
Abstract
To assess the role of brain antioxidant capacity in the pathogenesis of neonatal hypoxic-ischemic brain injury, we measured the activity of glutathione peroxidase (GPX) in both human-superoxide dismutase-1 (hSOD1) and human-GPX1 overexpressing transgenic (Tg) mice after neonatal hypoxia-ischemia (HI). We have previously shown that mice that overexpress the hSOD1 gene are more injured than their wild-type (WT) littermates after HI, and that H2O2 accumulates in HI hSOD1-Tg hippocampus. We hypothesized that lower GPX activity is responsible for the accumulation of H2O2. Therefore, increasing the activity of this enzyme through gene manipulation should be protective. We show that brains of hGPX1-Tg mice, in contrast to those of hSOD-Tg, have less injury after HI than WT littermates: hGPX1-Tg, median injury score = 8 (range, 0–24) versus WT, median injury score = 17 (range, 2–24), p < 0.01. GPX activity in hSOD1-Tg mice, 2 h and 24 h after HI, showed a delayed and bilateral decline in the cortex 24 h after HI (36.0 ± 1.2 U/mg in naive hSOD1-Tg versus 29.1 ± 1.7 U/mg in HI cortex and 29.2 ± 2.0 for hypoxic cortex, p < 0.006). On the other hand, GPX activity in hGPX1-Tg after HI showed a significant increase by 24 h in the cortex ipsilateral to the injury (48.5 ± 5.2 U/mg, compared with 37.2 ± 1.5 U/mg in naive hGPX1-Tg cortex, p < 0.008). These findings support the hypothesis that the immature brain has limited GPX activity and is more susceptible to oxidative damage and may explain the paradoxical effect seen in ischemic neonatal brain when SOD1 is overexpressed.
Similar content being viewed by others
Main
The developing brain faces unique challenges from oxidative stress, and has responses to injury and protective mechanisms that are different from the mature nervous system. The developmental profile of the activity of the endogenous antioxidant enzymes SOD1 and selenium-dependent GPX have been described for both rats (1–3) and mice (4). In the mouse brain, GPX activity is almost exclusively due to the classic, cellular glutathione peroxidase GPX1 (EC 1.11.1.9) (5). In the CD1 mouse, the outbred background strain used in this study, SOD1 activity decreases around the time of birth, and then remains unchanged to weaning. GPX activity, on the other hand, rises steeply around birth but declines sharply in the first few days of life and then remains low through weaning. Protein levels for both enzymes, however, show a steady increase during early development (4). We have shown that, in the CD1 mouse brain, adult levels of GPX activity are reached by weaning (6). Therefore, maturational differences may impact the response of the newborn brain to injury. It has been shown that the adult and juvenile brain attempts to compensate during oxidative stress in response to stroke (7) and traumatic brain injury (8) by either maintaining or increasing levels of the antioxidant enzymes SOD1 and GPX1. Increased SOD1 should protect the brain from increased production of superoxide that is a consequence of oxidative stress. Indeed, some models of adult stroke using rodents that overexpress hSOD1 have shown significant protection (9). However, in the same strain of hSOD1-Tg mice, we found worsened brain injury during the neonatal period (10). We hypothesized that downstream antioxidant enzymes were not sufficiently up-regulated in the neonatal brain, resulting in H2O2 accumulation and subsequent injury from generation of other reactive oxygen species like hydroxyl ions from the Fenton reaction. Indeed, we found that H2O2 accumulates and GPX activity declines in injured hSOD1 hippocampus after HI, whereas catalase activity remains unchanged (11,12). These findings suggest that the important enzyme for detoxification of H2O2 is GPX and not catalase. The accumulated H2O2 is toxic to immature neurons more so than mature neurons, and this toxicity can be reversed by addition of iron chelators and free radical scavengers (11,13).
Because it appeared that GPX, and not catalase, might be the critical downstream enzyme rescuing cells from H2O2 toxicity, and that GPX1 gene overexpression can be neuroprotective to adult animals after focal ischemia (14,15), we investigated whether an increase in GPX activity by overexpression of the hGPX1 transgene in rodent brain would result in protection during the neonatal period after HI. In the present study, we report that there is protection from HI injury in the hGPX1-Tg brains and show that increased cortical GPX activity is associated with this protection.
METHODS
Animals.
All experiments were approved by the Institutional Animal Care and Use Committee at the University of California San Francisco and carried out with the highest standards of care and housing, according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
hSOD1 transgenic pups.
Heterozygote transgenic mice carrying the human cytosolic SOD1 gene were bred with wild-type CD1 mice to produce pups that were both transgenic (Tg) and nontransgenic (WT) for hSOD1 (16,17). The heterozygotes expressed 3-fold higher SOD activity (18,19). Pups were identified at P6 by sampling tail blood using qualitative nondenaturing gel electrophoresis followed by nitroblue tetrazolium staining for the human SOD1 protein as previously described (17,19).
hGPX1 transgenic pups.
Heterozygote transgenic mice carrying the human GPX1 gene (20) were bred with wild-type CD1 mice to produce mixed litters of GPX1 overexpressing transgenic pups and WT littermates. Genotype was identified on P6 using tail clippings. PCR was performed with “platinum supermix” (Invitrogen, Carlsbad, CA), premixed Taq polymerase/dNTPs/MgCl2, primer sequence TCA AAG TTC CAG GCA ACA TCG TT [5′-3′] (Invitrogen), and 1 μL DNA. The PCR products were separated on a 2% agarose gel containing ethidium bromide for UV visualization.
Hypoxic-ischemic injury.
P7 mice were exposed to HI as previously described (10,21). Briefly, pups were anesthetized with 2.5% halothane, 30% nitrous oxide, and the right common carotid artery (CCA) was exposed and ligated by electrical coagulation. After a recovery period with the dam (1.5 h), the mice were placed in separate but interconnected chambers (two pups per chamber) partially submerged in a 37°C water bath, through which flowed a humidified gas mixture containing 8% oxygen/balance nitrogen for 45 min. Temperature was monitored by a skin surface thermometer probe during the procedure to maintain body surface temperature at 34°C. The sex of each animal was recorded and pups were weighed daily until sacrifice.
Histology.
For examination of the degree of injury after HI, animals were anesthetized with pentobarbital at either 24 h or 5 d after HI and euthanized. Brains were removed after perfusion fixation with 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Fifty-micron sections were cut on a vibrating microtome, and sections were stained with cresyl violet stain for injury scoring. Scoring analysis was performed on uniform brain sections at 5 d after insult with grading done on three regions of the cortex (anterior, mid, posterior), one region of the mid-caudate/striatum, four regions of the hippocampus (CA1, CA2, CA3, DG), and one region of the mid-thalamus. Analysis was performed in a blinded fashion. Regions were scored as follows: grade 0 = no evidence of injury; grade 1 = rare scattered pyknotic cells; grade 2 = single or small confluent areas of necrosis or apoptosis; grade 3 = large, multiple areas of necrosis or apoptosis, including involvement of entire thalamic nuclei, > 50% of the striatal or hippocampal area, or areas of cystic infarction in the cortex. Grades from each of the three cortical regions and from the four hippocampal regions were summated to give a cumulative grade for the entire brain. A median injury score >10 and <20 was considered moderate injury, whereas a score of 20 or greater was considered severe (16). WT (n = 14), hSOD1-Tg (n = 4), and hGPX1-Tg (n = 5) heterozygotes were used for histologic analysis at 24 h to show the extent of injury at this early stage in the progression of injury (Fig. 1). We have previously shown in detail the injury in the hSOD-Tg at 5 d after injury, hence the data are not repeated (10,22). Injury scores for the hGPX1-Tg versus WT are shown in Figure 2.

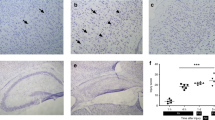
Photomicrographs of injury after HI in brain sections stained with cresyl violet. Representative injury 24 h (A, C, E) and 5 d after HI (B, D, F). (A, B) CD1 wild-type (WT). In (B), injury score = 17. (C, D) Human superoxide dismutase 1-transgenic (hSOD1-Tg). In (D), injury score = 20. (E, F) Human glutathione peroxidase-transgenic (hGPX1-Tg). In (F), injury score = 8. Arrows indicate small foci of injury. Scale bar = 500 μm.

Box plot of injury scores in hGPX1-Tg (n = 7) and WT (n = 14) littermates 5 d after HI. The horizontal line represents the median score. p < 0.01 by Mann-Whitney.
GPX enzymatic assay.
Tissue for GPX assay was obtained at 2 h or 24 h after HI, and at P7 and P8 for naive pups to correspond to the correct developmental age at sacrifice. Brains were removed by rapid decapitation onto a cold surface and dissected into right and left cortices and flash frozen. Tissue was stored at −70°C until assay. Tissue was homogenized with a Polytron in cold 0.1 M potassium phosphate, pH 7.4, and centrifuged for 10 min at 14,000 rpm. Selenium-dependent GPX activity was determined in a coupled test system in which reduced glutathione and tert-butyl hydroperoxide were used as substrate, and oxidized glutathione produced by GPX activity was measured by kinetically monitoring glutathione reductase-mediated NADPH oxidation at 340 nm. Activity was expressed as units per milligram of protein, where 1 unit (U) is defined as 1 nmol NADPH oxidized per minute (23,24). Protein content was determined with the Pierce BCA protein assay (Pierce, Rockford, IL.).
Statistical analysis.
Analyses were performed with Statview 4.51 (Abacus Concepts, Berkeley, CA). Ordinal data (injury score) was analyzed by Mann-Whitney test. Comparisons among groups were made by one-way ANOVA followed by Bonferroni-Dunn posthoc testing for multiple comparisons for continuous data. A p value of <0.05 was considered statistically significant. Data are presented as mean ± SD, or as percent of naive.
RESULTS
Histopathology.
Evaluation of brain injury induced by HI revealed that cortex, hippocampus, and striatum in the hemisphere ipsilateral to the CCA ligation were injured, whereas the contralateral hemisphere revealed no light microscopic damage, as previously described (10) (Fig. 1). The severity of injury varied between and within regions, with the hippocampus being the most affected region in all animals regardless of genotype. Twenty-four hours after HI, both cell death and edema were apparent in affected regions (Fig. 1, A, C, and E). At 5 d after injury, edema subsided and cell death evolved as visualized by Nissl staining (cresyl violet) (Fig. 1, B, D, and F). The histopathological injury in WT littermates of the hSOD-Tg and hGPX1-Tg animals was similar, as expected (Fig. 1, A and B; median injury score of 17 overall for the brain depicted in Fig. 1B). Previously, we showed that hSOD-Tg brains exhibited greater histopathological injury compared with WT at 5 d after HI (injury score = 20) (9). We confirmed that injury was also severe at 24 h after HI (Fig. 1C). In contrast, hGPX1-Tg brains were significantly protected (Fig. 1, E and F), with less injury compared with WT at 5 d after insult (Fig. 2): hGPX1-Tg median injury score = 8 (range, 0–24), WT median injury score = 19 (range, 2–24) (p < 0.01 by Mann-Whitney). There were no differences in degree of injury due to sex (male versus female, p > 0.43). There were also no differences in weight between WT and hGPX1-Tg (at P7, p > 0.13; at P12, p > 0.37).
GPX activity in WT after HI.
Baseline GPX activity was similar in WT brains from all litters. There were no differences between enzyme activities at P7 (31.1 ± 0.9 U/mg in cortex) and P8 (31.9 ± 2.0 U/mg) for the WT naive brains (p < 0.74), hence data were combined into “WT naive” (Fig. 3A). There were no differences in enzyme activities in WT cortices 2 h or 24 h after HI compared with naïve (p < 0.62 and p < 0.09, respectively). Neither the hypoxic cortex (contralateral to carotid ligation) nor the hypoxic-ischemic cortex (ipsilateral to carotid ligation) showed significant change in activity over the first 24 h.

GPX activity in WT, hSOD-Tg and hGPX1-Tg animals. Data are shown as a percentage of the value for uninjured animals. (A) WT littermates of hSOD-Tg and hGPX1-Tg. Values are combined, as there was no difference between the two groups of WT (n = 26 for naive; n = 15 for both 2 h after HI and 24 h after HI). (B) hSOD-Tg. There was a significant decrease 24 h after HI in both hemispheres, *p < 0.006, (n = 16 for naive; n = 5 for 2 h after HI; n = 7 for 24 h after HI). (C) hGPX1-Tg. There was a significant increase in the ipsilateral cortex 24 h after HI, *p < 0.008, (n = 13 for naïve; n = 7 for both 2 h after HI and 24 h after HI). GPX activities between hypoxic-ischemic and hypoxic cortex at both 2 h and 24 h are not significant. All by ANOVA and Bonferroni-Dunn posthoc.
GPX activity in hSOD-Tg after HI.
Baseline GPX activity in hSOD-Tg was significantly higher than in naive WT cortex (36.0 ± 1.2 U/mg protein for SOD, p < 0.004). However, after HI, GPX activity in both the hypoxic-ischemic and hypoxic hSOD-Tg cortices declined significantly by 24 h (HI cortex: 29.1 ± 1.7 U/mg, hypoxic cortex: 29.2 ± 1.22 U/mg, p < 0.006) (Fig. 3B) even though the contralateral hemisphere remained undamaged.
GPX activity in hGPX1-Tg pups after HI.
The baseline GPX enzyme activity in hGPX1-Tg naive cortex was higher than in WT cortex (37.2 ± 1.5 U/mg protein, p < 0.01). After HI, the activity increased 30% over naive hGPX1-Tg brain in the hypoxic-ischemic cortex (48.5 ± 5.16 U/mg protein, p < 0.008) but not in hypoxic cortex (39.6 ± 2.09 U/mg protein, p > 0.56) (Fig. 3C). This differential response correlated with improved histopathological outcome in these brains (Fig. 1, Fig. 2). However, the difference between hypoxic and hypoxic-ischemic hemispheres at 2 h and 24 h is not significant (p < 0.167 and 0.349, respectively).
GPX activity and injury state among groups.
Both hSOD-Tg and hGPX1-Tg naive brains have a small but significantly higher baseline GPX activity than the naive WT at P7 (WT versus hGPX1-Tg, p ≤ 0.011; WT versus hSOD-Tg, p ≤ 0.002). However, after HI, there was a significant increase in GPX activity in HI hGPX1-Tg cortex compared with HI WT (p < 0.02), whereas GPX activity declined over time in injured hSOD-Tg cortex (p < 0.002) (Fig. 4). There was not a significant difference between hSOD-Tg and WT at 24 h (p < 0.14).

GPX activity in WT, hSOD-Tg, and hGPX1-Tg cortex as a function of time. GPX activity is measured in the cortex of naive brain, and at 2 h and 24 h after HI. Naive, hSOD-Tg pups have significantly higher GPX activity than WT, *p < 0.002, as do hGPX1-Tg, *p < 0.011. 24 h after HI, hGPX1-Tg cortex has significantly higher activity than WT, †p < 0.02, and it is also higher than hSOD-Tg, §p < 0.002. Although there is a trend to difference between WT and hSOD-Tg at 24 h, it is not significant (p < 0.14). ANOVA with Bonferroni-Dunn posthoc for multiple comparisons.
DISCUSSION
The current study demonstrates that increased cortical GPX activity from gene overexpression is protective after a moderate HI insult. This protection may be due to a sustained increase in GPX activity during the period of oxidative stress. An increase in GPX activity might be expected after an ischemic insult as a compensatory mechanism in response to accumulation of H2O2, and this has been shown to be the case in adult models of stroke (7) and traumatic brain injury (6,8). However, in the neonatal brain subjected to HI, no compensatory change in GPX activity occurs in wild-type outbred CD-1 mouse brain. These data are similar to those reported for traumatic brain injury in immature brain (5). The increase in GPX activity in the injured cortex of hGPX1-Tg at 24 h was associated with protection, whereas the decrease in GPX activity in the hSOD-Tg at 24 h was associated with increased damage. The decrease in GPX activity bilaterally in hSOD-Tg brain suggests that oxidative stress occurs throughout the brain, even in regions that are not overtly damaged by the HI insult. In fact, our preliminary data show that reduced GSH content in naive hSOD-Tg cortex is significantly lower compared with naïve nonTg P7 cortex (0.98 ± 0.06 nmol/mg versus 1.27 ± 0.04 nmol/mg, p < 0.001). GSH levels remained low in the hypoxic-ischemic hSOD-Tg cortex in response to injury, whereas the contralateral hypoxic cortex maintained significantly higher levels (unpublished observation), suggesting that if GSH concentration is maintained, it may protect cells from apoptotic or necrotic cell death.
Our findings also show that although basal levels of GPX activity were similar in both hGPX1 and hSOD1 overexpressors, and were significantly higher than in WT brains, GPX activity after HI remained preserved only in hGPX1 and not in hSOD1 overexpressors. This decline in activity was also associated with consumption of GSH. Furthermore, a unilateral increase in GPX activity in the HI hemisphere, but not in the hypoxic hemisphere, indicates that substantial oxidative stress is needed to trigger a compensatory increase of GPX activity. We have previously shown that catalase activity does not change after HI in either wild-type or hSOD-Tg brains, so it is unlikely to be a major contributor to antioxidative capacity (12).
In the mature brain, studies with other strains of GPX1 overexpressing transgenic mice have found protection in different models of oxidative stress, such as adult focal cerebral ischemia/reperfusion (14,15,25,26) and transient hypoxia in hippocampal slices (27). This is the first study to report that GPX1 overexpression protects the neonatal brain after HI. Under HI conditions, an insufficient compensatory increase of GPX activity and, hence, detoxification of H2O2, may become a rate-limiting step, as we have previously reported in hSOD1-Tg mice (10). Our current results show that only conditions associated with increased GPX activity result in protection of the neonatal brain. Consistent with this idea, several studies have shown increased brain vulnerability to oxidative stress when either the GPX1 gene is knocked out or GSH is depleted. A direct involvement of GPX1 in protecting neurons from accumulation of reactive oxygen species (ROS) that occurs in both the cytosol and mitochondria has been shown by administration of the mitochondrial toxins, malonate and 3-nitropropionic acid or MPTP (1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine), which act either through inhibition of the respiratory chain in mitochondria or by the formation of ROS (28). Mature mice deficient in GPX1 have increased injury after administration of these agents (28), and adult mice with decreased GPX1 activity from GPX1 gene knockout have shown increased injury after ischemia-reperfusion (26,29). When GPX1 knock-outs are crossed with hSOD1 overexpressors, the resulting hSOD-Tg+/−/GPX1−/− adult hybrids show no protection after ischemia-reperfusion (30), suggesting that the benefit incurred by dismutation of superoxide in the mature brain is eliminated when H2O2 cannot be detoxified by GPX1. This hybrid cross mimics that which occurs developmentally in the newborn brain.
In neonatal rats, whereas pharmacological depletion of GSH, a substrate for GPX1, damages mitochondria and leads to mortality, systemic administration of a GSH precursor, glutathione monoethyl, reverses the injurious effect of GSH depletion (31). Low GSH levels lead to accumulation of intraparenchymal ROS, changes in cellular red-ox cell potential (32) and initiation of stress-activated signal transduction pathways (33–36). Studies in cultured neurons support in vivo data and show that diminished intracellular GSH concentrations affect the equilibrium between cytosolic and mitochondrial GSH and induce direct mitochondrial damage and neuronal death (37,38).
Recent studies show that GPX1 can also preserve GSH homeostasis by detoxifying peroxynitrite (ONOO-) (28,39) which, along with superoxide, inactivates glutathione reductase (40), an enzyme that is solely responsible for GSH resynthesis from its oxidized form. GPX1 overexpression has also been shown to attenuate activation of glial and microglial cells, protect endothelium, and diminish both apoptotic and necrotic neuronal death (15) after ischemia-reperfusion. It is not clear from these published data, however, whether these effects are the sole consequence of the preserved antioxidative homeostasis or whether other mechanisms contribute as well.
A variety of antioxidants have shown mixed results depending on age of animal, time of administration, and drug class. Studies of exogenously applied SOD1 in adult models of trauma and stroke have shown mixed protection depending on model and form of SOD1 used (41). SOD mimetics (42) have shown a reduction in infarct volume when given to neonatal rats before HI (43) and when given to adult gerbils after ischemia-reperfusion (44). The GPX mimic ebselen is protective in adult rats (45, 46) but is toxic in some circumstances (47). Antioxidants tested in the developing brain include allopurinol, mannitol, methionine, and deferoxamine. Allopurinol, a xanthine oxidase inhibitor, is protective after HI in piglets (48) and rats (49,50). Deferoxamine, an iron chelator, affords protection after HI in mice (22) and rats (51), presumably through a reduction in low molecular weight iron, and hence a decrease in the conversion of H2O2 to ˙OH through the Fenton reaction. Deferoxamine is protective against excitotoxic (NMDA) and oxidative (H2O2) damage in primary cultures of hippocampal neurons (13).
The developing brain responds differently to ischemic stress, therefore, a detailed understanding of the maturational changes in oxidative biology is necessary to develop appropriate strategies for prevention of HI injury (52,53). The present study, in the context of previous results from our and other laboratories, suggests that the relationships between H2O2 accumulation and the imbalance between the antioxidant enzymes SOD1 and GPX1 are involved in the capacity of the neonatal brain to protect itself after HI.
Acknowledgments.
The authors thank Charles Epstein, M.D., for the hSOD1-Tg and hGPX1-Tg mice and Alexandra Aminoff for technical assistance.
Abbreviations
- CCA:
-
common carotid artery
- GPX:
-
glutathione peroxidase
- GSH:
-
glutathione
- HI:
-
hypoxia-ischemia
- P:
-
postnatal day
- red-ox:
-
oxidation-reduction reaction
- ROS:
-
reactive oxygen species
- SOD1:
-
copper-zinc superoxide dismutase
- Tg:
-
transgenic
- WT:
-
wild-type
References
Aspberg A, Tottmar O 1992 Development of antioxidant enzymes in rat brain and in reaggregation culture of fetal brain cells. Brain Res Dev Brain Res 66: 55–58
Mavelli I, Rigo A, Federico R, Ciriolo M, Rotilio G 1982 Superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Biochem J 204: 535–540
Buard A, Clement M, Bourre J 1992 Developmental changes in enzymatic systems involved in protection against peroxidation in isolated rat brain microvessels. Neurosci Lett 141: 72–74
Khan JY, Black SM 2003 Developmental changes in murine brain antioxidant enzymes. Pediatr Res 54: 77–82
de Haan JB, Bladier C, Griffiths P, Kelner M, O'Shea RD, Cheung NS, Bronson RT, Silvestro MJ, Wild S, Zheng SS, Beart PM, Hertzog PJ, Kola I 1998 Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J Biol Chem 273: 22528–22536
Fan P, Yamauchi T, Noble LJ, Ferriero DM 2003 Age-dependent differences in glutathione peroxidase activity after traumatic brain injury. J Neurotrauma 20: 437–445
Guegan C, Ceballos-Picot I, Nicole A, Kato H, Onteniente B, Sola B 1998 Recruitment of several neuroprotective pathways after permanent focal ischemia in mice. Exp Neurol 154: 371–380
Goss JR, Taffe KM, Kochanek PM, DeKosky ST 1997 The antioxidant enzymes glutathione peroxidase and catalase increase following traumatic brain injury in the rat. Exp Neurol 146: 291–294
Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H 1994 Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 25: 165–170
Ditelberg J, Sheldon RA, Epstein CJ, Ferriero DM 1996 Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res 39: 204–208
Mischel RE, Kim YS, Sheldon RA, Ferriero DM 1997 Hydrogen peroxide is selectively toxic to immature murine neurons in vitro. Neurosci Lett 231: 17–20
Fullerton HJ, Ditelberg JS, Chen SF, Sarco DP, Chan PH, Epstein CJ, Ferriero DM 1998 Copper/zinc superoxide dismutase transgenic brain accumulates hydrogen peroxide after perinatal hypoxia ischemia. Ann Neurol 44: 357–364
Almli LM, Hamrick SE, Koshy AA, Täuber MG, Ferriero DM 2001 Multiple pathways of neuroprotection against oxidative stress and excitotoxic injury in immature primary hippocampal neurons. Brain Res Dev Brain Res 132: 121–129
Ishibashi N, Prokopenko O, Weisbrot-Lefkowitz M, Reuhl KR, Mirochnitchenko O 2002 Glutathione peroxidase inhibits cell death and glial activation following experimental stroke. Brain Res Mol Brain Res 109: 34–44
Weisbrot-Lefkowitz M, Reuhl K, Perry B, Chan PH, Inouye M, Mirochnitchenko O 1998 Overexpression of human glutathione peroxidase protects transgenic mice against focal cerebral ischemia/reperfusion damage. Brain Res Mol Brain Res 53: 333–338
Sheldon RA, Almli L, Ferriero DM 2002 Copper/zinc superoxide dismutase transgenic brain in neonatal hypoxia-ischemia. Methods Enzymol 353: 389–397
Epstein CJ, Avraham KB, Lovett M, Smith S, Elroy-Stein O, Rotman G, Bry C, Groner Y 1987 Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci U S A 84: 8044–8048
Chan PH, Epstein CJ, Li Y, Huang TT, Carlson E, Kinouchi H, Yang G, Kamii H, Mikawa S, Kondo T, et al. 1995 Transgenic mice and knockout mutants in the study of oxidative stress in brain injury. J Neurotrauma 12: 815–824
Huang TT, Raineri I, Eggerding F, Epstein CJ 2002 Transgenic and mutant mice for oxygen free radical studies. Methods Enzymol 349: 191–213
Mirault ME, Tremblay A, Furling D, Trepanier G, Dugre F, Puymirat J, Pothier F 1994 Transgenic glutathione peroxidase mouse models for neuroprotection studies. Ann N Y Acad Sci 738: 104–115
Rice JE III, Vannucci RC, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141
Sarco DP, Becker J, Palmer C, Sheldon RA, Ferriero DM 2000 The neuroprotective effect of deferoxamine in the hypoxic-ischemic immature mouse brain. Neurosci Lett 282: 113–116
Roveri A, Maiorino M, Ursini F 1994 Enzymatic and immunological measurements of soluble and membrane-bound phospholipid-hydroperoxide glutathione peroxidase. Methods Enzymol 233: 202–212
Flohé L, Günzler WA 1984 Assays of glutathione peroxidase. Methods Enzymol 105: 114–121
Ishibashi N, Prokopenko O, Reuhl K, Mirochnitchenko O 2002 Inflammatory response and glutathione peroxidase in a model of stroke. J Immunol 168: 1926–1933
Keller JN, Huang FF, Zhu H, Yu J, Ho YS, Kindy TS 2000 Oxidative stress-associated impairment of proteasome activity during ischemia-reperfusion injury. J Cereb Blood Flow Metab 20: 1467–1473
Furling D, Ghribi O, Lahsaini A, Mirault ME, Massicotte G 2000 Impairment of synaptic transmission by transient hypoxia in hippocampal slices: improved recovery in glutathione peroxidase transgenic mice. Proc Natl Acad Sci U S A 97: 4351–4356
Klivenyi P, Andreassen O, Ferrante RJ, Dedeoglu A, Mueller G, Lancelot E, Bogdanov M, Andersen JK, Jiang D, Beal MF 2000 Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J Neurosci 20: 1–7
Crack PJ, Taylor JM, Flentjar NJ, de Haan J, Hertzog P, Iannello RC, Kola I 2001 Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J Neurochem 78: 1389–1399
Crack PJ, Taylor JM, de Haan JB, Kola I, Hertzog P, Iannello RC 2003 Glutathione peroxidase-1 contributes to the neuroprotection seen in the superoxide dismutase-1 transgenic mouse in response to ischemia/reperfusion injury. J Cereb Blood Flow Metab 23: 19–22
Martensson J, Meister A, Mrtensson J 1991 Glutathione deficiency decreases tissue ascorbate levels in newborn rats: ascorbate spares glutathione and protects. Proc Natl Acad Sci U S A 88: 6898
Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H 1998 Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 17: 2596–2606
Wilhelm D, Bender K, Knebel A, Angel P 1997 The level of intracellular glutathione is a key regulator for the induction of stress-activated signal transduction pathways including Jun N-terminal protein kinases and p38 kinase by alkylating agents. Mol Cell Biol 17: 4792–4800
Kretz-Remy C, Mehlen P, Mirault M, Arrigo AP 1996 Inhibition of I kappa B-alpha phosphorylation and degradation and subsequent NF-kappa B activation by glutathione peroxidase overexpression. J Cell Biol 133: 1083–1093
Li X, Song L, Jope RS 1998 Glutathione depletion exacerbates impairment by oxidative stress of phosphoinositide hydrolysis, AP-1, and NF-kappa B activation by cholinergic stimulation. Brain Res Mol Brain Res 53: 196–205
Cho S, Hazama M, Urata Y, Goto S, Horiuchi S, Sumikawa K, Kondo T 1999 Protective role of glutathione synthesis in response to oxidized low density lipoprotein in human vascular endothelial cells. Free Radic Biol Med 26: 589–602
Drukarch B, Schepens E, Jongenelen C, Stoof JC, Langeveld CH 1997 Astrocyte-mediated enhancement of neuronal survival is abolished by glutathione deficiency. Brain Res 770: 123–130
Wüllner U, Seyfried J, Groscurth P, Beinroth S, Winter S, Gleichmann M, Heneka M, Loschmann P, Schulz JB, Weller M, Klockgether T 1999 Glutathione depletion and neuronal cell death: the role of reactive oxygen intermediates and mitochondrial function. Brain Res 826: 53–62
Sies H, Klotz LO, Sharov VS, Assmann A, Briviba K 1998 Protection against peroxynitrite by selenoproteins. Z Naturforsch 53: 228–232
Francescutti D, Baldwin J, Lee L, Mutus B 1996 Peroxynitrite modification of glutathione reductase: modeling studies and kinetic evidence suggest the modification of tyrosines at the glutathione disulfide binding site. Protein Eng 9: 189–194
Francis JW, Ren J, Warren L, Brown RH Jr, Finklestein SP 1997 Postischemic infusion of Cu/Zn superoxide dismutase or SOD: Tet451 reduces cerebral infarction following focal ischemia/reperfusion in rats. Exp Neurol 146: 435–443
Salvemini D, Wang ZQ, Zweier JL, Samouilov A, Macarthur H, Misko T, Currie MG, Cuzzocrea S, Sikorski JA, Riley DP 1999 A nonpeptidyl mimic of superoxide dismutase with therapeutic activity in rats. Science 286: 304–306
Shimizu K, Rajapakse N, Horiguchi T, Payne RM, Busija DW 2003 Neuroprotection against hypoxia-ischemia in neonatal rat brain by novel superoxide dismutase mimetics. Neurosci Lett 346: 41–44
Mollace V, Iannone M, Muscoli C, Palma E, Granato T, Modesti A, Nistico R, Rotiroti D, Salvemini D 2003 The protective effect of M40401, a superoxide dismutase mimetic, on post-ischemic brain damage in Mongolian gerbils. BMC Pharmacol 3: 8
Dawson DA, Masayasu H, Graham DI, Macrae IM 1995 The neuroprotective efficacy of ebselen (a glutathione peroxidase mimic) on brain damage induced by transient focal cerebral ischaemia in the rat. Neurosci Lett 185: 65–69
Imai H, Graham DI, Masayasu H, Macrae IM 2003 Antioxidant ebselen reduces oxidative damage in focal cerebral ischemia. Free Radic Biol Med 34: 56–63
Guerin PJ, Gauthier ER 2003 Induction of cellular necrosis by the glutathione peroxidase mimetic ebselen. J Cell Biochem 89: 203–211
Peeters-Scholte C, Braun K, Koster J, Kops N, Blomgren K, Buonocore G, Van Buul-Offers S, Hagberg H, Nicolay K, van Bel F, Groenendaal F 2003 Effects of allopurinol and deferoxamine on reperfusion injury of the brain in newborn piglets after neonatal hypoxia-ischemia. Pediatr Res 54: 516–522
Palmer C, Vannucci RC, Towfighi J 1990 Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr Res 27: 332–336
Palmer C, Towfighi J, Roberts RL, Heitjan DF 1993 Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr Res 33: 405–411
Palmer C, Roberts RL, Bero C 1994 Deferoxamine posttreatment reduces ischemic brain injury in neonatal rats. Stroke 25: 1039–1045
Whitelaw A, Thoresen M 2002 Clinical trials of treatments after perinatal asphyxia. Curr Opin Pediatr 14: 664–668
Hamrick SE, Ferriero DM 2003 The injury response in the term newborn brain: can we neuroprotect?. Curr Opin Neurol 16: 147–154
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by National Institutes of Health grant NS 33997.
Rights and permissions
About this article
Cite this article
Sheldon, R., Jiang, X., Francisco, C. et al. Manipulation of Antioxidant Pathways in Neonatal Murine Brain. Pediatr Res 56, 656–662 (2004). https://doi.org/10.1203/01.PDR.0000139413.27864.50
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000139413.27864.50
This article is cited by
-
Regionally Impaired Redox Homeostasis in the Brain of Rats Subjected to Global Perinatal Asphyxia: Sustained Effect up to 14 Postnatal Days
Neurotoxicity Research (2018)
-
Paraquat and Maneb Exposure Alters Rat Neural Stem Cell Proliferation by Inducing Oxidative Stress: New Insights on Pesticide-Induced Neurodevelopmental Toxicity
Neurotoxicity Research (2018)
-
Effect of Resveratrol on Oxidative Stress and Mitochondrial Dysfunction in Immature Brain during Epileptogenesis
Molecular Neurobiology (2018)
-
Acute maternal oxidant exposure causes susceptibility of the fetal brain to inflammation and oxidative stress
Journal of Neuroinflammation (2017)
-
Glucose and Intermediary Metabolism and Astrocyte–Neuron Interactions Following Neonatal Hypoxia–Ischemia in Rat
Neurochemical Research (2017)
