
Abstract
Tuberous sclerosis complex (TSC) presents in the pediatric population with a constellation of benign tumors that affect the brain, heart, kidney, lung, and skin. No therapy has been shown to halt disease progression or to prevent its onset. The pathogenesis of TSC stems from the inactivation of one of the two TSC genes, TSC1 and TSC2. A key function of these genes is to regulate the mammalian target of rapamycin (mTOR) pathway in response to cellular energy and nutrient and growth factor availability. Consequently, TSC-related tumors exhibit uncontrolled activation of mTOR and its effectors. Previous work has shown that a specific mTOR inhibitor, rapamycin, effectively down-regulated mTOR activity in renal tumors of Eker rats that carry a germline Tsc2 mutation. Using this model, we investigated the effects of rapamycin on pituitary and renal tumors. We observed that rats with pituitary tumors had significantly shorter survival than those without pituitary pathology. Treatment with rapamycin effectively improved their clinical state and prolonged their survival. Rapamycin also resulted in a significant decrease in the size of the Tsc2-related renal tumors. In both types of pathology, tumor response was accompanied by down-regulation of ribosomal S6 kinase activity, reduction in cell size, and induction of apoptosis. Evidence for drug resistance was found in a small percentage of lesions after prolonged therapy. When rapamycin was given before onset of disease, subsequent development of macroscopic renal tumors was reduced, but no effect on the number of microscopic precursor lesions was found. We conclude that rapamycin-sensitive mTOR activity was critical to tumor progression in the Eker rat model, but rapamycin is unlikely to eradicate all disease as a result of the development of drug resistance. Our data also suggest the role of a rapamycin-insensitive pathway during tumor initiation.
Similar content being viewed by others


Main
Tuberous sclerosis complex (TSC) is one of the most common forms of neurocutaneous disorders (1). This autosomal dominant disease typically presents in the pediatric age group with neuropsychiatric signs and symptoms of epilepsy, mental retardation, and autism (2). Other diagnostic features include cardiac rhabdomyoma, facial angiofibroma, and retinal hamartomas. In later life, renal cysts, angiomyolipomas (AMLs), and occasional carcinomas contribute to TSC-related morbidity and mortality. Virtually unique to the postpubescent female individual with TSC is the development of lymphangioleiomyomatosis in the lungs, which may lead to restrictive pulmonary failure. The expression and severity of these clinical manifestations vary considerably, and current therapy is limited to symptomatic control.
The underlying pathology is also highly variable with respect to histology, anatomic location, and growth behavior. In the case of cardiac rhabdomyoma, such lesions typically regress spontaneously with age. In contrast, renal AMLs often progress insidiously, leading to an increased risk for hemorrhage as they enlarge. Despite these differences, the tumor-like lesions of TSC share underlying abnormalities in cell size, number, shape, and geographic location. As such, the pathology reflects defects in cellular control of growth, proliferation, migration, and differentiation.
The molecular mechanism of disease has been the subject of recent investigations, which began with the identification of the two genes TSC1 and TSC2 through positional cloning (3,4). Their function has been linked to several pathways, among which mammalian target of rapamycin (mTOR) signaling has been the most intensely studied. On the basis of genetic analyses in Drosophila, mutants of dTsc1 and dTsc2 result in aberrant cell size, which is mediated by the dAkt/Tor pathway (5). In mammalian cells, growth factor stimulation negatively regulates the TSC2 product tuberin through its phosphorylation by Akt at multiple sites, including Ser939 and Thr147 (6,7). Conversely, when Akt is inactive, tuberin's GTPase activating protein (GAP) activity promotes GTP hydrolysis of Rheb, a Ras-like G-protein, which in turn inhibits mTOR signaling (8–10). Downstream effectors of mTOR, including p70S6 K and 4E-binding protein 1 (4E-BP1)/eIF4E, are shown to modulate cell size and cell proliferation (11). There is also evidence to suggest that tuberin is sensitive to cellular energy balance through the direct influence of AMP kinase (12). Under conditions of low energy (i.e. increased AMP), active AMP kinase phosphorylates tuberin at S1345 and promotes its activity. This reduces cell size and prevents apoptosis caused by glucose deprivation. Collectively, the data suggest an important role of tuberin in regulating cellular response to energy and growth factor availability.
In TSC lesions such as AML, loss of heterozygosity at either one of the TSC1 or TSC2 loci is commonly found, and, consequently, some tumors lose expression of hamartin or tuberin (13,14). These findings support the notion that the pathogenesis of TSC follows the “two-hit” paradigm in which the target cells undergo biallelic inactivation of one of the TSC genes (15). Biochemically, the initiated cells sustain deregulated activation of the mTOR pathway, leading to increased cell growth and proliferation. These findings have been verified in various pathologies derived from TSC patients and associated animal models (16,17). These discoveries give rise to the hypothesis that inhibition of mTOR activity may have therapeutic value in TSC. Rapamycin is an antifungal macrolide antibiotic that possesses high specificity against mTOR through binding with the cellular protein FKBP12. The drug is widely used in renal transplant recipients as an immunosuppressive agent, but its effects in TSC patients are unknown.
We previously examined the short-term consequences of rapamycin in an animal model of TSC, the Eker rat (17). This strain of rat carries a spontaneously occurring germline mutation of the Tsc2 gene that functions as a null equivalent (18). Phenotypically, the Tsc2Ek/+ carriers develop renal epithelial tumors that are expressed in 100% of animals by the age of 12 mo. The cortical location of the tumors and their generous sizes make them ideal for preclinical evaluation of experimental therapies. As with the human disease, these kidney tumors undergo “two-hit” inactivation of the Tsc2 locus and show activation of the mTOR pathway. After acute exposure (3 d) to rapamycin, significant down-regulation of the mTOR effectors was observed along with evidence of tumor apoptosis and a decrease in proliferation index (17). However, the long-term effects of this drug on tumor progression and initiation have not been studied and are the subject of this investigation.
Extending the findings of our short-term study, we predict that rapamycin, when given over a protracted period, will induce tumor regression, prolong survival, and prevent tumor development. To test these hypotheses, we treated the Tsc2Ek/+ rats with rapamycin at different periods to evaluate its effects on tumor progression and initiation. Furthermore, we tested the ability of this drug to prolong survival of rats that are symptomatic from Tsc2-related pituitary tumors in the Eker rat.
METHODS
Chemicals and reagents.
The following antibodies were purchased from the respective suppliers: rabbit anti–PHAS-I (4E-BP1) from Zymed (San Francisco, CA), mouse anti-vascular endothelial growth factor (VEGF) from Research Diagnostics Inc. (Flanders, NJ), and mouse anti-actin from Sigma Chemical Co. (St. Louis, MO). The anti-gelsolin antibody was a gift from David Kwiatkowski (Brigham and Women's Hospital, Boston, MA). All other antibodies were purchased from Cell Signaling Technologies (Beverly, MA). Rapamycin was purchased from Calbiochem (La Jolla, CA). A stock solution was prepared by diluting 1 mg of rapamycin in 100 μL of DMSO. An In-Situ Cell Death Detection Kit (peroxidase) with a 3,3′-diaminobenzidine substrate was obtained from Roche Diagnostics (Indianapolis, IN). Secondary antibodies and chemiluminescence reagents were purchased from Amersham Pharmacia Biotech (Piscataway, NJ). The mouse and rabbit Elite ABC kits, 3,3′-diaminobenzidine, and Hematoxylin QS were purchased from Vector Laboratories (Burlingame, CA). Eosin was obtained from Richard Allen Scientific (Kalamazoo, MI).
Animals and treatments.
Rats that carry the Eker Tsc2 mutation and were bred in various genetic backgrounds, including Fischer 344 and Long Evans, were used in this study, as previously described (19). Animals were housed in the same room at an American Association for Laboratory Animal Science (AALAC)-accredited facility at the University of Washington. All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC). At the stated ages, animals were treated with rapamycin or vehicle as placebo, given intraperitoneally using dose regimens designed for the various experiments described. Animals were killed by CO2 inhalation, and the kidneys were harvested for biochemical and histologic analyses.
In the tumor prevention studies, 2-mo-old Eker carriers were exposed to rapamycin or placebo for 2 mo and killed at 8 mo of age. Kidneys were harvested for histologic evaluation as outlined below.
In the pituitary tumor experiments, animals with symptoms of lethargy, weight loss, and altered gait were chosen to receive rapamycin. Their body weight was monitored twice weekly. When the rats were killed, the abnormal pituitary glands were harvested for biochemical and histologic analyses.
Ultrasound.
Animals were anesthetized with a mixture of ketamine:xylazine (5:1.6). Each animal was given 0.9 mg/kg of the mixture in 1 mL of saline via an intraperitoneal injection. Kidneys were visualized using one of two ultrasound instruments, a Sonosite 180, L10-5 MHz transducer (Sonosite, Inc., Bothell, WA) or the Terason 128, 10L5 transducer (Burlington, MA). Both kidneys were examined for tumors. When a tumor was visualized, its diameter was measured in a cross-section as well as transverse view. Some of these measurements were compared with actual sizes as determined when the rats were killed.
Immunohistochemistry.
Kidneys, bisected sagittally on the renal pelvis, and pituitary tumors were fixed in formalin and paraffin embedded. Five-micrometer sections were deparaffinized, rehydrated, and washed with PBS. After antigen retrieval in 10 mM of sodium citrate (pH 6.0) and quenching of endogenous peroxidase activity with 1% H2O2, samples were blocked with 5% normal goat serum before incubation with primary antibodies overnight at 4°C. Negative controls were treated with 5% normal goat serum without primary antibody. Signals were processed according to the supplied protocol (Elite ABC Kit). Slides were counterstained with Hematoxylin QS, dehydrated, and mounted using Permount (Fischer Scientific, Santa Clara, CA). For the tumor prevention studies, kidneys were fixed in formalin, sliced sagittally along two planes on each side of the renal pelvis, embedded in paraffin blocks, and sectioned for histologic evaluation. Morphometric analysis was performed on each kidney by counting the number of lesions according to size (<10 cells, 10–100 cells, and >1 mm). For each animal, two sections of each kidney were stained with gelsolin and phospho-S6 antibodies to highlight the small precursor lesions. The total number of renal lesions was based on an average of 30 nonoverlapping low-power fields per animal.
Slides that were stained with hematoxylin and eosin were deparaffinized and rehydrated, then placed in a working solution of eosin (0.5% acetic acid, 25% eosin, and 74.5% of 80% ethanol) for 30 s, rinsed, and counterstained with Hematoxylin QS, dehydrated, and mounted using Permount. Transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed on pituitary tumor sections using the In-Situ Cell Death Detection kit according to the manufacturer's protocol.
Immunoblotting.
Renal tumors were dissected from unaffected kidney tissue. Similarly, pituitary tumors and adjacent CNS were procured for protein extraction. Tissues were homogenized in ice-cold radioimmunoprecipitation assay (RIPA) buffer [1% NP40, 1% sodium deoxycholate, 0.1% SDS, 0.15 M of NaCl, 10 mM of Tris (pH 7.2), 0.025 M of β-glycophosphate (pH 7.2), 2 mM of EDTA, and 50 mM of sodium fluoride] with protease and kinase inhibitors [0.05 mM of 4-(2-aminoethyl)benzenesulfonyl fluoride, 10 μg/mL of pepstatin, 1 mM of orthovanadate, 10 μg/mL of leupeptin, and 1 mM of microcystin LR]. Protein concentrations were determined using the BCA Protein Assay (Pierce, Rockford, IL). Equal amounts of protein were separated by SDS-PAGE, transferred to Immobilin-P membranes (Millipore, Bedford, MA), and blotted with antibodies according to the manufacturer's recommendations.
Statistics.
Kaplan-Meier estimates were used to calculate survival of the rats with and without pituitary tumors. T tests (two-tail) were used to compare tumor multiplicity. A p < 0.05 was considered significant.
RESULTS
Hemorrhage in pituitary tumors contributes to premature deaths in the Tsc2Ek/+ rats.
A previous study reported that 58% of adult Tsc2Ek/+ rats developed pituitary adenomas (20). However, the clinical significance of these lesions has not been examined in detail. Here, we analyzed a cohort of 70 Tsc2Ek/+ rats up to 16 mo of age and found pituitary tumors in 30 (43%) of 70; this incidence was not significantly different from that of our initial report (20). Among those that died prematurely (<16 mo of age), pituitary lesions were found in 22 (78.6%) of 28 animals. In comparison, only 8 of 42 (19.0%) rats that harbored pituitary lesions animals were alive at 16 mo (p < 0.0001, Fisher's exact test). Of the eight rats with pituitary tumors, three were moribund at the time when they were killed; thus, only five (12.8%) animals with pituitary pathology seemed healthy at 16 mo of age.
The association between premature deaths and pituitary pathology was independent of the genetic background carrying the Tsc2Eker mutation. Rats of Fischer 344 and Long Evans strains had similar presentations with respect to the pituitary lesions. Figure 1 illustrates the influence of pituitary tumors on the survival of the Eker rats in the Long Evans strain. The hazard ratio of death during the 16-mo period was 6.4 times higher (95% confidence interval 2.4–16.7) in the pituitary tumor group compared with those without pituitary tumors. Similarly, the median survival was significantly shortened in animals with pituitary lesions (10 mo versus not yet reached at 16 mo for the nonpituitary group).

Kaplan-Meier estimates of survival of the Eker rats in the Long Evans strain with (n = 18) and without (n = 26) pituitary tumors. All surviving animals were killed at 16 mo of age.
In this study, pituitary tumors were more common in male than in female rats. Eighty-one percent of those with pituitary lesions were male, and 64% of the male Tsc2Ek/+ carriers developed these tumors, whereas only 15% of female rats harbored pituitary adenomas upon necropsy (p < 0.0001, Fisher's exact test). There was a lack of correlation between the size of kidney tumors and the presence of pituitary lesions. In the Long Evans background, the mean renal tumor size was 4.4 mm among those without pituitary lesions and 1.8 mm in those with pituitary tumors. In contrast, kidney tumors in the Fischer background were larger (mean 12.8 mm) in those with pituitary tumors compared with those without (mean 7.9 mm). Importantly, renal tumor size did not correlate with survival of the Tsc2Ek/+ carriers.
Before death, the animals with pituitary adenomas commonly exhibited loss of balance, reduced responsiveness to stimuli, and progressive cachexia. The period between the onset of these symptoms and death was often short (within days). At necropsy, all of the Tsc2Ek/+ rats with this constellation of signs and symptoms were found to have abnormal pituitary glands consisting of gross tumors that were accompanied by hemorrhage (Fig. 2A). The pituitary masses ranged from 3 to 13 mm in diameter (mean 7.9 mm). Gross examination of the remaining brain did not show structural abnormalities such as tumors or hydrocephalus. When examined under the microscope, hematoxylin and eosin–stained sections showed densely cellular epithelioid neoplasms with faintly lobular pattern (Fig. 2B). The lobules were variable in size and occupied by sheets of cells with occasional vascular structures. The neoplastic cells have variable amounts of granular to vacuolated cytoplasm and generally large nuclei with well-defined nuclear borders and prominent nucleoli (imparting a distinctly endocrine appearance). Mitotic figures were uncommon, but apoptotic cells were numerous. On the basis of these observations, we conclude that pituitary lesions and their complications (e.g. hemorrhage) were the predominant contributor to premature deaths in the Tsc2Ek/+ rats.

Gross and histologic appearance of the pituitary tumors. (A) Photograph of pituitary mass (PT) with associated hemorrhage at necropsy. ON, optic nerves. (B) High-power transmitted light photomicrograph showing a representative field of a hematoxylin and eosin (H&E)-stained section demonstrating a faintly lobulated adenoma composed of relatively cytoplasm-rich cells with enlarged nuclei. Magnification: ×400.
mTOR pathway is up-regulated in the pituitary tumors.
Our previous study showed evidence of loss of heterozygosity in these pituitary tumors, suggesting a “two-hit” pathogenetic mechanism (20). This suggests the loss of tuberin function in the tumors, resulting in the aberrant activation of the mTOR pathway similar to that of other TSC pathology. Here, we examined the functional consequences of Tsc2 inactivation in these lesions by evaluating the activities of downstream effectors of the mTOR pathway. Indeed, phosphorylation of 4E-BP1 and p70S6 K activity, as reflected by the level of phosphorylated ribosomal S6 protein, were found to be highly elevated in the pituitary lesions compared with adjacent CNS (Fig. 3A, columns 1 and 2). Immunohistochemical analyses confirmed that pituitary tumor tissue expressed abundant phospho-S6 (Fig. 3B). Thus, activated mTOR in the pituitary lesions would serve as a potential target for therapy.

Activation of the mTOR pathway in the pituitary tumors and their response to rapamycin. (A) Western blot analysis of lysates derived from pituitary tumors and adjacent CNS of rapamycin-treated and untreated animals. The multiple bands of 4E-BP1 reflect the various phosphorylation states of the protein. The upper, slower migrating band represents the more highly phosphorylated protein. (B) Histologic evaluation of pituitary tumors with and without rapamycin exposure. H&E staining highlights the differences in cell size on the basis of the distribution density of the nuclei. Anti–phospho-S6 (Ser235/236) staining illustrates the effects of rapamycin on mTOR inhibition in vivo. TUNEL assay stained cells undergoing apoptosis. Magnification: ×400.
Rapamycin reverses clinical deterioration and prolongs survival of the Tsc2Ek/+ rats.
As a specific inhibitor of mTOR, we tested the effects of rapamycin in Tsc2Ek/+ rats with clinical manifestation of pituitary tumors. Three animals with altered gait, abnormal responses to stimuli, and progressive cachexia were treated with daily intraperitoneal injections of rapamycin or an inactive drug (Table 1). Animals ranged from 13 to 16 mo of age (median 14.5). Within 1 wk of initiation of treatment, animals that received rapamycin but not the placebo-treated rat showed clinical improvement as demonstrated by a return to normal gait and responsiveness to stimuli. Furthermore, their resumption of a normal appetite resulted in progressive weight gain after treatment, unlike the placebo-treated animal, which showed continued weight loss (Table 1). The change in body weight was not due to nonspecific effects of the drug because asymptomatic Tsc2Ek/+ rats did not show evidence of significant weight gain after rapamycin treatment. To further demonstrate a cause–effect relationship, rapamycin was withdrawn after clinical recovery of the “sick” animals. Within 2 wk of stopping treatment, the two animals deteriorated in their neurologic status and began to lose weight. Re-initiation of treatment with rapamycin promptly reversed the clinical signs and symptoms. Neither of the two animals that received rapamycin died while on treatment, and their overall survival from the onset of clinical deterioration was between 45 and 55 d; this was significantly longer than the expected 1–7 d previously observed in the nontreated symptomatic Tsc2Ek/+ carriers.
At necropsy, each of the symptomatic animals had gross evidence of pituitary tumors despite rapamycin treatment. This suggests that clinical improvement occurred without complete eradication of the tumors. Although we could not accurately measure the size of the pituitary lesions in vivo before and after rapamycin therapy, pathologic analyses revealed dramatic responses in cell size and cell death (Fig. 3B). Compared with untreated pituitary tumors, rapamycin treatment was associated with a decrease in cell size as shown by the increased “crowding” of the nuclei in the treated samples. These latter cells also stained positively in the TUNEL assay, suggesting activation of an apoptotic pathway. This is supported by increased levels of cleaved caspase 3 in the tumors (Fig. 3A). Conversely, the levels of phospho-S6 in the treated lesions became undetectable, whereas changes in 4E-BP1 phosphorylation were less apparent (Fig. 3A). Together, we analyzed seven untreated and two treated animals by immunocytochemistry and four untreated and six treated tumors by Western blotting; all showed a dramatic reduction in phospho-S6 in the treated compared with the untreated samples. Therefore, we deduce that the clinical improvement after rapamycin may be attributed in part to reduced tumor cell size and increased cell death secondary to down-regulation of p70S6 K activity.
Renal tumors respond to rapamycin.
To further evaluate the impact of rapamycin on Tsc2-related tumors in vivo, we examined its antitumor effects on renal tumors in the Eker rats. Our previous study showed significant biochemical down-regulation of the mTOR pathway in the kidney tumors after a brief (3 d) exposure over a broad dose range (0.1–2.0 mg · kg−1 · d−1) (17). Here, we monitored tumor size in response to rapamycin using serial noninvasive imaging. Percutaneous ultrasound was performed on 12-mo-old Tsc2Ek/+ rats to identify those with easily visible solid lesions in the kidneys before initiation of treatment. The ultrasound measurements were found to be highly reproducible and correlated closely with actual tumor sizes (Fig. 4A). Animals were exposed to rapamycin for durations ranging from 2 wk to 2 mo. Serial ultrasound assessment was able to detect significant reduction in tumor volumes (Fig. 4B). Treatment for up to 2 mo resulted in >90% tumor volume regression (Table 2). Hence, rapamycin was highly effective in reversing renal tumor growth in adult Tsc2Ek/+ rats.

Noninvasive imaging of kidneys by serial ultrasound to document gross tumor response to rapamycin. (A) Comparison of ultrasound with gross pathologic measurement of an Eker rat kidney tumor. (B) Serial images of a kidney tumor taken before and after a 45-d treatment with rapamycin (0.2 mg/kg). Numbers in the ultrasound images represent diameter measurements of the tumor. Histogram shows tumor response expressed as tumor volume (based on average diameter from three measurements and applying the formula 4/3πr3, where r represents radius).
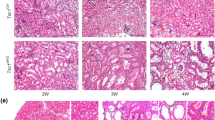
Microscopically, extensive tumor necrosis was apparent in some tumors after 2 wk of rapamycin treatment (Fig. 5A). In those that were treated for longer periods, the sites of disease in the kidneys have been largely replaced by stromal “scars” and reactive tissues, with most of the tumor debris having been cleared (Fig. 5A). The remaining nontumor kidney tissue seemed unaffected histologically. This is consistent with the absence of clinical toxicity or treatment-related death in the treated cohort. It seems that rapamycin has selective cytotoxic effects on the Tsc2-related tumors.

Renal tumor responses to rapamycin. (A) Microscopic changes of tumors after 2 wk (magnification: ×100) and 2 mo (magnification: ×200) of treatment. H&E staining. (B) Biochemical responses after 2 d, 2 wk, and 2 mo of rapamycin, compared with placebo controls. Dosages are as labeled. Lysates of residual tumors (T) and adjacent kidney tissue (N) were subjected to immunoblot analysis using the indicated antibodies. Actin serves as a loading control.
To evaluate the mechanisms of tumor response, we examined biochemical alterations after rapamycin (Fig. 5B). Three sets of animals were treated for various durations (2 d, 2 wk, and 2 mo) with rapamycin or placebo. Consistent changes in the phosphorylation of ribosomal S6 and 4E-BP1 were observed over different treatment periods. Phosphorylation of ribosomal S6 was strongly suppressed with all treatment regimens. Compared with pituitary tumors, dephosphorylation of 4E-BP1 was more pronounced in the treated renal tumors; this may reflect tissue-specific differences in the regulation of 4E-BP1. Tumor VEGF expression was significantly down-regulated by rapamycin, whereas the level of cleaved caspase 3 increased with the inhibitor. Of the cell-cycle proteins, cyclin D1 remained relatively unchanged as previously reported (17), but p27kip1 accumulated in the tumor cells, especially after prolonged rapamycin exposure.
In vivo resistance to rapamycin.
Although tumor response was often dramatic in the rapamycin-treated kidneys, there were rare lesions that seemed unaffected. These tumors not only lacked evidence of necrosis or apoptosis but also continued to express high levels of phospho-S6 (Fig. 6A, tumor 2), whereas other tumors in the same kidneys responded appropriately to the drug (Fig. 6A, tumor 1). That they were found at the time of ongoing rapamycin treatment suggests an intrinsic resistance to the drug. These “nonresponsive” tumors varied in size and location throughout the kidneys but were, in general, small (microscopic). Nonetheless, the heterogeneous response of the Tsc2Ek/+ rat kidney tumors to rapamycin supports the existence of in vivo intrinsic drug resistance.

In vivo resistance of renal tumors to rapamycin. (A) Persistent up-regulation of p70S6 K activity in a kidney tumor (tumor 2) stained with anti–phospho-S6 antibodies. H&E staining showed viable tumor cells after 4 wk of rapamycin treatment. By comparison, a tumor from the same kidney showing response to the drug (tumor 1) was stained with H&E and anti–phospho-S6 antibodies. (B) Residual phospho-S6–positive tumor cells after 8 wk of rapamycin treatment and a 2-wk recovery period. This tumor was significantly reduced in size but showed emergence of abnormal cells with high p70S6 K activity. Magnification: ×200.
We also observed that in rapamycin-sensitive tumors, residual tumor cells persisted despite protracted treatment for 2 mo. Staining of a serial section from phosphorylated S6 protein taken from an apparent “tumor scar” (see Fig. 5A) revealed abnormal cells that reacted strongly to anti–phospho-S6 antibodies (Fig. 6B). These “activated” cells may have acquired resistance to rapamycin within a lesion that was otherwise responsive to the inhibitor. Such acquired resistance has been reported in other mammalian cells (21). Collectively, our data showed that pharmacologic inhibition of mTOR was effective in inducing tumor regression and prolonging survival in adult Tsc2Ek/+ rats with macroscopic renal and pituitary tumors. However, our studies documented resistance to rapamycin, which may have prevented complete eradication of disease.
Can rapamycin prevent tumor formation?. If mTOR inhibition could halt tumor progression as shown above, then we reasoned that rapamycin exposure before tumor development may effectively prevent their formation. For testing this hypothesis, 10 Tsc2Ek/+ rats were randomly assigned to receive rapamycin (n = 6) or placebo (n = 4) over a 2-mo period between the ages of 2 and 4 mo. Previous studies have shown that this is a vulnerable period for target cells in the kidneys to undergo tumor initiation (inactivation of the second Tsc2 allele) (22,23). At the end of the 2-mo treatment, animals were left untreated for 4 mo and killed at 8 mo of age. The kidneys were examined for evidence of macroscopic tumors and microscopic precursor lesions on the basis of gelsolin and phospho-S6 staining as previously described (24). Both of these methods correlated well, but “background” staining of nontumor structures such as distal renal tubules with anti-gelsolin antibodies made quantitative evaluation of precursor lesions more difficult. The number of lesions was tabulated according to their sizes [tumors with <10 cells, 10–100 cells, and macroscopic (≥1 mm) tumors]. For microscopic disease, the data were expressed as number of lesions per low-power field.
At 8 mo of age, none of the rapamycin-treated animals developed macroscopic (≥1 mm) lesions, compared with a mean of 1.25 lesions per animal in the control group (p = 0.02, t test). However, this trend was not observed with the microscopic lesions when analyzed according to sex. Unlike a previous study that reported a greater number of microscopic tumors in the male than in the female rats (25), we found the opposite to be true in this cohort of animals. As a group, treated and untreated female rats had significantly more precursor lesions of <10 cells (mean 7.2 versus 1.4 per low-power field per animal; p < 0.0001) and lesions with 10–100 cells (mean 3.8 versus 0.5 per low-power field per animal; p < 0.0001) compared with the male carriers. The most significant difference between these studies lies in the genetic background of the rats carrying the Eker mutation (Wistar versus Long Evans). Unfortunately, there were too few female rats to make valid comparisons between control and treated groups. For the male rats, there were 0.4 ± 0.1 lesions of 10–100 cells per low-power field per animal in the control group compared with 0.5 ± 0.3 lesions per low-power field per animal in the rapamycin group (p = 0.3, t test; Table 3). Similarly, there was no significant difference between the number of lesions <10 cells (control 1.0 ± 0.3 versus rapamycin 1.5 ± 0.4; p = 0.3, t test). These observations suggest that rapamycin was effective in reducing the incidence of macroscopic tumors but did not significantly reduce the number of precursor lesions.
DISCUSSION
Recent studies have implicated an important role of the TSC genes in mediating mTOR activity by sensing cellular energy balance and nutrient and growth factor availability (26). The consequences of the loss of TSC1/2 function include the multiorgan development of benign tumors in humans with highly variable clinical presentations. Some patients manifest early in infancy with epilepsy or cardiac outflow tract obstruction, whereas others present in adulthood with complications of renal AMLs and pulmonary lymphangioleiomyomatosis. Despite the diverse pathologies, the underlying lesions share features of aberrant regulation of cell growth, proliferation, differentiation, and migration. It remains to be determined whether all of these processes can be explained on the basis of the disrupted regulation of the mTOR pathway alone.
The availability of a specific mTOR inhibitor, rapamycin, and a well-characterized rodent model of TSC provides an ideal setting to examine the role of mTOR activity in spontaneously derived Tsc2-related tumors in vivo. The aims of this study were to determine the effects of mTOR inhibition on tumor progression and initiation. We previously showed in short-term studies that rapamycin given over a wide dose range effectively down-regulated mTOR activities in renal tumors of the Tsc2Ek/+ rats (17). In this study, we showed that rapamycin treatment induced significant regression of established tumors of the kidney and pituitary without histologic evidence of nontumor cytotoxicity. The data also show that complication from pituitary tumors (e.g. hemorrhage) was a major cause of premature deaths of the Tsc2Ek/+ rats and that rapamycin could prolong the survival of these animals. The findings suggest that elevated mTOR activity contributes significantly to the progression of these tumors, and the inhibition of this pathway effectively reversed the growth of the lesions through a number of mechanisms, including reduction in cell size and induction of cell death. The selective tumor cytotoxicity of rapamycin in rat Tsc2-related tumors argues in favor of its safe application in the clinical setting.
The development of pituitary tumors in the Eker rat model has been described in our previous study showing their occurrence with loss of heterozygosity at the Tsc2 locus (20). The current immunohistologic and biochemical data provide further evidence that the loss of Tsc2 function in these lesions is accompanied by the activation of the mTOR pathway. Thus, pituitary tumorigenesis follows a “two-hit” mechanism similar to that in other TSC lesions. However, there are significant species-related differences because the incidence of pituitary pathology in human TSC patients is very low, although case reports of hyperprolactinemia, diabetes insipidus, and acromegaly have been documented (27). Nonetheless, our findings illustrate the involvement of the mTOR pathway in pituitary adenomas of the rat and may have relevance to the understanding of sporadic human pituitary tumor development.
In rare instances, the presence of hyperphosphorylated ribosomal S6 proteins in persistent renal tumors after rapamycin treatment strongly suggests the existence of in vivo drug resistance. This suggests that complete eradication of lesions in TSC patients may not be realistic with rapamycin treatment alone. Studies in yeast and mammalian cells have highlighted some of the mechanisms related to rapamycin resistance, including mutations of various components of the signaling pathway such as FKBP-12 and mTOR itself (21). In addition, posttranslational regulation of 4E-BP1 has been implicated in cellular sensitivity to rapamycin. Modulation of other pathways, including p53 and IGF-1, also affects the degree of drug-induced apoptosis. Strategies to overcome resistance await further understanding of the underlying mechanisms.
The lack of change in the frequency of precursor lesions in the kidneys after rapamycin treatment was unexpected given its effects on macroscopic tumors. Whereas the 2-mo drug treatment at a young age was effective in preventing the development of lesions ≥1 mm, the number of microscopic tumors did not show a trend in reduction. This was true for lesions <10 cells as well as those between 10 and 100 cells. The inhibitor was given between the ages of 2 and 4 mo, when “second” hit events occur on the basis of the ability of chemical and radiation carcinogens to increase the number of renal lesions in a dose-dependent manner during this period. Although the paucity of effect of rapamycin on microscopic tumor multiplicity may be secondary to inadequate drug concentration and/or duration of exposure, this seems unlikely because of the in vivo effects observed for macroscopic disease. We conclude that whereas mTOR activation may be critical to tumor progression, it may not be necessary for tumor initiation. This suggests the participation of alternative pathways. In a recent study, Brugarolas et al. (28) suggested an mTOR-independent link between TSC2 and VEGF expression. They found that high levels of VEGF secretion by Tsc2−/− cells in vitro were only modestly reduced by rapamycin, whereas a histone deacetylase inhibitor, trichostatin A, was more effective in normalizing the level. However, in our spontaneous renal tumors, VEGF expression was dramatically down-regulated after short- and long-term rapamycin treatments (see Fig. 5B). Our data are consistent with mTOR-mediated VEGF expression in vivo and support the hypothesis that the development of macroscopic disease depends on VEGF-directed tumor angiogenesis. Other alternative mechanisms that may play a role in renal tumor initiation include the β-catenin pathway. We have previously shown that Wnt/β-catenin signaling is also under the regulation of TSC1/TSC2 (29). Coexpression of hamartin and tuberin was found to inhibit Wnt-induced β-catenin–dependent transcriptional activation, and the endogenous TSC proteins interact with the GSK3/Axin degradation complex. Future studies will address the contribution of this pathway in tumor initiation in animal models of TSC.
Findings from this study have several clinical implications: 1) The impressive antitumoral response to rapamycin of the macroscopic lesions in the Eker rat would predict similar effects in TSC patients. We await the results of an ongoing phase I/II study in TSC individuals with AMLs for confirmation. On the basis of its extensive use in renal transplant patients, much is known about the toxicity profile of rapamycin and its pharmacokinetics and drug monitoring to ensure safe delivery. 2) The lack of effect in reducing the number of precursor lesions argues in favor of its use for symptomatic and/or macroscopic disease rather than in disease prevention. 3) The potential problem of drug resistance calls for continued search for alternative therapies. 4) The use of rapamycin in CNS manifestations (e.g. cortical tubers, subependymal giant cell astrocytomas) remains untested. Its ability to cross the blood-brain barrier and its pharmacodynamics in the CNS have not been well characterized, especially in the pediatric population.
In summary, the mTOR pathway plays an important role in Tsc2 tumor progression, but its contribution to the early tumorigenic events is less certain. Nonetheless, rapamycin was highly effective in reducing tumor size and prolonging survival of symptomatic Tsc2Ek/+ rats and thus represents the first mechanism-based therapy for this disease. The anticipated problem of drug resistance may lessen its success, especially in long-term therapy. Future clinical trials are needed to determine its proper indication, dose scheduling, and duration of treatment. Further investigation of TSC1/TSC2 function will also provide additional targets for other hypothesis-driven therapies in TSC-related diseases.
Acknowledgments.
We thank David Flum, M.D., for valuable statistical advice; Donald Born, M.D., for pathologic interpretation of the pituitary tumors; Marla Paun for technical assistance with ultrasound; and members of the Yeung laboratory for suggestions and critical reading of the manuscript.
Abbreviations
- AML:
-
angiomyolipoma
- mTOR:
-
mammalian target of rapamycin
- TSC:
-
tuberous sclerosis complex
- TUNEL:
-
transferase-mediated dUTP nick-end labeling
- VEGF:
-
vascular endothelial growth factor
- 4E-BP1:
-
4E-binding protein 1
References
Kandt RS 2003 Tuberous sclerosis complex and neurofibromatosis type 1: the two most common neurocutaneous diseases. Neurol Clin 21: 983–1004
Gomez MR 1999 Definition and criteria for diagnosis. Gomez MR, Sampson JR, Whittemore VH Tuberous Sclerosis Complex. Oxford University Press, New York pp 10–23 Gomez MR 1999 Definition and criteria for diagnosis. In: Gomez MR, Sampson JR, Whittemore VH (eds) Tuberous Sclerosis Complex. Oxford University Press, New York, pp 10–23
Consortium 1993 Identification and characterization of the tuberous sclerosis gene on chromosome 16. The European Chromosome 16 Tuberous Sclerosis Consortium. Cell 75: 1305–1315
van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, Lindhout D, van den Ouweland A, Halley D, Young J, Burley M, Jeremiah S, Woodward K, Nahmias J, Fox M, Ekong R, Osborne J, Wolfe J, Povey S, Snell RG, Cheadle JP, Jones AC, Tachataki M, Ravine D, Sampson JR, Reeve MP, Richardson P, Wilmer F, Munro C, Hawkins TL, Sepp T, Ali JB, Ward S, Green AJ, Yates JR, Kwiatkowska J, Henske EP, Short MP, Haines JH, Jozwiak S, Kwiatkowski DJ 1997 Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277: 805–808
McManus EJ, Alessi DR 2002 TSC1-TSC2: a complex tale of PKB-mediated S6K regulation. Nat Cell Biol 4: E214–E216
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC 2002 Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10: 151–162
Inoki K, Li Y, Zhu T, Wu J, Guan KL 2002 TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657
Stocker H, Radimerski T, Schindelholz B, Wittwer F, Belawat P, Daram P, Breuer S, Thomas G, Hafen E 2003 Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol 5: 559–565
Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D 2003 Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol 5: 578–581
Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J 2003 Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol 13: 1259–1268
Fingar DC, Richardson CJ, Tee AR, Cheatham L, Tsou C, Blenis J 2004 mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol 24: 200–216
Inoki K, Zhu T, Guan KL 2003 TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: 577–590
Green AJ, Johnson PH, Yates JR 1994 The tuberous sclerosis gene on chromosome 9q34 acts as a growth suppressor. Hum Mol Genet 3: 1833–1834
Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, Hornigold N, van Slegtenhorst M, Welsh CT, Kwiatkowski DJ 1996 Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet 59: 400–406
Knudson AG 2001 Two genetic hits (more or less) to cancer. Nat Rev Cancer 1: 157–162
Goncharova EA, Goncharov DA, Eszterhas A, Hunter DS, Glassberg MK, Yeung RS, Walker CL, Noonan D, Kwiatkowski DJ, Chou MM, Panettieri RA Jr Krymskaya VP 2002 Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol Chem 277: 30958–30967
Kenerson HL, Aicher LD, True LD, Yeung RS 2002 Activated mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res 62: 5645–5650
Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG 1994 Predisposition to renal carcinoma in the Eker rat is determined by germ- line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci USA 91: 11413–11416
Yeung RS, Gu H, Lee M, Dundon TA 2001 Genetic identification of a locus, Mot1, that affects renal tumor size in the rat. Genomics 78: 108–112
Yeung RS, Katsetos CD, Klein-Szanto A 1997 Subependymal astrocytic hamartomas in the Eker rat model of tuberous sclerosis. Am J Pathol 151: 1477–1486
Huang S, Bjornsti MA, Houghton PJ 2003 Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther 2: 222–232
Walker C, Goldsworthy TL, Wolf DC, Everitt J 1992 Predisposition to renal cell carcinoma due to alteration of a cancer susceptibility gene. Science 255: 1693–1695
Hino O, Klein-Szanto AJ, Freed JJ, Testa JR, Brown DQ, Vilensky M, Yeung RS, Tartof KD, Knudson AG 1993 Spontaneous and radiation-induced renal tumors in the Eker rat model of dominantly inherited cancer. Proc Natl Acad Sci USA 90: 327–331
Onda H, Lueck A, Marks PW, Warren HB, Kwiatkowski DJ 1999 Tsc2(+/-) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J Clin Invest 104: 687–695
Eker R, Mossige J, Johannessen JV, Aars H 1981 Hereditary renal adenomas and adenocarcinomas in rats. Diagn Histopathol 4: 99–110
Harris TE, Lawrence JC Jr 2003 TOR signaling. Sci STKE re15
Zimmerman D 1999 The endocrine system in tuberous sclerosis complex. Gomez MR, Sampson JR, Whittemore VH Tuberous Sclerosis Complex. Oxford University Press New York 218–227 Zimmerman D 1999 The endocrine system in tuberous sclerosis complex. In: Gomez MR, Sampson JR, Whittemore VH (ed) Tuberous Sclerosis Complex. Oxford University Press, New York, pp 218–227
Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG Jr 2003 TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4: 147–158
Mak BC, Takemaru K, Kenerson HL, Moon RT, Yeung RS 2003 The tuberin-hamartin complex negatively regulates beta-catenin signaling activity. J Biol Chem 278: 5947–5951
Author information
Authors and Affiliations
Additional information
This study was supported by grants from the National Institutes of Health (CA61889, CA102662).T.A.D. is currently affiliated with Department of Cell Biology, University of North Carolina, Chapel Hill, NC 27599.
Rights and permissions
About this article
Cite this article
Kenerson, H., Dundon, T. & Yeung, R. Effects of Rapamycin in the Eker Rat Model of Tuberous Sclerosis Complex. Pediatr Res 57, 67–75 (2005). https://doi.org/10.1203/01.PDR.0000147727.78571.07
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000147727.78571.07
This article is cited by
-
An update on the central nervous system manifestations of tuberous sclerosis complex
Acta Neuropathologica (2020)
-
A brain proteomic investigation of rapamycin effects in the Tsc1 +/− mouse model
Molecular Autism (2017)
-
mTOR promotes pituitary tumor development through activation of PTTG1
Oncogene (2017)
-
mTOR inhibitor reverses autistic-like social deficit behaviours in adult rats with both Tsc2 haploinsufficiency and developmental status epilepticus
European Archives of Psychiatry and Clinical Neuroscience (2017)
-
Exosomes mediate the acquisition of the disease phenotypes by cells with normal genome in tuberous sclerosis complex
Oncogene (2016)
