE DEVELOPING MOUSE LUNG LEADS TO NEONATAL DEATH RESULTING FROM SEVERE LUNG DEFECTS
In this phenotype, branching of secondary bronchi is altered and the number of small peripheral alveolar ducts and terminal saccules is markedly reduced (150). In addition, the epithelium fails to undergo proper distal differentiation, lacking the expression of pro-SP-C protein and Vascular endothelial growth factor a, the latter correlating with a reduction in alveolar capillaries. So far, inactivation of only two WNT ligands have resulted in lung defects. Wnt7b is normally expressed in epithelial cells of the lung periphery. Wnt7b−/− mice exhibit perinatal death due to respiratory failure. Defects were observed in proliferation of the lung mesenchyme resulting in lung hypoplasia. In addition, Wnt7b−/− embryos and newborn mice exhibit severe defects in the smooth muscle component of the major pulmonary vessels, with increased apoptosis of the vascular smooth muscle cells (VSMC), resulting in rupture of the major blood vessels and hemorrhages in the lungs after birth. Wnt5a is expressed at high levels in the distal lung mesenchyme. Wnt5a−/− mice die perinatally from lung defects including truncation of the trachea, overexpansion of the peripheral airways and delayed lung maturation. Absence of WNT5a activity in the mutant lungs leads to increased cell proliferation and up-regulation of the expression of Fgf10, Bmp4, Shh, and Ptc. On the other hand, hyperactive Wnt signaling changes the developmental potential of embryonic lung endoderm to express markers of gut endoderm differentiation (151).
DICKKOPF REVEALS FIBRONECTIN AS AN IMPORTANT MATRIX TARGET OF WNT SIGNALING IN LUNG MORPHOGENESIS
Our recent experiments show that early embryonic mouse lung organ cultures treated with Dickkopf (DKK1), a potent and specific diffusible inhibitor of Wnt action that is also endogenously secreted by the distal lung epithelium, display impaired branching, characterized by failed cleft formation and enlarged terminal buds. The DKK1-treated lung explants show reduced α-smooth muscle actin (α-SMA) expression and defects in the formation of the pulmonary vascular network. These defects coincide with a pattern of decreased Fibronectin (FN) deposition and Platelet derived growth factor-A (Pdgf-a) expression. All of the DKK1-induced morphogenetic defects can be recapitulated by inhibition of FN with an antifibronectin antibody and conversely can be rescued by addition of exogenous FN (149). This points out the importance of correct orientation of the extracellular matrix in response to growth factor signaling. It also suggests that Fibronectin is a downstream target of Wnt signaling.
FUNCTIONAL INTEGRATION OF KEY GROWTH FACTOR SIGNALING PATHWAYS IN LUNG BUD OUTGROWTH, BUD ARREST, AND BUD BRANCHING
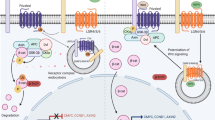
Our current concepts are diagrammed in Figure 1. We postulate that the dynamically changing relative activity of SHH, FGF10, and mSPRY2 may impart automaticity to the branching process. SHH is high and FGF10 is correspondingly low where branching is not supposed to take place. In contrast, SHH is suppressed locally by PTC and HIP, so that FGF10 is therefore high where a branch is supposed to occur. FGF10 in turn dynamically induces its inhibitor mSpry2 as branches lengthen. Thus, the net relative activities between SHH, FGF10, and mSPRY2 may determine FGF signal strength in the epithelium and hence the relative rate of bud outgrowth rate at a given point and hence interbranch length. As a bud begins to elongate toward a mesenchymal source of FGF10, mSpry2 begins to be expressed in the distal tip. During subsequent elongation, Fgf10 continues to be expressed in the distal mesenchyme and the level of mSpry2 gradually increases as the bud lengthens. When the bud finally approaches the pleura, the Fgf10 expression domain adjacent to the distal tip appears to thin out and some of it appears to be pushed laterally to lie between adjacent branch tips. At the time, mSpry2 expression in the distal tip is at its highest level, perhaps mediating bud outgrowth arrest. A tip-splitting event then occurs. Of note is that mSpry2 expression is extinguished between the daughter bud tips, but continues to be expressed within the tips of the daughter bud epithelia. This cycle of interaction is then repeated during subsequent branching events. BMP4 and VEGF signaling, as discussed above, appear to play necessary but complementary roles to accelerate this overall process.

The figure models the functional integration of key growth factor signaling pathways in lung bud outgrowth, bud arrest, and bud branching. Panel A depicts the function of FGF10 to stimulate bud outgrowth. Fgf10 is expressed in the distal mesenchyme so that a decreasing gradient of FGF10 acts to stimulate chemotaxis of the bud tip toward the subpleural source of FGF10. Heparan sulfation is also important for FGF function. Panel B depicts the function of BMP4 to stimulate lung branch tip outgrowth together with FGF10. FGF10 is shown stimulating BMP4 expression, whereas the ligand binding proteins Gremlin (GRE) and Chordin (CHO) exert negative modulation on BMP4. Panel C depicts the functional interaction of SHH and Hip with FGF10. SHH inhibits Fgf10 expression away from the branch tip. However at the branch tip, Hip inhibits SHH, releasing the SHH mediated inhibition of Fgf10 expression. Panel D superimposes the functional integration of Fgf10, Bmp4, and Shh to mediate the delicate balance between chemotaxis and proliferation leading to bud induction versus inhibition of bud outgrowth. Panel E depicts the events that may determine interbranch length by leading to arrest of bud outgrowth. FGF10 induces SPRY2, which in turn inhibits epithelial outgrowth. Meanwhile, in more proximal regions suppression of branching is mediated by SHH, which inhibits Fgf10 expression outside the peripheral mesenchyme. Panel F depicts a potential mechanism for bud tip splitting in which WNT signaling drives Fibronectin (FN) depostion between the branch tips, leading to epithelial cleft formation. Meanwhile, Dickkopf (DKK1) inhibits Wnt signaling away from the cleft, leading to lower levels of FN deposition where clefting does not occur.
MOLECULAR ORIGIN OF THE EPITHELIAL TIP-SPLITTING EVENT
Because tip-splitting events occur widely in nature where genes are not present, in such processes as viscous fingering, electrolysis of metals, river deltas, and oil field deposits, it is not clear whether physical forces are primary or secondary to growth factors in lung development (152). Certainly, physical force can modulate the rate of branching, inasmuch as tracheal ligation, which doubles the intraluminal pressure in embryonic lung, significantly accelerates branching (153). However, the numerous null mutants now available show that growth factors such as FGF10 and SHH are essential and these null mutations can abrogate the influence of increased force. FGF10 clearly is capable of inducing a strong chemotactic response of denuded lung epithelial bud tips toward and indeed to engulf an FGF10 soaked bead. Moreover, terminal buds appear to migrate into and toward the FGF10 epitopes within the embryonic lung mesenchyme. However, one of many puzzles has been why Spry2 expression, which is controlled by FGF10, is focally extinguished in a small stretch of epithelium right in the cleft between two new daughter buds. Another plausible explanation for tip splitting is the “rock in the stream” hypothesis, wherein a solid bar of something in the mesenchyme acts like a rock to divide the flowing stream of epithelium into two as it chemotaxes toward FGF10. Fibronectin and myofibroblasts have been suggested as candidates to play the role of the rock. Certainly our new data on Wnt signaling, using DKK as a means to abrogate the canonical Wnt pathway, suggest that Fibronectin deposition is indeed a good candidate to function as the rock in the stream, whereas Wnt signaling appears to control its deposition. Netrins, deposited within the extracellular matrix, may also play a role in regulating epithelial behavior in response to FGF signaling during branching morphogenesis in the embryonic lung (154).
POSTNATAL LUNG DEVELOPMENT
Many of the same genes that are responsible for lung organogenesis also play key roles in postnatal lung development, although many are also silent by that time. Null mutation studies have revealed essential roles for PDGF-A chain and for FGFR3 and FGFR4 as well as elastin in the induction of alveolar ridges and the correct orientation of elastic fibers in the postnatal lung. This is interesting because elastin expression by myofibroblasts, in response to PDGF and FGF signaling, has been adduced as a critical event during the alveolar phase of lung development. Because VEGF signaling by the epithelium to the endothelium is also critical for normal alveolarization, it is interesting to speculate that hydraulic force within the capillary vasculature may also be important. Certainly, abrogation of VEGF signaling by inducible misexpression of a dominant negative VEGF receptor under the control of the Sp-C promoter abrogates alveolarization as well as peripheral capillary morphogenesis, underscoring that epithelial to endothelial crosstalk is also an important mechanism in lung alveolar morphogenesis. After delivery, particularly premature delivery, exposure to endotoxin, oxygen, and/or barotrauma, with the resulting induction of cytokines including excessive amounts of TGF-β activity, adversely affects alveolarization and hence may induce the human pathobiological condition termed BPD or infantile chronic lung disease.
Abbreviations
- BMP:
-
bone morphogenetic protein
- DKK:
-
Dickkopf
- EGF (R):
-
epidermal growth factor (receptor)
- ERK:
-
extracellular regulated kinase
- FGF (R):
-
fibroblast growth factor (receptor)
- FN:
-
fibronectin
- LRP:
-
lipoprotein receptor-related proteins
- MAP:
-
membrane-associated protein
- PDGF:
-
platelet-derived growth factor
- RAR:
-
retinoic acid receptor
- sFRP:
-
secreted Frizzled-related protein
- SHH:
-
Sonic hedgehog
- Sp-C:
-
surfactant protein C
- TGF-α (β):
-
transforming growth factor alpha (beta)
- VEGF (R):
-
vascular endothelial growth factor (receptor)
References
Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV 2000 The molecular basis of lung morphogenesis. Mech Dev 92: 55–81
Affolter M, Bellusci S, Itoh B, Shilo B, Thiery JP, Werb Z 2003 Tube or not tube: remodeling epithelial tissues via branching morphogenesis. Dev Cell 4: 11–18
Rudnick D 1933 Developmental capacities of the chick lung in chorioallantoic grafts. J Exp Zool 66: 125–153
Alescio T, Cassini A 1962 Induction in vitro of tracheal buds by pulmonary mesenchyme grafted on tracheal epithelium. J Exp Zool 150: 83–94
Taderara JV 1967 Control of lung differentiation in vitro. Dev Biol 16: 489–512
Spooner BS, Wessells NK 1970 Mammalian lung development: interactions in primordium formation and bronchial morphogenesis. J Exp Zool 175: 445–454
Wessells NK 1970 Mammalian lung development: interactions in formation and morphogenesis of tracheal buds. J Exp Zool 175: 455–466
Shannon JM 1994 Induction of alveolar type II cell differentiation in fetal tracheal epithelium by grafted distal lung mesenchyme. Dev Biol 166: 600–614
Shannon JM, Nielsen LD, Gebb SA, Randell SH 1998 Mesenchyme specifies epithelial differentiation in reciprocal recombinants of embryonic lung and trachea. Dev Dyn 212: 482–494
Shannon JM, Hyatt BA 2004 Epithelial-mesenchymal interactions in the developing lung. Annu Rev Physiol 66: 625–645
Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L 1994 Targeted expression of a dominant negative FGF receptor blocks branching morphogenesis and epithelial differentiation of mouse lung. EMBO J 13: 3296–3301
Celli G, LaRochelle WJ, Mackem S, Sharp R, Merlino G 1998 Soluble dominant-negative receptor uncovers essential roles for fibroblast growth factors in multi-organ induction and patterning. EMBO J 17: 1642–1655
Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS 1998 Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev 12: 3156–3161
Park WY, Miranda B, Lebeche D, Hashimoto G, Cardoso WV 1998 FGF-10 is a chemotactic factor for distal epithelial buds during development. Dev Biol 201: 125–134
Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S 1999 Fgf10 is essential for limb and lung formation. Nat Genet 21: 138–141
Ang SL, Rossant J 1994 HNF-3 beta is essential or node and notochord formation in mouse development. Cell 78: 561–574
Chisaka O, Capecchi MR 1991 Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene hox-1.5. Nature 350: 473–479
Manley NR, Capecchi MR 1995 The role of Hoxa-3 in mouse thymus and thyroid development. Development 121: 1989–2003
Aubin J, Lemieux M, Tremblay M, Berard J, Jeannotte L 1997 Early postnatal lethality in Hoxa-5 mutant mice is attributable to respiratory tract defects. Dev Biol 192: 432–445
Kinkead R, LeBlanc M, Gulemetova R, Lalancette-Hebert M, Lemieux M, Mandeville I, Jeannotte L 2004 Respiratory adaptations to lung morphological defects in adult mice lacking Hoxa5 gene function. Pediatr Res 56: 553–562
Chytil F 1985 Vitamin A and lung development. Pediatr Pulmonol 1( 3 suppl): S115–S117
Mendelsohn C, Lohnes D, Decimo D, Lufkin T, LeMeur M, Chambon P, Mark M 1994 Function of the retinoic acid receptors (RARs) during development (II). Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 120: 2749–2771
Motoyama J, Liu J, Mo R, Ding Q, Post M, Hui CC 1998 Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat Genet 20: 54–57
Desai TJ, Malpel S, Flentke GR, Smith SM, Cardoso WV 2004 Retinoic acid selectively regulates Fgf10 expression and maintains cell identity in the prospective lung field of the developing foregut. Dev Biol 273: 402–415
Perl AK, Wert SE, Nagy A, Lobe CG, Whitsett JA 2002 Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc Natl Acad Sci U S A 99: 10482–10487
Supp DM, Witte DP, Potter SS, Brueckner M 1997 Mutation of an axonemal dynein affects left-right symmetry in inversus viscerum mice. Nature 389: 963–966
Lin CR, Kioussi C, O'Connell S, Briata P, Szeto D, Liu F, Izpisua-Belmonte JC, Rosenfeld MG 1999 Pitx2 regulates lung asymmetry, cardiac positioning and pituitary and tooth morphogenesis. Nature 401: 279–282
Kimura S, Hara Y, Pineau T, Fernandez-Salguero P, Fox CH, Ward JM, Gonzalez FJ 1996 The T/ebp null mouse: thyroid-specific enhancer-binding protein is essential for the organogenesis of the thyroid, lung, ventral forebrain, and pituitary. Genes Dev 10: 60–69
Minoo P, Hamdan H, Bu D, Warburton D, Stepanik P, deLemos R 1995 TTF-1 regulates lung epithelial morphogenesis. Dev Biol 172: 694–698
Litingtung Y, Lei L, Westphal H, Chiang C 1998 Sonic hedgehog is essential to foregut development. Nat Genet 20: 58–61
Pepicelli CV, Lewis PM, McMahon AP 1998 Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol 8: 1083–1086
Mahlapuu M, Enerback S, Carlsson P 2001 Haploinsufficiency of the forkhead gene Fox f1, a target for sonic hedgehog signaling, causes lung and foregut malformations. Development 128: 2397–2406
Miller LA, Wert SE, Clark JC, Xu Y, Perl AK, Whitsett JA 2004 Role of Sonic hedgehog in patterning of tracheal-bronchial cartilage and the peripheral lung. Dev Dyn 231: 57–71
Li Y, Zhang H, Choi SC, Litingtung Y, Chiang C 2004 Sonic hedgehog signaling regulates Gli3 processing, mesenchyme proliferation, and differentiation during mouse lung organogenesis. Dev Biol 270: 214–231
Crisera CA, Maldonado TS, Longaker MT, Gittes GK 2000 Defective fibroblast growth factor signaling allows for nonbranching growth of the respiratory-derived fistula tract in esophageal atresia with tracheoesophageal fistula. J Pediatr Surg 35: 1421–1425
Kim PC, Mo R, Hui Cc C 2001 Murine models of VACTERL syndrome: role of sonic hedgehog signaling pathway. J Pediatr Surg 36: 381–384
Spilde T, Bhatia A, Ostlie D, Marosky J, Holcomb G III, Synder CL, Gittes GK 2003 A role for sonic hedgehog signaling in the pathogenesis of human tracheoesophageal fistula. J Pediatr Surg 38: 465–468
Spilde TL, Bhatia AM, Mehta SS, Hembree MJ, Preuett BL, Ostlie DJ, Prasadan K, Li Z, Synder CL, Gittes GK 2004 Aberrant fibroblast growth factor receptor 2 signalling in esophageal atresia in tracheoesophageal fistula. J Pediatr Surg 39: 537–539
Cardoso WV 2000 Lung morphogenesis revisted: old facts, current ideas. Dev Dyn 219: 121–130
Hogan BL 1999 Morphogenesis. Cell 96: 225–233
Warburton D, Bellusci S 2004 The molecular genetics of lung morphogenesis and injury repair. Paediatr Respir Rev 5( suppl A): S283–S287
Michos O, Panman L, Vintersten K, Beier K, Zeller R, Zuniga A 2004 Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development 131: 3401–3410
Weaver M, Yingling JM, Dunn NR, Bellusci S, Hogan BL 1999 Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development 126: 4005–4015
Bellusci S, Henderson R, Winnier G, Oikawa T, Hogan BL 1996 Evidence from normal expression and targeted misexpression that bone morphogenetic protein-4 (Bmp-4) plays a role in mouse embryonic lung morphogenesis. Development 122: 1693–1702
Takahashi H, Ikeda T 1996 Transcripts for two members of the transforming growth factor superfamily BMP-3 and BMP-7 are expressed in developing rat embryos. Dev Dyn 207: 439–449
King JA, Marker PC, Seung KJ, Kingsley DM 1994 BMP5 and the molecular, skeletal, and soft-tissue alterations in short ear mice. Dev Biol 166: 112–122
Chen CK, Kuhnlein RP, Eulenberg KG, Vincent S, Affolter M, Schuh R 1998 The transcription factors KNIRPS and KNIRPS RELATED control cell migration and branch morphogenesis during Drosophila tracheal development. Development 125: 4959–4968
Ribeiro C, Ebner A, Affolter M 2002 In vivo imaging reveals different cellular functions for FGF and Dpp signaling in tracheal branching morphogenesis. Dev Cell 2: 677–683
Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA III, Loyd JE, Nichols WC, Trembath RC 2000 Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet 26: 81–84
Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R 1995 Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature 376: 337–341
Miettinen PJ, Warburton D, Bu D, Zhao JS, Berger JE, Minoo P, Koivisto T, Allen L, Dobbs L, Werb Z, Derynck R 1997 Impaired lung branching morphogenesis in the absence of functional EGF receptor. Dev Biol 186: 224–236
Goldin GV, Opperman LA 1980 Induction of supernumerary tracheal buds and the stimulation of DNA synthesis in the embryonic chick lung and trachea by epidermal growth factor. J Embryol Exp Morphol 60: 235–243
Warburton D, Seth R, Shum L, Horcher PG, Hall FL, Werb Z, Slavkin HC 1992 Epigenetic role of epidermal growth factor expression and signalling in embryonic mouse lung morphogenesis. Dev Biol 149: 123–133
Seth R, Shum L, Wu F, Wuenschell C, Hall FL, Slavkin HC, Warburton D 1993 Role of epidermal growth factor expression in early mouse embryo lung branching morphogenesis in culture: antisense oligodeoxynucleotide inhibitory strategy. Dev Biol 158: 555–559
Zhao J, Chen H, Wang YL, Warburton D 2001 Abrogation of tumor necrosis factor-alpha converting enzyme inhibits embryonic lung morphogenesis in culture. Int J Dev Biol 45: 623–631
Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL 1997 Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development 124: 4867–4878
Weaver M, Dunn NR, Hogan BL 2000 BMP4 and Fgf10 play opposing roles during lung bud morphogenesis. Development 127: 2695–2704
Tefft JD, Lee M, Smith S, Lienwand M, Zhao J, Bringas P Jr, Crowe DL, Warburton D 1999 Conserved function of mSpry-2, a murine homolog of Drosophila sprouty, which negatively modulates respiratory organogenesis. Curr Biol 9: 219–222
Lebeche D, Malpel S, Cardoso WV 1999 Fibroblast growth factor interactions in the developing lung. Mech Dev 86: 125–136
Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier E, Gao G, Goldfarb M 1995 Receptor specificity of the fibroblast growth factor family. J Biol Chem 271: 15292–15297
Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L 1994 Targeted expression of a dominant negative FGF receptor block branching morphogenesis and epithelial differentiation of the mouse lung. EMBO J 13: 3296–3301
Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P 1998 Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci U S A 95: 5082–5087
Xu X, Weinstein M, Li C, Naski M, Cohen RI, Ornitz DM, Leder P, Deng C 1998 Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development 125: 753–765
De Moerlooze L, Spencer-Dene B, Revest J, Hajihosseini M, RosewelI I, Dickson C 2000 An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signaling during mouse organogenesis. Development 127: 482–492
Revest JM, Spencer-Dene B, Kerr K, De Moerlooze L, Rosewell I, Dickson C 2001 Fibroblast growth factor receptor 2-IIIb acts upstream of Shh and Fgf4 and is required for limb bud maintenance but not for the induction of Fgf8, Fgf10, Msx1, or Bmp4. Dev Biol 231: 47–62
Hokuto I, Perl AK, Whitsett JA 2003 Prenatal, but not postnatal, inhibition of fibroblast growth factor receptor signaling causes emphysema. J Biol Chem 278: 415–421
Ornitz DM, Itoh N 2001 Fibroblast growth factors. Genome Biol 2: REVIEWS3005
Chailley-Heu B, Boucherat O, Barlier-Mur AM, Bourbon JR 2004 FGF18 is up-regulated in the postnatal rat lung and enhances elastogenesis in myofibroblasts. Am J Physiol Lung Cell Mol Physiol 288: L43–L51
Park WY, Miranda B, Lebeche D, Hashimoto G, Cardoso WV 1998 FGF-10 is a chemotactic factor for distal epithelial buds during development. Dev Biol 201: 125–134
Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS 1998 Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev 12: 3156–3161
Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S 1999 Fgf10 is essential for limb and lung formation. Nat Genet 21: 138–141
Ohuchi H, Hori Y, Yamasaki M, Harada H, Sekine K, Kato S, Itoh N 2000 FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse organ development. Biochem Biophys Res Commun 277: 643–649
Mailleux AA, Tefft D, Ndiaye D, Itoh N, Thiery JP, Warburton D, Bellusci S 2001 Evidence that SPROUTY2 functions as an inhibitor of mouse embryonic lung growth and morphogenesis. Mech Dev 102: 82–94
Sutherland D, Samakovlis C, Krasnow MA 1996 branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell 87: 1091–1101
Ahmad SM, Baker BS 2002 Sex-specific deployment of FGF signaling in Drosophila recruits mesodermal cells into the male genital imaginal disc. Cell 109: 651–661
Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA 1998 sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell 92: 253–263
Casci T, Vinos J, Freeman M 1999 Sprouty, an intracellular inhibitor of Ras signaling. Cell 96: 655–665
Tefft D, Lee M, Smith S, Crowe DL, Bellusci S, Warburton D 2002 mSprouty2 inhibits FGF10-activated MAP kinase by different binding to upstream target proteins. Am J Physiol Lung Cell Mol Physiol 283: L700–L706
Hadari YR, Kouhara H, Lax I, Schlessinger J 1998 Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol Cell Biol 18: 3966–3973
Schaeffer HJ, Weber MJ 1999 Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol Cell Biol 19: 2435–2444
Minowada G, Jarvis LA, Chi CL, Neubuser A, Sun X, Hacohen N, Krasnow MA, Martin GR 1999 Vertebrate Sprouty genes are induced by FGF signaling and cause chondrodysplasia when overexpressed. Development 126: 4465–4475
Lee MK, Zhao J, Smith SM, Tefft JD, Bringas P, Hwang C, Warburton D 1998 The Shc66 and 46 kD isoforms are differentially downregulated at parturition in the fetal mouse lung. Pediatr Res 44: 850–859
Lim J, Wong ES, Ong SH, Yusoff P, Low BC, Guy GR 2000 Sprouty proteins are targeted to membrane ruffles upon growth factor receptor tyrosine kinase activation. Identification of a novel translocation domain. J Biol Chem 275: 32837–32845
Yigzaw Y, Cartin L, Pierre S, Scholich K, Patel TB 2001 The C terminus of sprouty is important for modulation of cellular migration and proliferation. J Biol Chem 276: 22742–22747
Wong ES, Lim J, Low BC, Chen Q, Guy GR 2001 Evidence for direct interaction between Sprouty and Cbl. J Biol Chem 276: 5866–5875
Hashimoto S, Nakano H, Singh G, Katyal S 2002 Expression of Spred and Sprouty in developing rat lung. Mech Dev 119( suppl 1): S303–S309
Miyoshi K, Wakioka T, Nishinakamura H, Kamio M, Yang L, Inoue M, Hasegawa M, Yonemitsu Y, Komiya S, Yoshimura A 2004 The Sprouty-related protein, Spred, inhibits cell motility, metastasis, and Rho-mediated actin reorganization. Oncogene 23: 5567–5576
Nobuhisa I, Kato R, Inoue H, Takizawa M, Okita K, Yoshimura A, Taga T 2004 Spred-2 suppresses aorta-gonad-mesonephros hematopoiesis by inhibiting MAP kinase activation. J Exp Med 199: 737–742
Kato R, Nonami A, Taketomi T, Wakioka T, Kuroiwa A, Matsuda Y, Yoshimura A 2003 Molecular cloning of mammalian Spred-3 which suppresses tyrosine kinase-mediated Erk activation. Biochem Biophys Res Commun 302: 767–772
Smith CI, Webster EH, Nathanson MA, Searls RI, Hilfer SR 1990 Altered patterns of proteoglycan deposition during maturation of the fetal mouse lung. Cell Differ Dev 32: 83–96
Spooner BS, Bassett KE, Spooner BS Jr 1993 Embryonic lung morphogenesis in organ culture: experimental evidence of a proteoglycan function in the extracellular matrix. Trans Kans Acad Sci 96: 46–55
Toriyama K, Muramatsu H, Hoshino T, Torii S, Muramatsu T 1997 Evaluation of heparin-binding growth factors in rescuing morphogenesis of heparitinase-treated mouse embryonic lung explants. Differentiation 61: 161–167
Izvolsky KI, Zhong L, Wei L, Yu Q, Nugent MA, Cardoso WV 2003 Heparan sulfates expressed in the distal lung are required for Fgf10 binding to the epithelium and for airway branching. Am J Physiol Lung Cell Mol Physiol 285: L838–L846
Izvolsky KI, Shoykhet D, Yang Y, Yu Q, Nugent MA, Cardoso WV 2003 Heparan sulfate-FGF10 interactions during lung morphogenesis. Dev Biol 258: 185–200
Shannon JM, McCormick-Shannon K, Burhans MS, Shangguan X, Srivastava K, Hyatt BA 2003 Chondroitin sulfate proteoglycans are required for lung growth and morphogenesis in vitro. Am J Physiol Lung Cell Mol Physiol 285: L1323–L1336
van Tuyl M, Post M 2000 From fruitflies to mammals: mechanisms of signaling via the Sonic hedgehog pathway in lung development. Respir Res 1: 30–35
Pepicelli CV, Lewis PM, McMahon AP 1998 Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol 8: 1083–1086
Bellusci S, Furuta Y, Rush MG, Henderson R, Winnier G, Hogan BL 1997 Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development 124: 53–63
Chuang PT, McMahon AP 1999 Vertebrate Hedgehog signaling modulated by induction of a Hedgehog-binding protein. Nature 397: 617–621
Massague J 1998 TGF-beta signal transduction. Ann Rev Biochem 67: 753–791
Pelton RW, Saxena B, Jones M, Moses HL, Gold LI 1991 Immunohistochemical localization of TGF beta 1, TGF beta 2, a TGF beta 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol 115: 1091–1105
Millan FA, Denhez F, Kondaiah P, Akhurst RJ 1991 Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development 111: 131–143
Schmid P, Cox D, Bilbe G, Maier R, McMaster GK 1991 Differential expression of TGF beta 1, beta 2 and beta 3 genes during mouse embryogenesis. Development 111: 117–130
McLennan IS, Poussart Y, Koishi K 2000 Development of skeletal muscles in transforming growth factor-beta 1 (TGF-beta1) null-mutant mice. Dev Dyn 217: 250–256
Bragg AD, Moses HL, Serra R 2001 Signaling to the epithelium is not sufficient to mediate all of the effects of transforming growth factor beta and bone morphogenetic protein 4 on murine embryonic lung development. Mech Dev 109: 13–26
Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, Speer C, Poelmann RE, Gittenberger-de Groot AC 2001 Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation 103: 2745–2752
Kaartinen V, Voncken W, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J 1995 Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet 11: 415–421
Buckley S, Bui KC, Hussain M, Warburton D 1996 Dynamics of TGF-beta 3 peptide activity during rat alveolar epithelial cell proliferative recoevery from acute hyperoxia. Am J Physiol 271: L54–L60
Zhao J, Sime PJ, Bringas P Jr, Tefft JD, Buckley S, Bu D, Gauldie J, Warburton D 1999 Spatial-specific TGF-beta1 adenoviral expression determines morphogenetic phenotypes in embryonic mouse lung. Eur J Cell Biol 78: 715–725
Zhou L, Dey CR, Wert SE, Whitsett JA 1996 Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-beta 1 chimeric gene. Dev Biol 175: 227–228
Serra R, Pelton RW, Moses HL 1994 TGF beta 1 inhibits branching morphogenesis and N-myc expression in lung bud organ cultures. Development 120: 2153–2161
Zhao J, Bu D, Lee M, Slavkin HC, Hall FL, Warburton D 1996 Abrogation of transforming growth factor-beta type II receptor stimulates embryonic mouse lung branching morphogenesis in culture. Dev Biol 180: 242–257
Zhao J, Sime PJ, Bringas P Jr, Gauldie J, Warburton D 1999 Adenovirus-mediated decorin gene transfer prevents TGF-beta1-induced inhibition of lung morphogenesis. Am J Physiol 277: L412–L422
Zhao J, Crowe DL, Castillo C, Wuenschell C, Chai Y, Warburton D 2000 Smad7 is a TGF-beta inducible attenuator of Smad2/3-mediated inhibition of embryonic lung morphogenesis. Mech Dev 93: 71–81
Zhao J, Lee M, Smith S, Warburton D 1998 Abrogation of Smad3 and Smad2 or of Smad 4 gene expression positively regulates murine embryonic lung branching mophogenesis in culture. Dev Biol 194: 182–195
Zhao J, Shi W, Chen H, Warburton D 2000 Smad7 and Smad6 differentially modulates transforming growth factor beta-induced inhibition of embryonic lung morphogenesis. J Biol Chem 275: 23992–23997
Gauldie J, Galt T, Bonniaud P, Robbins C, Kelly M, Warburton D 2003 Transfer of the active form of transforming growth factor-beta 1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am J Pathol 163: 2575–2584
Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J 1997 Adenovector-mediated gene transfer of active transforming growth factor-beta 1 induces prolonged severe fibrosis in rat lung. J Clin Invest 100: 768–776
Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr, Datto MB, Frederick JP, Wang XF, Warburton D 2002 Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol 282: L585–L593
Gebb SA, Shannon JM 2002 Tissue interactions mediate early events in pulmonary vasculogenesis. Dev Dyn 217: 159–169
Miquerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A 1999 Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol 212: 307–322
Acarregui MJ, Penisten SC, Goss KL, Ramirez K, Snyder JM 1999 Vascular endothelial growth factor gene expression in human fetal lung in vitro. Am J Respir Cell Mol Biol 20: 14–23
Maede S, Suzuki S, Suzuki T, Endo M, Moriya T, Chida M, Kondo T, Sasano H 2002 Analysis of intrapulmonary vessels and epithelial-endothelial interactions in the human developing lung. Lab Invest 82: 293–301
Brown KR, England KM, Goss KL, Snyder JM, Acarregui MJ 2001 VEGF induces airway epithelial cell proliferation in human fetal lung in vitro. Am J Physiol Lung Cell Mol Physiol 281: L1001–L1010
Kasahara Y, Tuder RM, Taraseviciene-Stewart L, Le Cras TD, Abman S, Hirth PK, Waltenberger J, Voelkel NF 2000 Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest 106: 1311–1319
Zeng X, Wert SE, Federici R, Peters KG, Whitsett JA 1998 VEGF enhances pulmonary vasculogenesis and disrupts lung morphogenesis in vivo. Dev Dyn 211: 215–227
Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N 1999 VEGF is required for growth and survival in neonatal mice. Development 126: 1149–1159
Corne J, Chupp G, Lee CG, Homer RJ, Zhu Z, Chen Q, Ma B, Du Y, Roux F, McArdle J, Waxman AB, Elias JA 2002 IL-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J Clin Invest 106: 783–791
Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P 2002 Loss of HIF-2alpha and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8: 702–710
Farnebo F, Piehl F, Lagercrantz J 1999 Restricted expression pattern of vegf-d in the adult and fetal mouse: high expression in the embryonic lung. Biochem Biophys Res Commun 257: 891–894
Makinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, Kauppinen R, Jackson DG, Kubo H, Nishikawa S, Yla-Herttuala S, Alitalo K 2001 Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med 7: 199–205
Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen MG, Stacker SA, Alitalo K 2001 Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J 20: 1223–1231
Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi JH, Claesson-Welsh L, Alitalo K 1998 Vascular endothelial growth factor C induces angiogenesis in vivo. Proc Natl Acad Sci U S A 95: 14389–14394
Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC 2000 An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 407: 535–538
Tamai K, Semenov M, Kato Y, Spokony R, Liu C, Katsuyama Y, Hess F, Saint-Jeannet JP, He X 2000 LDL-receptor-related proteins in Wnt signal transduction. Nature 407: 530–535
Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S 2000 arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature 407: 527–530
Heisenberg CP, Tada M, Rauch GJ, Saude L, Concha ML, Geisler R, Stemple DL, Smith JC, Wilson SW 2000 Silberblick/Wnt11 mediates convergent extension movements during zebrafish gastrulation. Nature 405: 76–81
Torres MA, Yang-Snyder JA, Purcell SM, DeMarais AA, McGrew LL, Moon RT 1996 Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early Xenopus development. J Cell Biol 133: 1123–1137
Kuhl M, Geis K, Sheldahl LC, Pukrop T, Moon RT, Wedlich D 2001 Antagonistic regulation of convergent extension movements in Xenopus by Wnt/beta-catenin and Wnt/Ca2+ signaling. Mech Dev 106: 61–76
Ishitani T, Kishida S, Hyodo-Miura J, Ueno N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J, Matsumoto K 2003 The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol Cell Biol 23: 131–139
Mao B, Wu W, Davidson G, Marhold J, Li M, Mechler BM, Delius H, Hoppe D, Stannek P, Walter C, Glinka A, Niehrs C 2002 Kremen proteins are Dickkopf receptors that regulate Wnt/beta-catenin signalling. Nature 417: 664–667
Kawano Y, Kypta R 2003 Secreted antagonists of the Wnt signaling pathway. J Cell Sci 116: 2627–2634
Mao B, Niehrs C 2003 Kremen2 modulates Dickkopf2 activity during Wnt/LRP6 signaling. Gene 302: 179–183
Hoang BH, Kubo T, Healey JH, Yang R, Nathan SS, Kolb EA, Mazza B, Meyers PA, Gorlick R 2004 Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma cells by modulating the Wnt-beta-catenin pathway. Cancer Res 64: 2734–2739
Tebar M, Destree O, de Vree WJ, Ten Have-Opbroek AA 2001 Expression of Tcf/Lef and sFrp and localization of beta-catenin the developing mouse lung. Mech Dev 109: 437–440
DasGupta R, Fuchs E 1999 Multiple roles for activated LEF/TCF transcription complexes during hair follicle development and differentiation. Development 126: 4557–4568
Maretto S, Cordenonsi M, Dupont S, Braghetta P, Broccoli V, Hassan AB, Volpin D, Bressan GM, Piccolo S 2003 Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A 100: 3299–3304
Derynck R, Zhang YE 2003 Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature 425: 577–584
De Langhe S, Sala FG, Del Moral P-M, Fairbanks TJ, Yamada KM, Warburton D, Burns RC, Bellusci S 2005 Dickopf-1 (DKK1) reveals that fibronectin is a major target of Wnt signaling in branching morphogenesis of the mouse embryonic lung. Dev Biol 277: 316–331
Mucenski ML, Wert SE, Nation JM, Loudy DE, Huelsken J, Birchmeier W, Morrisey EE, Whitsett JA 2003 Beta-Catenin is required for specification of proximal/distal cell fate during lung morphogenesis. J Biol Chem 278: 40231–40238
Okubo T, Hogan BL 2004 Hyperactive Wnt signaling changes the developmental potential of embryonic lung endoderm. J Biol 3: 11
Fleury V 1999 Two symmetries linking biological and physical branching morphogenesis. In: Fleury V, Gouyet J-F, Leonetti M (eds) Branching in Nature: Dynamics and Morphogenesis of Branching Structures, from Cell to River Networks. Springer, Paris
Blewett CJ, Zgleszewski SE, Chinoy MR, Krummel TM, Cilley RE 1996 Bronchial ligation enhances murine fetal lung development in whole-organ culture. J Pediatr Surg 31: 869–877
Liu Y, Stein E, Oliver T, Li Y, Brunken WJ, Koch M, Tessier-Lavigne M, Hogan BL 2004 Novel role for Netrins in regulating epithelial behavior during lung branching morphogenesis. Curr Biol 14: 897–905
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by grants HL44060, HL44977, HL60231, HL75773, HL73014 to D.W.; grants ALA 9PB-0082, HL074832, HL74862, HL74862 to S.B.; CNRS to V.F.; and grants HL68597 and HL61286 to W.S.
Rights and permissions
About this article
Cite this article
Warburton, D., Bellusci, S., De Langhe, S. et al. Molecular Mechanisms of Early Lung Specification and Branching Morphogenesis. Pediatr Res 57, 26–37 (2005). https://doi.org/10.1203/01.PDR.0000159570.01327.ED
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000159570.01327.ED
This article is cited by
-
State of the art on lung organoids in mammals
Veterinary Research (2021)
-
Complete lung agenesis caused by complex genomic rearrangements with neo-TAD formation at the SHH locus
Human Genetics (2021)
-
In smokers, Sonic hedgehog modulates pulmonary endothelial function through vascular endothelial growth factor
Respiratory Research (2017)
-
Looking ahead: where to next for animal models of bronchopulmonary dysplasia?
Cell and Tissue Research (2017)
-
Expression of T-box transcription factors 2, 4 and 5 is decreased in the branching airway mesenchyme of nitrofen-induced hypoplastic lungs
Pediatric Surgery International (2017)
