
Abstract
RNA polymerases (POL) are integral constituents of the protein synthesis machinery, with POL I and POL III coding for ribosomal RNA and POL II coding for protein. POL I is located in the nucleolus and transcribes class I genes, those that code for large ribosomal RNA. It has been reported that the POL system is seriously affected in perinatal asphyxia (PA) immediately after birth. Because POL I is necessary for protein synthesis and brain protein synthesis was shown to be deranged after hypoxic-ischemic conditions, we aimed to study whether POL derangement persists in a simple, well-documented animal model of graded global PA at the activity, mRNA, protein, and morphologic level until 8 d after the asphyctic insult. Nuclear POL I activity was determined according to a radiochemical method; mRNA steady state and protein levels of RPA4O—an essential subunit of POL I and III-were evaluated by blotting methods; and the POL I subunit polymerase activating factor-53 was evaluated using immunohistochemistry. Silver staining and transmission electron microscopy were used to examine the nucleolus. At the eighth day after PA, nuclear POL I decreased with the length of the asphyctic period, whereas mRNA and protein levels for RPA4O were unchanged. The subunit polymerase activating factor-53, however, was unambiguously reduced in several brain regions. Dramatic changes of nucleolar morphology were observed, the main finding being nucleolar disintegration at the electron microscopy level. We suggest that severe acidosis and/or deficient protein kinase C in the brain during the asphyctic period may be responsible for disintegration of the nucleolus as well as for decreased POL activity persisting until the eighth day after PA. The biologic effect may be that PA causes impaired RNA and protein synthesis, which has been already observed in hypoxic-ischemic states.
Similar content being viewed by others

Main
In eukaryotes, three different POL transcribe nuclear genes. POL I transcribes class I genes, those that code for large ribosomal RNA (rRNA). It is located in the nucleolus, represents 50–70% of polymerase activity per cell, and as many as 40,000 copies per cell are present (1). This high number of POL I molecules transcribes as few as about 100 genes, which is appropriate as the rRNA precursor is long (i.e. 6,000–15,000 nucleotides), and as many as 50 polymerase molecules may transcribe each rRNA gene simultaneously. This intense transcriptional activity reflects the need of the cell for rRNA; 10 million copies of rRNA are required (2) to form the 10 million ribosomes required for a single cell division. rRNA genes, transcribed by POL I, form the primary RNA transcript, the 45S-rRNA. Before release from the nucleus as ribosomal particles, 45S-rRNA is cleaved to a copy of 28S-rRNA, 18S-rRNA, and 5.8S-rRNA each. POL III transcribes class III genes, those that encode a number of small RNA molecules, including 5S-rRNA. It is located in the nucleoplasm, makes up 10% of the polymerase activity of the cell, and about 20,000 copies are present per cell (3). These transcribed genes are assembled with other rRNA products transcribed by different POL to form ribosomes. This process, ribosome biogenesis, is described in detail in excellent reviews (4, 5). The complex mechanism of rRNA—transcription by POL—has not been fully elucidated yet, but the principles are well established and given elsewhere (6, 7). The regulation of transcription by POL is described in a series of publications (4).
Information about RNA polymerases in hypoxic-ischemic states is limited (8–10) and data from adult models of ischemia/hypoxia representing stroke rather than PA are of limited value for the explanation and the understanding of pathophysiology and biochemistry of PA, as it is widely accepted that metabolism and disease mechanisms are different in the immature system (11–14).
In a recent report using a simple, well-documented, and noninvasive model of PA that resembles the clinical situation (15, 16), we have shown that the POL I system in PA was affected as soon as within minutes after the asphyctic insult (17). It was the aim of this study to show whether this finding would be transient or persisting, and we indeed observed that POL derangement as reflected by decreased POL activity, an impaired POL subunit at the immunohistochemical and nucleolar disintegration at the electron microscopy level, was found approximately a week after the asphyctic insult.
METHODS
Animals and Experimental Design
Asphyxia was induced in pups delivered by cesarean section on pregnant Sprague-Dawley rats. Within the last day of gestation as evaluated by estabularium protocols, animals were killed by neck dislocation and hysterectomized. The uterine horns, still containing the fetuses, were extirpated and placed in a water bath at 37°C for various periods from 5 to 20 min. Cesarean-delivered control and asphyctic pups were obtained from the same mother, inasmuch as each rat delivered approximately 10–14 pups.
After the asphyctic period, i.e. incubation at 37°C, the uterus horns were rapidly opened and the pups removed. Pups were cleaned, the umbilical cord was ligated, and the animals were allowed to recover in a hood. Only litters with pups weighing >4.5 g at the time of delivery were used in the experiments. Ten pups per group (normoxia for 10 min and 20 min of asphyxia) were studied and six pools of three pup brains per group (normoxia, 10 min of asphyxia, 20 min of asphyxia) each were used for the POL I activity determinations. In the group with normoxia and 10 min of PA, 100% survival was be found and in the group with 20 min of asphyxia, 74% survived. At 21 min of asphyxia, only 10% of the pups survived.
Rat pups were given to surrogate mothers and were well accepted. The animal studies were carried out according to the rules from the American Physiology Society and approved by the local animal committee. At the age of 8 d, rats were killed for the experiments. Animals used for biochemical studies were killed by decapitation and the brain was kept at −80°C until biochemical assays were performed. Brains used for POL I activity studies were prepared immediately as given below. Brains used for morphologic studies were taken from fixation-perfused rats as given below.
mRNA Isolation from Brain, Northern Blots, and Dot Blots
The organs were obtained at autopsy and taken immediately into liquid nitrogen. Frozen brain samples were ground and mRNA extraction was performed using the Quick Prep Micro mRNA Purification kit (Amersham Pharmacia Biotech AB, Uppsala, Sweden) (18). Subsequently, 6 μg mRNA each, dissolved in 6 μL diethylpyrocarbonate (DEPC) water, 11.8 μL of DMSO, 2.4 μL 0.1 M NaHPO4 buffer, and 3.5 μL deionized 40% glyoxal were incubated for 1 h at 50°C and subsequently cooled on ice. To this solution, 6.3 μL of a mixture of 50% glyoxal, 10 mM sodium phosphate, and 0.4% bromphenol blue were added. The sample was applied onto 1.2% agarose gel and electrophoresed at 3–4 V/cm for 2.5 h in circulating 0.01 M phosphate buffer, pH 7.0. RNA was then transferred to a positively charged nylon membrane (Hybond N+, DuPont, Bad Homburg, Germany) by capillary blotting and fixed with 0.05 N sodium hydroxide for 5 min at room temperature and finally equilibrated at pH 7.0 with three washes in 2× standard saline citrate (SSC). The cDNA probe for β-actin was purchased (ATCC 9800, American Type Culture Collection, Manassas, VA, U.S.A.), cDNA for RNA polymerase subunit RPA40 was constructed, and these probes were used for Northern and dot blots.
Preparation and labeling of the probe for RPA40 (EMBO, Heidelberg, Germany) was done as follows: The RPAC 40 homolog was isolated from AA8 OHO cells (ATCC 4433) and amplified using the plasmid pSPORT1 in Escherichia coli XL1-Blue MRF (Stratagene, Heidelberg, Germany) in Liria Broth (LB) medium containing 100 μg ampicillin/mL overnight at 37°C. Isolation was carried out with Wizard Minipreps (Promega, Madison, WI, U.S.A.). cDNA RPA40 was cut from 5 μg of the plasmid by the use of Eco R I (5 U) and Hin dIII (5 U), separated on 1% TAE agarose gel electrophoresis, and eluted by QIAquick (QIAGEN, Valencia, CA, U.S.A.) from the gel. One hundred nanograms of the RPAC 40 homolog from AA8 were added to 5 μL 10× SSC buffer, 1 μL 20 mM dNTP-Mix, 1 μL Taq-DNA polymerase (5 U/μL, Amersham Pharmacia Biotech AB), and 1 μL 5′-CCTTCAATAGGAGACAAG-3′ (18-mer, temperature 38.3°C, 100 pmol/μL, amino acid sequence in AA8RPA:LSPTEG) or 5′-CGCCGAGATCCCAACTCT-3′ (18-mer, temperature 52.3°C, 100 pmol/μL, amino acid sequence in AA8RPA: AEIPTL) and 5 μL fluorescein-nucleotide mix (Renaissance random primer fluorescein labeling kit, DuPont). This mixture was used for the amplification in 40 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s (19).
Labeling of the β-actin probe was performed during PCR and after the human β-actin control amplimer set (CLONTECH, Palo Alto, CA, U.S.A.), adding 5 μL of fluorescein-nucleotide mix (Renaissance random primer fluorescein labeling kit). PCR products were separated in a 1% Tris acetate-EDTA (TAE)-agarose gel and eluted.
After fixation of bound RNA, the nylon membrane was incubated in prehybridization solution [0.25 M phosphate buffer, pH 7.2, containing 5% SDS wt/vol, 1 mM EDTA, and 0.5% blocking reagent (from Dupont NEL 203)] for 12 h at 65°C in a hybridization oven. The blots were hybridized overnight at 65°C with the labeled probes each (50 ng/mL of prehybridization buffer).
After hybridization, nonspecifically bound material was removed by posthybridization washes with 0.5× and 0.1× prehybridization buffer twice at 10 min each at 65°C. The 0.5× and 0.1× prehybridization buffer was brought up to 65°C before use, and the second wash was performed at room temperature.
Hybridized blots were blocked with 0.5% blocking reagent in 0.1 M Tris HCl, pH 7.2, and 0.15 M NaCl for 1 h at room temperature. Membranes were then incubated with antifluorescein horseradish peroxidase antibody (DuPont) at a 1:1000 dilution in the solution given above for 1 h under constant shaking. Membranes were washed four times for 5 min each in the solution given above.
The nucleic acid chemiluminescence reagent (DuPont) was added to the membranes and incubated for 1 min. Excess detection reagent was removed by the use of filter paper, the membrane was placed in plastic wrap and exposed to autoradiography reflection films (DuPont) for 15 min at room temperature.
Dot blots were performed according to the method of White and Bancroft (20). One microgram of mRNA in 10 μL diethylpyrocarbonate (DEPC) H2O was added to 30 μL of a mixture of 500 μL deionized formamide, 162 μL 37% formaldehyde, and 100 μL 10 × MOPS. This solution was brought to 65°C for 5 min and cooled on ice for 3 min.
A nylon membrane (Hybond N, Amersham Pharmacia Biotech AB) was moistened in 10× MOPS and put into two layers of moistened Whatman 3MM filter paper (BioRAD, Vienna, Austria) in the dot blot apparatus. The chamber was closed tightly and 40 μL of each sample was applied under vacuum. Under vacuum, 0.05 M NaOH was added, the nylon membrane was removed from the dot blot apparatus, and it was again incubated with 0.05 M NaOH for 5 min. Finally, the nylon membrane was equilibrated in 2× SSC to reach a pH of 7.5. Prehybridization was performed by 0.1 mL of prehybridization solution [5 × SSC, 0.1% (wt/vol) SDS, 0.5% (wt/vol) blocking reagent (DuPont), 5% dextran sulfate, dissolved at 60°C, and used at room temperature] per square centimeter of membrane and the volume of the probe was 20 ng/mL, wrapped in a plastic bag. Subsequent washing steps and detection of the products was performed according to the manufacturer's instructions. Densitometry of films was performed using the Elscript 400 densitometer (Hirschmann, Berlin, Germany).
Western Blots for the Determination of RPA40 Immunoreactive Protein
Snap frozen brains were thawed on ice and mixed 1:5 (wt/vol) with homogenization solution (0.25 M sucrose, 1 mM EDTA, 3 mM imidazole, 0.1% ethanol, pH 7.2) (21). Samples were homogenized for 30 s (six strokes) at 440 rev/min in a Potter-Elvehjem homogenizer on ice. The homogenate was centrifuged for 5 min at 3000 ×g and 4°C. The supernatant was used for the determination of protein and immunoblotting. Using ice-cold homogenization solution, the supernatants were brought to equivalent protein concentrations. To these supernatants, equal amounts of sample buffer were added. Samples were heated at 95°C and loaded onto a 7.5% SDS-polyacrylamide gel. After separation proteins were transferred electrophoretically to nitrocellulose membranes for 1 h at 100 V in blotting buffer (25 mM Tris, 192 mM glycine, 20% methanol), membranes (Hybond-P, Amersham Pharmacia Biotech AB) were washed and blocked in PBS (137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4 × 12 H2O, 1.5 mM KH2PO4) containing 0.05% Tween-20 and 1% BSA for 1 h at room temperature, followed by probing of the membranes with a RPA4O antibody generously provided by I. Grummt, German Cancer Research Center, Heidelberg, Germany. This antibody was diluted 1:1000 (vol/vol) with PBS containing 0.05% Tween-20. Membranes were washed four times for 15 min in PBS containing 0.05% Tween-20 and 1% BSA before incubation at room temperature with horseradish peroxidase-conjugated goat-anti-rabbit IgG (Sigma Chemical, St. Louis, MO, U.S.A.), diluted 1:16.000 (vol/vol) in PBS/Tween-20. Membranes were washed again four times for 15 min and the blots were developed with the ECL Western blotting System (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, UK). Densitometry was performed as described above.
Determination of Brain Tissue pH
Brain tissue pH was measured 8 d after the asphyctic period by inserting the tip of a tissue pH electrode of 2 mm diameter (Wissenschaftlich-Technische Werkstaetten, Vienna, Austria) into the thawed whole tissue kept at −80°C. Measurements were done in triplicate with pepsin-distilled water rinsing cycles (16).
Determination of Nuclear POL I
Preparation and isolation of brain cell nuclei.
Rat brain was homogenized and nuclei were isolated according to the method given by McEwen and Zigmond (22). Animals were killed by decapitation and brains were rapidly removed, placed on ice, and minced. Pools of three neonatal brains (average weight 200 mg) were transferred to a precooled 5-mL glass homogenizer with a Teflon pestle with ice-cold buffer I (0.32 M sucrose, 1 mM KH2PO4, 1 mM MgCl2, pH 6.5). The tissue was homogenized with 20 gentle strokes and transferred to a chilled tube. The homogenate was filtered through two layers of sterile cheese cloth into 35-mL centrifuge tubes and centrifuged for 10 min at 850 ×g at 4°C (Beckman J2-21 centrifuge, Beckman Coulter, Inc., Fullerton, CA, U.S.A.). The supernatants were removed, and the pellet was resuspended in 20 mL of buffer I, mixed, and centrifuged again for 10 min under the same conditions. Procedures were repeated with 20 mL of buffer I and centrifugation at 650 ×g for 10 min at 4°C. The supernatant was discarded and the pellet was resuspended in 4 mL of buffer I and vigorously vortexed. Twenty-five milliliters of buffer II (2.39 M sucrose, 1 mM KH2PO4, 1 mM MgCl2, pH 6.5) were added and mixed thoroughly with the nuclear suspension.
This mixture was centrifuged for 60 min at 63,600 ×g in a SW28 rotor of a Beckman L8–55 ultracentrifuge (Beckman Coulter, Inc.). After carefully removing the supernatant and swabbing the interior walls of the tubes with tissue paper, the purified nuclear pellets were resuspended in 5 mL solution containing 10 mM Tris-HCl pH 7.5, 1 mM MgCl2, 1 mM dithiothreitol (DTT), and 10% (vol/vol) glycerol.
The quality of each nuclear suspension was optically controlled by phase contrast microscopy (Nikon TMS, Tokyo, Japan). The total amount of nuclei of each preparation was counted using a cell counter (Sysmex Microcellcounter CC 108, TOA Medical Electronics Corp., Japan).
Measurement of protein, DNA, and RNA concentrations.
The brain nuclei suspension was analyzed for protein content by the method of Lowry et al.(23) using BSA as standard. The concentration of DNA and RNA was measured using an analyzer (GeneQuant II, Amersham Pharmacia Biotech).
Measurement of POL I activity.
POL I activity was measured according to the methods described by Kokko et al.(24) and Nakamura et al.(25) with slight modifications. Isolated brain nuclei (15–25 μg of DNA) were suspended in incubation medium containing 140 mM Tris-HCI, pH 7.9, 3.5 mM DTT, 7 mM MgCl2, 9 mmol/L NaF, 0.6 mM each of guanosine 5′-triphosphate, ATP, and cytidine 5′-triphosphate, 10 μmol/L nonlabeled uridine triphosphate (UTP), and 1 μCi [31H]UTP (12.2 Ci/mmol, Amersham Pharmacia Biotech) with 1 μg α-amanitin (Sigma Chemical). All assays were performed in triplicate. Samples were incubated at 37°C for 15 min; blank samples were kept on ice for the same period of time. The reaction was stopped by cooling the tubes to 0°C and adding 100 μL of 0.4 mol/L Na-pyrophosphate and 400 μL of 25% ice-cold trichloroacetic acid (TCA) solution.
After keeping all tubes in ice for 15 min and centrifugation at 10,000 ×g for 15 min at 4°C, the supernatants were discarded and the pellets were washed twice with 800 μL of ice-cold TCA. The precipitate was dried at 37°C for 15 min and dissolved in 100 μL of formic acid and 5 mL scintillation fluid (Ready Safe liquid scintillation cocktail, Beckman Coulter, Inc.). The radioisotope activity was measured with a liquid scintillation analyzer (2000 CA TM-Carb, Packard, Netherlands).
RNA polymerase activity was measured in six pools (three brains per pool) of each group (normoxia, 5 min, 10 min, 15 min, and 20 min of asphyxia followed by a 10-min recovery period) and expressed as the difference between the sample and the blanks. Results were given as counts per milligram DNA and alternatively per milligram protein.
Histologic and Morphometric Examinations
Fixation and embedding.
A syringe was placed into the aorta of anesthetized animals and a fixative consisting of 2% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, was used for the perfusion of animals for 5 min. For electron microscopy investigations, a second fixative (3.0% glutaraldehyde in the identical buffer) was perfused for 5 min. Perfused corpses were placed at 4°C and subsequently brains were removed from the skull, postfixed for 2 h in the same fixative, washed three times in PBS, dehydrated, and embedded in paraffin for light microscopy and embedded in LR-White (Eppelheim, Germany) for electron microscopy studies. For light microscopy, paraffin sections of 5 μm were mounted on positively charged slides (DAKO, Carpinteria, CA, U.S.A.). We used LR-White semithin sections of 0.5 μm for morphometric analysis and 0.1 μm for electron microscopy observations.
Immunohistochemistry
Because the antibody against RPA40 did not stain paraffin-embedded brain sections, we decided to use an antibody against the essential POL I subunit PAF53 (kindly supplied by I. Grummt, DKFZ, Heidelberg, Germany).
Six-micrometer-thick paraffin sections were mounted on adhesive slides, dewaxed, rehydrated, and washed twice in PBS. For antigen retrieval, sections were subsequently incubated in PBS containing 0.2 M EDTA, autoclaved at 121°C for 2 min, and allowed to cool down for 3 h. Sections were rinsed briefly in distilled water, washed twice in PBS, and preincubated with FCS and 0.05% Tween-20 (Sigma Chemical) for 30 min at room temperature. The primary antibodies were diluted 1:300 in DAKO antibody diluent. The incubation took place overnight at room temperature. Optimized standardization of all further steps (secondary reagent incubation, section washings, and signal development) was performed using an automated immunostainer using DAKO immunoperoxidase kit (DAKO TechMate designed for Horizon immunostainer). The immunostained slices were dehydrated in ascending ethanol series and two changes of xylene, permanently mounted with a coverslip, and examined on a Nikon Microphot light microscope.
Silver Staining of Nucleoli and Nuclear and Nucleolar Morphometry
For the visualization of nucleoli, slides were dewaxed by three changes of xylene for 10 min each, rehydrated in a graded series of ethanol, and incubated in 0.1 M citric acid adjusted to pH 6.0 at 121°C in an autoclave for 20 min and allowed to cool overnight. The silver-staining procedure was performed as described by Lindner (26) and counterstaining was with 0.1% methyl green.
Morphometry of nuclear- and nucleolar-size micrographs of silver-stained brain sections was done on a Nikon Microphot FMX. Kodak technical pan films (100 ISO) developed for 6 min in Ifotec HC at 20°C were used. Prints were enlarged to a final size of ×2500 and the images scanned into a computer. The size of nuclei and nucleoli were measured using the computer program Scan (Sigma, St. Louis, MO, U.S.A.). We compared normoxic with graded perinatally asphyctic pups. In a second approach, we stained semithin sections of exactly 0.5 μm thickness with toluidine blue, which preferentially stains nucleolar ribonucleoproteins (27). The results obtained reflect the average nucleolar portion within the cell nucleus (28).
Electron Microscopical Studies
Thin sections were mounted on 300-mesh copper grids stained with 1% uranyl acetate for 3 min and 1% lead citrate for 1 min, rinsed in five changes of distilled water, air dried, and examined in a transmission electron microscope (Jeoul 1200, Tokyo, Japan).
The observed structural differences in the arrangement of nucleolar components that we named “compact” and “fragmented-nucleolonema type” were quantified in a blinded manner as follows: Observed nucleoli were assigned to one of the defined types, compact or fragmented-nucleolonema type, by two independent observers in the normoxia group and the asphyctic groups from 5 to 20 min.
Statistical Methods
ANOVA with subsequent Kruskal-Wallis test, or t test when appropriate, was applied. Statistical significance was considered at the p < 0.05 level.
RESULTS
Biochemical Results
Biochemical results are presented in the Table 1. For mRNA levels, a single band was observed in the Northern blots, indicating specificity of the hybridization technique. mRNA steady state levels for the essential subunit RPA4O were normalized versus the housekeeping gene β-actin. No significant differences between animals subjected to normoxia and animals subjected to asphyctic periods of 10 or 20 min were observed in the dot blots.
For protein and immunoreactive RPA4O levels, a single band at 40 kD revealed the specificity of the antibody. The levels of RPA immunoreactivity were not significantly different between normoxic and asphyctic rat brains. Nuclear POL I activity significantly decreased with the length of the asphyctic period when normalized versus protein or RNA. Brain pH was comparable between asphyctic and normoxic brain.
Morphologic Results
Immunohistochemistry.
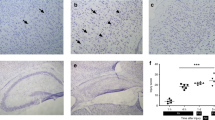
As shown in Figue 1, positive staining for PAF53 was observed in three brain regions: frontal cortex, striatum, and hippocampus CA1. In all brain regions of asphyctic pups, the signal was found in the nucleolus and was consistently decreased. Under identical staining conditions, most cells from PA revealed weak or no contrast. Quantification of PAF53 immunoreactivity in frontal cortex is shown in Table 2.

Reduced immunostaining for PAF53 is found in three areas at 20 min of perinatal asphyxia (right panel) compared with normoxia (left panel). The (nucleolar) staining pattern in frontal cortex (a,b), striatum (c,d), hippocampus CA1 (e,f) revealed a fuzzy, washed out appearance in the PA group [×1600, the bar represents 40 μm (a–e) and 100 μm (e–f)].
Silver staining.
As shown in Figure 2, nuclei and nucleoli were well contrasted by silver impregnation and nuclear counterstaining in normoxic animals. Ten minutes of asphyxia were correlated with a reduction in silver staining, and in all brains subjected to 20 min of asphyxia, neurons had a “fuzzy,” washed-out appearance, and the majority of the nuclei and nucleoli were reduced in size (Fig. 2).

Silver-staining pattern in three brain regions (right, PA;left, normoxia). (a,b) Frontal cortex neurons after silver staining for nucleolar visualization. A dense, well-contrasted staining is shown in brain of normoxic animals, whereas nucleoli of animals after 20 min of asphyxia were reduced in size and showed fuzzy, washed-out staining. Identical staining patterns were found in striatum (c,d) and hippocampus CA1 (e,f) (×1600, the bar represents 40 μm).
Morphometry revealed a significantly reduced total nucleolar size. The average nucleolar fraction per cell was also reduced, indicating that the reduction in nuclear size was not secondary to a reduction in cell size (Table 3).
Electron microscopical findings.
Under normoxic conditions, the nucleolar dense fibrillar component forms strands arranged around the fibrillar center, forming together the fibrillar complex (29, 30). This fibrillar complex is associated with and embedded in the granular component (Fig. 3a).

Electron microscopicy demonstration of nucleolar changes in PA. The nucleolus from a frontal cortex neuron of a normoxic rat pup (left) in comparison with a nucleolus from an animal asphyctic for 20 min (right) at a magnification of ×30,000. The most prominent difference is the fragmented, disintegrated appearance of the strands of the dense fibrillar component (df). gc, granular component. Bar represents 200 nm.
In PA, distinct rearrangements—fragmentations of nucleolar structure—were found.
Under asphyctic conditions, the granular component dispersed and disappeared. Strands of dense fibrillar component revealed a deconvoluted, fragmented appearance. Only a small portion was attached to a fibrillar center, a major portion appeared in loosely arranged fragments and was clearly distinguishable from possible remains of granular component (Fig. 3b).
Morphometry of the two morphologically different nucleolar types showed the prevalence of the fragmented nucleolar type in frontal cortex of animals with PA (Table 4).
DISCUSSION
In a previous study, we showed deterioration of RNA polymerase and nucleolar changes within minutes after PA (17). Here, we show that deficient POL I activity along with decreased essential POL subunit PAF53 protein and disintegration of nucleolar structures still can be detected 8 d after the asphyctic insult. We proposed that decreased POL I activity in brain of asphyctic animals was caused by acidosis, and, indeed, irreversible inactivation of POL I and II by low pH has been described (31). This could serve as an explanation for impaired POL I activity at the time point of 8 d in this study. Another possibility is that POL I activity and even nucleolar integrity are affected by dramatic reduction of protein kinases as, for example, protein kinase C (PKC). In brain and in heart, we detected PKC and PKA decreasing with the length of the asphyctic period in the identical animal model used in the present study (15, 16). This proposed tentative mechanism is supported by previously published observations that a) POL I shows protein kinase activity itself (32), b) POL I is phosphorylated by nuclear protein kinases (33, 34), and c) it has been shown that POL I activity is strongly linked to PKC activity (35) and that the interaction between PKC and POL is valid also for other RNA polymerases (36, 37).
The essential POL I subunit RPA40 was preserved in terms of protein levels, thus arguing against a simple loss of cells and protein. The essential subunit PAF53, however, was remarkably reduced in asphyctic brain as shown by immunohistochemistry. We suggest that this subunit was disappearing, disintegrating from POL I, degraded, or its synthesis simply not recovering during the period of 8 d. Disintegration from the POL I complex is less likely as, during conditions like serum starvation, actinomycin treatment, and mitosis, PAF53 remains attached to the transcriptional machinery (38). It is also unclear whether deficient or disintegrated PAF53 would have contributed to impaired POL I activity, as Hanada et al.(39) have shown that it is rather involved in the formation of the initiation complex at the promoter by mediating the interaction between POL I and UBF, the upstream binding factor for active rRNA synthesis. Decreased immunoreactive PAF53 in asphyctic brain samples may therefore help to explain decreased RNA, which was described in neurons to persist until at least 3 mo after PA (17). Decreased POL I activity along with decreased PAF53 may well cause decreased RNA in the postasphyctic period. Decreased RNA synthesis after ischemic-hypoxic insults has been described already (8, 10, 40), and the consequence of decreased RNA on decreased protein synthesis has been addressed by several groups (41–45).
In addition, synthesis and turnover of rRNA in the brain are low, even under physiologic conditions (46), supporting our findings of delayed recovery of postasphyctic POL functions and RNA synthesis in neuronal and glial cells during early postnatal development is decreasing (47).
Disintegration of the nucleolus in our present study is most probably linked to impaired POL I activity but could lead to decreased RNA and protein synthesis. Nucleolar changes, as revealed by silver staining, were found already in heart after PA in the identical animal model, as published previously (19). Here, we describe the underlying ultrastructural findings of disintegration—rearrangement of nucleolar structural components. The granular component was dispersed/disappearing, and the strands of nucleolar dense fibrillary component of the nucleolus were fragmented. As at the time point of 8 d after PA, pH returned to physiologic levels, nucleolar changes—most probably due to severe acidosis at the time of the asphyctic insult—may not have been repaired within 8 d. It is widely accepted that nucleolar changes as evaluated by silver staining reflect nucleolar activity (48, 49); that the dense fibrillar component, here shown to be altered in the postasphyctic brain, contains DNA (50); and that this DNA is actively described (51, 52) and is mandatory for early processing steps of nascent transcripts (53). mRNA steady state levels for RPA40 were unchanged at the time point of 8 d, but we cannot exclude that determination of the transcriptional rate would have shown decreased transcriptional function. The possibility that nucleolar disintegration in addition to POL I inhibition was originally induced by severe acidosis is highly likely, as Barsotti et al.(54) found nucleolar fragmentation by α-amanitin-mediated blocking of POL II. The ATP depletion, found in the early phase of PA in our model, may have contributed to the disintegration of nucleolar structure as well, because the integrity of the nucleolus depends on ATP (55, 56).
We conclude that deficient POL I and nucleolar changes persist until at least d 8 of the postasphyctic period. Both phenomena may be the result of severe acidosis, ATP depletion, and probably impaired phosphorylation of POL I at the time of the asphyctic insult. Because of several factors such as irreversible inhibition of POL I by acidosis, low RNA synthesis, and turnover in the postnatal brain, recovery of the RNA polymerase machinery was delayed. The consequences of long-lasting deficient POL I and nucleolar changes are obviously impaired RNA and protein synthesis in the postasphyctic brain.
Abbreviations
- PA:
-
perinatal asphyxia
- PAF53:
-
polymerase activating factor-53
- POL:
-
RNA polymerase
- RPA4O:
-
RNA polymerase subunit-40
References
Chambon P 1975 Eucaryotic nuclear RNA polymerases. Annu Rev Biochem 44: 613–638
Sentenac A 1985 Eucaryotic RNA polymerases. Crit Rev Biochem 18: 31–91
Geiduschek EP, Tocchini-Valentini GP 1988 Transcription by RNA polymerase. Annu Rev Biochem 57: 873–914
Larson DE, Zahradka P, Seils BH 1991 Control points in eucaryotic ribosome biogenesis. Biochem Cell Biol 69: 5–22
Long EO, Dawid IB 1980 Repeated genes in eukaryotes. Annu Rev Biochem 49: 727–764
Kerppola TK, Kane CM 1991 RNA polymerase: regulation of transcript elongation and termination. FASEB J 5: 2822–2842
Sollner-Webb B, Tower J 1986 Transcription of cloned eukaryotic ribosomal RNA genes. Annu Rev Biochem 55: 801–830
Yanagihara T 1978 Experimental stroke in gerbils: effect on translation and transcription. Brain Res 158: 435–444
Gottlieb M, Chavko M, Marsala J 1981 Effect of ischemia on the activity of DNA dependent RNA polymerase and DNA polymerase. J Neurochem 37: 255–257
Dienel GA, Cruz NF, Rosenfeld SJ 1985 Temporal profiles of proteins responsive to transient ischemia. J Neurochem 44: 600–610
Raju TNK 1992 Some animal models for the study of perinatal asphyxia. Biol Neonate 62: 202–214
Blennow M, Ingvar M, Lagercrantz H, Stone-Elander S, Eriksson L, Forssberg H, Ericson K, Flodmark O 1995 Early [18F] FDG positron emission tomography in infants with hypoxic-ischemic encephalopathy shows hypermetabolism during the postasphyctic period. Acta Paediatr 84: 1289–1295
Abdel-Rahman A, Parks JK, Devereaux MW, Sokol RJ, Parker WD, Rosenberg AA 1995 Developmental changes in newborn lamb brain mitochondrial activity and postasphyxial lipid peroxidation. Proc Soc Exp Biol Med 209: 170–177
Hallgren B, Sourander P 1958 The effect of age on the nonhaemin iron in the human brain. J Neurochem 3: 41–51
Lubec B, Dell'Anna E, Fang-Kircher S, Marx M, Herrera-Marschitz M, Lubec G 1997 Decrease of brain protein kinase C, protein kinase A and cyclin dependent kinase correlating with pH precedes neuronal death in neonatal asphyxia. J Invest Med 45: 284–294
Lubec B, Marx M, Herrera-Marschitz M, Labudova O, Hoeger H, Gille L, Nohl H, Mosgoelier W, Lubec G 1997 Decrease of heart protein kinase C and cyclin dependent kinase precedes death in perinatal asphyxia of the rat. FASEB J 11: 482–491
Mosgoeller W, Kastner PH, Fang-Kircher S, Hoeger H, Lubec G, Lubec B 2000 Brain RNA polymerase and nucleolar structure in perinatal asphyxia of the rat. Exp Neurol 161: 174–182
Hardmeier R, Hoeger H, Fang-Kircher S, Khoshsorur A, Lubec G 1997 Transcription and activity of superoxide dismutase, catalase and glutathione peroxidase following irradiation in radiation resistant and radiation sensitive mice. Proc Natl Acad Sci U S A 94: 7572–7576
Hoeger H, Labudova O, Mosgoeller W, Herrera-Marschitz M, Fuerst G, Kitzmueller E, Lubec B 1998 Deficient transcription of subunit RPA 40 of RNA polymerase I and III in heart of rats with neonatal asphyxia. Life Sci 62: 275–282
White BA, Bancroft FC 1982 Cytoplasmic dot hybridization. Simple analysis of mRNA levels in multiple small cell or tissue samples. J Biol Chem 257: 8569–8575
Labudova O, Krapfenbauer K, Moenkemann H, Rink H, Kitzmueller E, Cairns N, Lubec G 1998 Decreased transcription factor junD in brain of patients with Down syndrome. Neurosci Lett 252: 159–162
McEwen BS, Zigmond RE 1972 Isolation of brain cell nuclei. In: Marks N, Rodnight R (eds) Research Methods in Neurochemistry, Vol l, Plenum Press, New York, 139–161.
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ 1951 Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265–275
Kokko E, Isomaa V, Jaenne O 1977 Progesterone-regulated changes in transcriptional events in rabbit uterus. Biochim Biophys Acta 479: 354–366
Nakamura H, Yokota T, Akamizu T, Mori T, Imura H 1987 Triiodothyronine effects on RNA polymerase activities in isolated neuronal and glial nuclei of the mature brain cortex. Metabolism 36: 931–934
Lindner LE 1993 Improvements in the silver staining technique for nucleolar organizer regions (agNOR). J Histochem Cytochem 41: 439–445
Smetana K, Schoefer C, Mosgoeller W, Wachtler F, Schwarzacher HG, Jiraskowa I, Ochs R 1993 Cytochemistry of satellite nucleoli in human lymphocytes. Acta Histochem 95: 228–231
Burck HC 1981 Histologische Technik. 4th ed. Georg Thieme Verlag, Stuttgart, Germany
Hozak P, Zatsepina OV, Likovsky Z 1990 In situ separation of nucleolar components by hypotonic treatment of cells. Biol Cell 68: 167–170
Hozak P, Geraud G, Hernandez-Verdun D 1992 Revealing nucleolar architecture by low ionic strength treatment. Exp Cell Res 203: 128–133
Bull P, Martial J, Teilez R, Venegas A, Valenzuela P 1980 The pH dependence of rat liver polymerases I and II. Arch Biol Med Exp 13: 265–269
Rose KM, Stetler DA, Jacob ST 1981 Protein kinase activity of RNA polymerase I purified from a rat hepatoma: probable function of Mr 42,000 and 24,600 polypeptides. Proc Natl Acad Sci U S A 78: 2833–2837
Hirsch J, Mortelo OJ 1976 Phosphorylation of rat liver ribonucleic acid polymerase I by nuclear protein kinases. J Biol Chem 251: 5408–5413
Duceman BW, Rose KM, Jacob ST 1981 Activation of purified hepatoma RNA polymerase I by homologous protein kinase NII. J Biol Chem 256: 10755–10758
Allo SN, McDermott PJ, Carl LL, Morgan HE 2009 1991 Phorbol ester stimulation of protein kinase C activity and ribosomal DNA transcription. J Biol Chem 266: 22003–22009
Xu G, Gai Q, James OB 1998 Protein kinase C inhibits transcription from the RNA polymerase III promoter of the human c-myc gene. Cancer Lett 123: 199–205
Chuang LF, Zhao FK, Chuang RY 1989 Isolation and purification of protein kinase C from human leukemia ML-1 cells phosphorylation of human leukemia RNA polymerase II in vitro. Biochim Biophys Acta 992: 87–95
Seither P, Zatsepina O, Hoffmann M, Grummt I 1997 Constitutive and strong association of PAF53 with RNA polymerase I. Chromosoma 106: 216–225
Hanada K, Song CZ, Yamamoto K, Yano K, Maeda Y, Yamaguchi K, Muramatsu M 1996 RNA polymerase I associated factor 53 binds to the nucleolar transcription factor UBF and functions in specific rDNA transcription. EMBO J 15: 2217–2226
Dienel GA, Pulsinelli WA, Duffy TE 1980 Regional protein synthesis in rat brain following acute hemispheric ischemia. J Neurochem 35: 1216–1226
Cooper HK, Zalewska I, Kawakami S, Hossmann KA, Kleihues P 1977 The effect of ischemia and recirculation on protein synthesis in the rat brain. J Neurochem 28: 929–934
Bodsch W, Barbier A, Oehmichen M, Grosse Ophoff B, Hossmann KA 1986 Recovery of monkey brain after prolonged ischemia. J Cereb Blood FIow Metab 6: 22–33
Kleihues P, Hossmann KA 1971 Protein synthesis in the cat brain after prolonged cerebral ischemia. Brain Res 35: 409–418
Raley-Susman KM, Lipton P 1990 In vitro ischemia and protein synthesis in the rat hippocampal slice: the role of calcium and NMDA receptor activation. Brain Res 515: 27–38
DeGarcia DJ, O‘NeiI BJ, Frisch C, Krause GS, Skjaerlund JM, White BC, Grossman LI 1993 Studies of the protein synthesis system in the brain cortex during global ischemia and reperfusion. Resuscitation 25: 161–170
Stoykova AS, Dudov KP, Dabeva MD, Hadjiolov AA 1983 Different rates of synthesis and turnover of ribosomal RNA in rat brain and liver. J Neurochem 41: 942–949
Serra I, Avola R, Vanella A, Lombardo B, Agodi A, Giuffrida AM 1984 RNA synthesis in neuronal and glial cell nuclei from rat cerebral hemispheres during early postnatal development. Neurochem Res 9: 1051–1062
Derenzini M, Farabegoli F, Trere D 1992 Relationship between interphase AgNOR distribution and nucleolar size in cancer cells. Histochem J 24: 951–956
Andersen L 1990 Number, volume and size distribution of nucleoli in rat neurosecretory cells with suppressed and stimulated secretion. Acta Anat 137: 311–315
Mosgoeller W, Schoefer O, Derenzini M, Steiner M, Maier U, Wachtler F 1993 Distribution of DNA in human Sertoli cell nucleoli. J Histochem Cytochem 41: 1487–1493
Schoefer C, Mueller M, Leitner MD, Wachtler F 1993 The uptake of uridine in the nucleolus occurs in the dense fibrillar component. Immunogold localization of incorporated digoxigenin-UTP at the electron microscopic level. Cytogenet Cell Genet 64: 27–30
Hozak P, Cook PR, Schoefer O, Mosgoeller W, Wachtler F 1994 Site of transcription of ribosomal RNA and intranucleolar structure in HeLa cells. J Cell Sci 107: 639–648
Mosgoeller W, Steiner M, Hozak P, Penner E, Weiserska-Gadek J 1996 Nuclear architecture and ultrastructural distribution of poly (ADP-ribosyl) transferase, a multifunctional enzyme. J Cell Sci 109: 409–418
Barsotti P, Derenzini M, Novello M, Pession-Brizzi A, Marinozzi V 1980 Pretreatment of rats with cycloheximide prevents nucleolar fragmentation induced by alpha-amanitine. Biol Cell 39: 159–162
Langst G, Becker PB, Grummt I 1998 TTF-l determines the chromatin architecture of the active rDNA promoter. EMBO J 17: 3135–3145
Langst G, Blank TA, Becker PB, Grummt I 1997 RNA polymerase I transcription on nucleosomal templates: the transcription termination factor TTF-I induces chromatin remodeling and relieves transcriptional repression. EMBO J 17: 760–768
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by the Red Bull Company, Salzburg, Austria.
Rights and permissions
About this article
Cite this article
Kastner, P., Mosgoeller, W., Fang-Kircher, S. et al. Deficient Brain RNA Polymerase and Altered Nucleolar Structure Persists until Day 8 after Perinatal Asphyxia of the Rat. Pediatr Res 53, 62–71 (2003). https://doi.org/10.1203/00006450-200301000-00013
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200301000-00013
