
Abstract
Pediatric Hematology Oncology as a specialty was possible because of the evolution of the science of Hematology, which developed microscopy for describing blood cell morphology and methods for quantitation of these elements. Before pediatric blood diseases could be defined, it was necessary to establish the normal blood values of infancy and childhood. The unique features of the blood of the newborn were the focus of many of the early studies. After normal values were established, specific blood disease and hematologic syndromes of children began to be described in Europe and the United States. Pediatric Hematology Oncology is a broad and complex area that encompasses perturbations of the several-formed elements of the blood and their precursors in the bone marrow, as well as the coagulation-fibrinolytic systems in the plasma, the reticuloendothelial system, and malignancies of the blood and solid tissues and organs. The interactions of the blood and nutrition have long been important areas of study. Advances in Pediatric Oncology have been particularly spectacular in the last 50 years. Using multi-modal therapy including combination chemotherapy, more than 80% of children with cancer can now be cured. During the last 50 years, Pediatric Hematology Oncology has increasingly used tools of the “new biology”: immunology, biochemistry, enzymology, genetics and molecular genetics, and others. During the last century, many diseases have been recognized and defined by biochemical and genetic mechanisms, and in some instances they have been prevented or cured.
Similar content being viewed by others
Main
The hematology oncology of infancy and childhood is a relatively recent area of study whose development depended upon the evolution of the science of Hematology and, especially, upon methods to study the blood and its elements. As Wintrobe has pointed out, the development of the field of Hematology has been driven by technology. He divided the early evolution of Hematology into two general areas: morphology, which relied on the development of microscopy and quantitation of the elements of the blood, which came later (1).
The invention of the microscope enabled identification of the blood cells. Antonj van Leeuwenhoek working in Delft, Holland, constructed a primitive microscope from a minute biconcave lens mounted between two metal plates attached to a screw that permitted focusing. Leeuwenhoek's publication in 1674 contained the first accurate description of the red blood corpuscles (2): The blood is composed of exceedingly small particles, named globules, which in most animals are red in color…. These particles are so minute that 100 of them placed side by side would not equal the diameter of a common grain of sand. In the centuries following Leeuwenhoek, the development of compound microscopes with two lenses greatly increased magnification and minimized spherical aberration permitting more accurate descriptions of the blood cells. Dr. William Hewson who often has been designated as one of the Fathers of Hematology noted that the red cells were flat rather than globular and also described the leukocytes for the first time and showed that coagulation occurred in the plasma (3). The last of the formed elements of the blood, the platelet, was independently recognized by several investigators. The most definitive early work on the platelet was done by Julius Bizzozero. His monograph, published in 1882 clearly recognized these cells as being distinct from red and white blood cells He suggested that they should be called platchen and assigned a hemostatic function to them (4). Bizzozero also identified the bone marrow as the site of blood formation. William Osler, early in his illustrious career, also described platelets accurately although he believed that they might be infectious agents, perhaps analogous to bacteria (5).
With improvements in microscopy, the morphology of the fixed blood cells began to be examined using thin films of blood, spread and dried on glass slides, which were then stained with aniline dyes that differentially stained the nuclei and granules of the leukocytes. Staining of peripheral blood smears was developed by Paul Ehrlich in 1877 while he was still a medical student. He identified the neutrophils, basophils and acidophils (eosinophils) on the basis of the staining of their granules (6). The development of supravital dyes provided a method for assessment of erythropoiesis by reticulocyte counts. These techniques permitted the flowering of morphologic hematology and many blood diseases such as the leukemias and the various types of anemia were described on the basis of typical morphologic findings.
Hematology as a quantitative discipline began with the development of practical and reliable methods to accurately enumerate the numbers of the various blood cells. These methods used gridded chambers of uniform depth (hemacytometers) into which precisely diluted suspensions of blood were placed. The numbers of cells in the chamber were counted and when combined with the known dilutions, the actual numbers of cells per cubic milliliter in the patient's blood could be calculated. Hb levels were estimated by comparing the density of color in fixed dilutions of hemolyzed blood with colorimetric standards, and later by spectroscopy. For many years, Hb values were reported as a % of normal, and because the definition of normal was often different, there was considerable variability from study to study. In 1929, Maxwell Wintrobe described his method for obtaining the hematocrit or packed red cell volume (PCV) by centrifugation of blood in a glass tube (7). He then defined red cell indices, the mean corpuscular volume (MCV), mean corpuscular Hb (MCH), and mean corpuscular Hb concentration (MCHC), which proved of enormous value in classifying the various forms of anemia (8). The latest advance in blood cell quantitation began in 1956 with the invention by Wallace H. Coulter of a sophisticated, computer driven electronic instrument that very accurately measured Hb, the numbers of all of the blood cells, as well as the red cell indices and the RDW (red cell distribution width) (9). Some instruments now also provide automated differential counts of the leukocytes.
EARLY AMERICAN PEDIATRIC HEMATOLOGY
The earliest American textbooks of Pediatrics gave very scant attention to hematologic problems of children. Dr. W.P. DeWees's 1825 A Treatise on the Physical and Medical Treatment of Children, arguably the first American Pediatric textbook, and Dr. Job Lewis Smith's 1869 A Treatise on the Diseases of Infancy and Childhood gave only passing notice of blood conditions such as neonatal jaundice and hemorrhage (10, 11). However, the monumental text of Dr. L. Emmett Holt, Sr., The Diseases of Infancy and Childhood, first published in 1897, contained a chapter on the diseases of the newborn, including hemorrhagic disease of the newborn, as well as a 20-page chapter on diseases of the blood (12). Holt described the normal Hb level, red and white blood cell and differential counts from birth through puberty. He also discussed the clinical features, blood findings and treatments (such as they were) of simple anemia, chlorosis, pseudo-leucemia of infancy, pernicious anemia, leucemia, hemophilia, purpura, and enlargement of the spleen. The chapter included color drawings of the blood cells in leukemia and pernicious anemia. The material Holt presented was obtained from European sources, as well as his personal experiences.
In the early part of the 20th century, both American and European pediatricians began to study the blood of newborn infants and children. Before identification of the many blood diseases of infancy and childhood was possible, age related normal values of all of the elements of the blood had to be established. Many of the early studies were done on newborns. Dr. William P. Lucas practiced and did research at the Children's Department of the University of California, San Francisco. In 1924 Lucas described extensive studies of the blood of 150 infants at birth and during the first 2 months of life (13). The relative polycythemia of the newborn, early physiologic anemia and changes in leukocytes and coagulation were clearly defined. In 1924, Dr. H.J. Lippman of the University of Minnesota also published detailed studies of the hematology of the first 48 hours after birth (14). Hematologic investigations in older children were complicated by concomitant nutritional diseases such as rickets, scurvy, and endemic infectious disease including tuberculosis and syphilis. In 1924, Dr. Lucas and his associate, Dr. E.C. Fleichner, wrote six chapters on the blood in Dr. I. Abt's eight-volume, multi-authored text Pediatrics, the first comprehensive American publication on pediatric hematology (15). Drs. A.H. Washburn in Denver and G.M. Guest in Cincinnati performed studies of the blood values of normal infants and children (16, 17). With definition of normal value, it became easier to identify specific hematologic diseases.
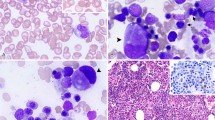
An extension of diagnosis from the blood into the bone marrow was provided by Dr. K. Kato of Chicago who in 1937 reported findings in bone marrow aspirates of 51 normal infants and children. Kato's report included excellent camera lucida drawings (18), At about the same time Dr. P. Vogel in New York reported marrow findings, illustrated by photomicrographs, in a variety of pediatric blood disorders including leukemia, Gaucher's disease, and metastatic neuroblastoma (19). Definition of the normal marrow morphology was furthered by the work of Dr. Philip Sturgeon in Los Angeles who performed and assessed 72 marrow aspirations on 50 normal infants and children (20).
EUROPEAN PEDIATRIC HEMATOLOGY
Many of the early European investigators of pediatric hematology, like their American contemporaries, gave special attention to the blood findings in the newborn period. In 1889 in one of the oldest hematological texts, Du Sang et de ses Alterations Anatomiques, Dr. G. Hayem in Paris described the blood picture at birth in detail, He noted the numbers of red and white blood cells and attributed the high Hb level at birth to hyperactivity of the bone marrow (21). Also in 1889, Dr. R von Jaksch from Prague published his paper Anemia Pseudoleucaemica Infantum describing a group of children who had chronic anemia, splenomegaly and leukocytosis (22). This important report is often considered to mark the beginning of pediatric hematology. The diagnosis of von Jaksch's anemia was widely used in Europe, but it ultimately was recognized as a diagnostic wastebasket that included the response of infants to the horrendous combination of nutritional and infectious insults so common at that time (23).
Pediatric hematology began to be more extensively studied in Europe during the early 20th century (23, 24). In Bern, Switzerland Dr. E. Glanzmann described the bleeding disorder, hereditary hemorrhagic thrombasthenia, which still bears his eponym. Glanzmann also conducted studies in anaphylactoid purpura (25, 26). In Zurich, Dr. Guido Fanconi and his Oberarzts, Drs. H. Willi, Conrad Gasser and Walter Hitzig published studies of the clinical, blood and bone marrow findings in leukemia, thrombocytopenia, various anemias, and neotropenias. The most notable of these was Professor Fanconi's description of the syndrome of aplastic anemia and multiple congenital anomalies that bears his eponym (27). Conrad Gasser's 1951 monograph entitled Die Hamolytischen Syndrome des Kindersalter was a significant contribution to the understanding of the hemolytic anemias of children (28).
An important early English pediatric hematologist was Sir Leonard Parsons of Birmingham who, in 1933, was among the first to recognize the hemolytic nature of erythroblastosis fetalis (29). Parsons trained a number of notable associates who made significant contributions to pediatric hematology including Dr. I.A.B. Cathie. Cathie coined the term, Erythrogenesis Imperfecta, for congenital, hypoplastic anemia (30). Centers of excellence in pediatric hematology were also developed in Scandinavia by Dr. B. Valquist and in Amsterdam by Dr. E. van Crevald where notable contributions were made in hemophilia.
The first European textbooks devoted to pediatric hematology were Ematologia Infantile Normale e Patologica published by Dr. Ferruchio Zibordi of Modena, Italy, in 1924, and Die Klinishe Haemtologie des Kindesalters published in Vienna by Drs. H. Baar and Stransky in 1928. An expanded edition of the Baar and Stransky text in English was published in 1963 (31–33).
In 1968, on the initiative of Dr. Walter Hitzig of Zurich, a group of European pediatricians particularly involved in hematology founded the European Society of Pediatric Hematology and Immunology. This was followed by the Italian Association of Pediatric Hematology and Oncology, The Nordic Society of Pediatric Hematology and Oncology, and similar organizations in Latin America and Japan (24).
In the late 1950s, a small unit devoted to pediatric oncology was established in Paris under Dr. Odile Schweisguth who is considered to be the pioneer pediatric oncologist in Europe. Most European universities now have established pediatric hematology oncology units that share common protocols and conferences.
PIONEERS OF AMERICAN PEDIATRIC HEMATOLOGY
A number of men made important contributions to the emerging specialty of American Pediatric Hematology in the first half of the 20th century. Four of them, Drs. Thomas B. Cooley, Louis K. Diamond, Carl H. Smith, and Wolf W. Zuelzer merit special mention. All of them were essentially self trained; all of them published extensively; and all were deeply committed to training. Personal sketches and anecdotes about these giants are included in Dr. Zuelzer's Historical Perspective of Pediatric Hematology published in Nathan and Oski's Blood Diseases of Infancy and Childhood and have particular insights and poignancy because Zuelzer knew all of them well (23).
A most important early contribution to Pediatric Hematology, and one that marked the birth of the specialty in America, was made by Dr. Thomas B. Cooley (1871–1945) of Detroit in 1925 and 1927 when he reported a new hematologic syndrome in a group of young children with anemia, splenomegaly, erythroblastosis, and marked abnormalities of the skeleton and facies (34, 35). He noted that all of the patients were of Italian or Greek ethnicity. Cooley designated his new syndrome, Erythroblastic Anemia, which was rapidly given the eponym, Cooley's Anemia or Mediterranean Anemia and later Thalassemia. Wolf Zuelzer later described Cooley's meager research resources: a part time technician, Pearl White, an ancient monocular microscope and a small filing cabinet (36). Cooley continued his work in pediatrics, genetics and hematology at Detroit's Children's Hospital of Michigan and the Child Research Center of Michigan until 1945, the year of his death.
Dr. Louis K. Diamond (1902–1999), who has been called the Father of American Pediatric Hematology, attended Harvard College and Medical School and worked for most of his life at the Boston Children's Hospital. He entered pediatric training under the mentorship of Dr. Kenneth D. Blackfan who was Professor and Chief of Pediatrics. Blackfan was interested in children's blood diseases and encouraged Diamond to pursue this as a specialty. Diamond stated that he became hooked on hematology as a junior house officer when he cared for a child with glandular fever (probably infectious mononucleosis) who had lymphadenopathy and unusual lymphocytes in the blood. Blackfan suggested that Diamond should spend six (unpaid!) years in clinical and research training as preparation for this career. He was appointed an Assistant Professor of Pediatrics on the Harvard faculty in 1933 and concentrated his practice on the blood diseases of infancy and childhood for the next 50 years (37, 38). Dr. Diamond's contributions were legion. In 1932, while still a trainee, he largely wrote, but coauthored with Blackfan and Dr. James M. Baty, a long and incisive paper stating that three conditions previously believed to be separate entities, fetal hydrops, icterus gravis familiaris, and anemia of the newborn, were all manifestation of an unknown underlying disease process, which they termed, erythroblastosis fetalis (39). In 1938, Diamond and Blackfan described congenital hypoplastic anemia, which still bears their eponym (40). In 1944 the Atlas of the Blood of Children was published by Blackfan, Diamond and C.M Leister (41). Despite the listing of Blackfan, who died in 1941, as the first author, Diamond was the principal force behind this remarkable book. It contained 70 magnificent, hand painted color illustrations of peripheral blood smears in a wide variety of blood diseases drawn by Dr. Leister, who was a practicing pediatrician. This book was far more than a color atlas. It described normal values for all of the blood cells from birth through adolescence as well as all of the known pediatric blood conditions of the time. Interestingly, there is no mention made in the Atlas of bone marrow findings in these diseases, for at the Boston Children's Hospital only surgical biopsies of the bone marrow were permitted (42). In addition to his work in erythroblastosis, Diamond made important contributions to blood serology and blood banking. During World War II he served for 3 years as the first director of the National Blood Program of the American Red Cross. Diamond had a long lasting influence on pediatric hematology in America through his mentorship of a generation of pediatric hematologists who trained with him at the Boston Children's Hospital. Diamond's trainees, sometimes called Diamond Chips, became leaders in pediatric hematology, hematologic research, and pediatrics in America for a half a century. At least eight of them became departmental chairmen. In 1968, Dr. Diamond became Emeritus and was succeeded by Dr. David G. Nathan. Although trained in Internal Medicine, Nathan became deeply involved in clinical and research aspects of the blood diseases of children, made numerous contributions to pediatric hematology and continued the Diamond tradition of training many pediatric hematologists. Nathan, with Dr. Frank A. Oski also published Hematology of Infancy and Childhood, a preeminent textbook now in its fifth edition (43).
Dr. Wolf W. Zuelzer (1909–1987) was born in Berlin Germany (23, 44). His father was a prominent physician and clinical investigator who was the first to demonstrate — many years before Banting and Best — that a pancreatic extract could temporarily reverse diabetic ketoacidosis. Dr. Zuelzer originally pursued studies in literature and art, but when he was within 6 months of completing a PhD, he abruptly switched to medicine. He took preclinical years at the University of Bonn, and then transferred to the University of Berlin. However, in 1933 Hitler came into power and began systematic elimination of non-Aryan professors and students from the medical schools and hospitals. Zuelzer was partly Jewish and fearing for his career (as well as for his life), he went to Czechoslovakia and received his medical degree at the University of Prague. He then emigrated to the United States and after a pediatric internship at the Massachusetts General Hospital served a year as an unpaid volunteer in pathology at the Boston Children's Hospital under Dr. Sidney Farber, who became his adoptive father, mentor, and role model. After a specially designed combined fellowship in pathology and pediatrics under Dr. Joseph Brenneman at the Chicago Children's Hospital and aided by a strong recommendation from Dr. Farber, Dr. Zuelzer became head of pathology and ultimately the head of the entire laboratory medicine service at the Children's Hospital of Michigan overlapping with Dr. Thomas Cooley. In 1955 he was appointed as director of the Child Research Center where he was able to combine his clinical and research interests. He published investigations in many areas of pediatric hematology, including a description of the megaloblastic anemia of infancy due to folic acid deficiency, for which he received the first E. Mead Johnson Award for Pediatric Research in 1949 (45). He also made important contributions to the understanding of ABO hemolytic disease of the newborn. Zuelzer trained many hematology fellows over more than 35 years.
Dr. Carl H. Smith (1895–1971) was a native New Yorker and a graduate of Cornell Medical School. After a pediatric residency at the New York Nursery and Child's Hospital, he entered the private practice of Pediatrics, which he continued for the rest of his life. He became interested in hematology and founded a clinic at the New York Hospital while in private practice. He established Pediatric Hematology at the Cornell Medical School, where he held a clinical, rather than a full time, faculty appointment. Dr. Smith established a Blood Research Foundation with gifts from grateful patients that he used to support training and research. Because of the large Italian and Greek communities in New York City, he had a large thalassemia service and in a series of publications, contributed enormously to the understanding and clinical management of this disease. He was one of the earliest to recognize the syndrome of overwhelming and frequently fatal infections that occurred after splenectomy (46). Smith trained a number of fellows who had illustrious careers in pediatric hematology. In 1960, long before word processors and electronic literature searches, Smith single-authored Blood Diseases of Infancy and Childhood, the first American textbook devoted to Pediatric Hematology Oncology (47). This comprehensive and exhaustively referenced text, had three re-editions. Its successor, now in its sixth edition, is edited by Dr. Dennis Miller and remains an important resource. Dr. Smith's textbook was notable for its inclusion of chapters on childhood cancers because pediatric hematologists who were managing childhood leukemia were also treating other malignancies with chemotherapy.
THE EMERGING SUBSPECIALTY OF AMERICAN PEDIATRIC HEMATOLOGY
Following World War II with release from wartime restraints, American Departments of Pediatrics experienced dramatic expansion in their research and training activities, fueled primarily by National Institutes of Health (National Institutes of Health) grants. Most medical school departments established subspecialty divisions of Pediatric Hematology Oncology.
Venues for presentation and discussion of pediatric hematologic topics began to be established (24). Beginning in the 1950s the Pediatric Blood Club held, and continues to hold, a scientific meeting during the annual meetings of the American Pediatric Society and the Society for Pediatric Research (APS, SPR). The Blood Club had no formal membership and kept no records. For a while, the costs of the room and projection equipment were raised by passing the hat at the meetings. In 1958, the SPR joined shortly by the APS, began simultaneous scientific sessions devoted to pediatric subspecialties, including hematology oncology, as a part of their annual meetings.
A Pediatric Hematology Oncology sub-committee was established by the American Society of Hematology (ASH) in 1975 under the chairmanship of Dr. Gerald S. Gilchrist, and its first scientific session was held in during the 1976 ASH annual meeting. This was an informal organization without by-laws or membership requirements, whose only function was to present an annual scientific program. A Section of Pediatric Hematology Oncology was established by the American Academy of Pediatrics (AAP) in 1975 that held scientific sessions during the annual AAP meetings. The Blood Club and the ASH and AAP organizations did not consider economic issues, which were believed by many hematologists to be increasingly important at a time when major changes in financing of medical care and reimbursement were occurring in the United States.
SUBSPECIALTY CERTIFICATION
In 1972 and 1973, the American Board of Internal Medicine (ABIM) offered two separate certifying examinations in medical Hematology and medical Oncology for internists. Some pediatric hematologists became concerned that third party payers, including state Crippled Children's Services, might decide to reimburse only certified sub-specialists and that certified internist hematologists and oncologists might be designated to care for children with these diseases. In 1970, Dr. Irving Schulman sent a letter to pediatrician members of ASH announcing an ad hoc meeting to discuss certification. The meeting was held at the annual ASH meeting in San Juan Puerto Rico in December 1970 and was chaired by Dr. Schulman. The group voted to ask the American Board of Pediatrics (ABP) to set up a process for sub-board certification and submitted the names of 15 leading pediatric hematologist oncologists for consideration by the ABP to serve as a certifying committee (48). Eight of these were chosen to serve as a Special Competency Committee in Pediatric Hematology Oncology with Dr. Schulman as chairman. The Committee was charged with specifying requirements and writing the first certifying examination. Unlike Internal Medicine, the Committee decided that there should be a single certification for both Pediatric Hematology and Oncology, and that the content of the certifying examination would take into consideration the oncology field. Prior certification in general pediatrics was a made a requirement. Applicants for the first two examinations in 1974 and 1976 were required to submit evidence of having had 2 years of full-time specialty training, 5 years of clinical practice of pediatric hematology oncology or some combination of training and practice.
The Committee members believed that it would be inappropriate for them to receive grandfather certification and insisted that they should be tested before writing a certifying examination for everyone else. Dr. Frederick Burg, who was Associate Executive Secretary of the ABP and also the Associate Director of the National Board of Medical Examiners (NBME) was appointed as test development officer for a certifying examinationination. The ABP and Dr. Burg obtained permission from the ABIM to use test questions from the previous ABIM examinations that pertained to pediatric hematology oncology. These questions, unknown to any Committee member, were organized by Dr. Burg into the same format projected for the pediatric examination, and these became the qualifying examination for the Committee. All of the members of the Committee passed this examination and were declared eligible for ABP certification. It was the usual practice of the ABP that the chairman of a first subspecialty committee should receive the first ABP certificate. Dr. Schulman, in a gracious gesture, insisted that Dr. Louis K. Diamond, also a Committee member, should receive ABP Hematology Oncology certificate #l. The Committee spent more than a year writing multiple-choice test questions and were greatly assisted by Dr. Burg and the resources and experience of the NBME and ABP. About 80% of the questions of the first certifying examination concerned hematologic subjects.
The first ABP certifying examination in Pediatric Hematology Oncology was given simultaneously in Chicago, Philadelphia, and San Francisco on November 2, 1974. The sub-board Committee specified in advance that the examination proctors should permit the examinees to move up to the viewing screen during the slide presentation of pictorial materials perhaps in recognition of the fact that many of the more senior people taking the examination might have trouble focusing! The passing mark for the examination was set as one SD below the mean score. Three hundred two pediatric hematologist oncologists sat for the examination and 211 (70%) of them were certified.
Thirteen ABP examinations were given between 1974 and 2000 that certified 1602 Pediatric Hematology Oncologists. Beginning in 1988, certification was time-limited to 7 years and recertification examinations were given in 1996 and 2001. About 90% of those whose certificates had expired sought recertification, and over 98% of those taking the recertifying examinations passed.
The American Board of Medical Specialties approved the sub-specialty of Pediatric Hematology Oncology in 1973. However, the requirements for training were not approved by the Committee on Graduate Medical Education (COGME) until 1983 and Residency Review Committee (RRC) accreditation for training programs was established in 1984. In 1987 training in an RRC accredited program was made necessary for eligibility In 1978, 2 years of full-time training graduate training in pediatric hematology oncology were required for sub-board eligibility and in 1986, 3 years of fellowship were required. In 1988 active participation in basic or clinical research was made a mandatory component of approved fellowship training.
THE AMERICAN SOCIETY OF PEDIATRIC HEMATOLOGY ONCOLOGY AND THE AMERICAN JOURNAL OF PEDIATRIC HEMATOLOGY ONCOLOGY
The American Society of Pediatric Hematology Oncology (ASPHO) was founded in 1981 largely through the organizing initiatives of Dr. Carl Pochedly who served as secretary-treasurer for 14 years. ASPHO held meetings in conjunction with the SPR/APS and Blood Club from 1982–1987, but in August, 1988 had its first independent scientific session (49). At this meeting, an annual Distinguished Career Award was established to:give recognition of outstanding service and significant scientific contributions to the understanding and treatment of blood disease and cancer in children. Meetings of ASPHO have recently again been held at the time of the annual spring meetings of the Pediatric Academic Societies.
In 1979, the American Journal of Pediatric Hematology Oncology, which became the official organ of ASPHO, was established by Dr. Carl Pochedly who was editor for 14 years. In 1994, in recognition of its international circulation and content, it became the Journal of Pediatric Hematology Oncology. Initially published as a quarterly, the Journal began bi-monthly editions in 1997.
IMPORTANT AREAS OF PEDIATRIC HEMATOLOGY/ONCOLOGY
Neonatal Hematology
The hematology of the neonate has been an almost separate area, shared, sometimes uncomfortably, by pediatric hematologists and neonatologists. In 1960 Drs. Frank A. Oski and J. Lawrence Naiman published Hematologic Problems in the Newborn, the first American text devoted to this subject (50).
Erythroblastosis fetalis.
As recently as 1946, erythroblastosis fetalis, or hemolytic disease of the newborn, affected between 0.5–1.0% of fetuses and newborns in the United States. Affected newborns had a 50% mortality and significant neurologic complications occurred in many survivors (42). As previously mentioned, Diamond, Blackfan and Batey were the first to unify three neonatal hematologic syndromes as manifestations of erythroblastosis, but they did not identify a pathophysiologic mechanism (39). In 1938, Dr. Ruth Darrow, a pathologist who had several of her own children die of kernicterus and severe erythroblastosis advanced a brilliant inductive hypothesis about its cause (51). Assembling all of the available information, as well as her own tragic personal experience, she noted the usual sparing of the first child and the progressively more severe involvement of subsequent children. She also recognized that the hematological and histopathological findings indicated severe hemolysis. She concluded that: the mother is actively immunized against fetal red cells or some component of them. . .The antibodies formed in the maternal organism may then pass to the child through the placenta. The offending red cell component was discovered in 1940 by Drs. Carl Landsteiner and Alexander Weiner and given the name Rh (rhesus) because of an antibody they produced by injection of RBC of rhesus monkeys into rabbits. This antibody agglutinated the RBC of 85% of normal individuals who were designated as being Rh positive (52). Interestingly, Landsteiner's discovery of Rh was accomplished 50 years after he had discovered the ABO blood groups, for which he had been awarded the Nobel Prize in Medicine in 1930 (53). Dr. Philip Levine described a transfusion reaction in a postpartum woman who was given a compatible blood transfusion of her husband's blood shortly after she delivered a baby with severe erythroblastosis fetalis. Levine demonstrated Rh antibodies in the mother, defining the etiology of erythroblastosis fetalis (54).
Newborns with erythroblastosis frequently developed hyperbilirubinemia and kernicterus. The treatment of icterus gravis by exsanguination transfusion was first reported in 1925 by Dr. A.P. Hart at Toronto's Hospital for Sick Children (55). After discovery of the Rh factor, exchange transfusion evolved as a way to remove circulating antibody, sensitized RBC, and later bilirubin. The treatment was spearheaded by Drs. A. Weiner and H. Wallerstein in New York and Dr. Louis K. Diamond in Boston. Wallerstein's method involved infusion of blood into the longitudinal sinus (56). Weiner's method involved transection of the radial artery and systemic heparinization (57). Diamond's more practical technique used the umbilical vein to alternately remove and infuse Rh-negative RBC (58). Diamond and his associates developed practical guideline for the pre and postnatal management of Rh sensitized pregnancies that reduced neonatal mortality from 50% to 5%, intrauterine deaths from 21% to less than 10% and kernicterus was virtually eliminated (59).
The penultimate advances in erythroblastosis fetalis were made by Dr. A.W. Liley of New Zealand who devised a method of spectroscopic analysis of amniotic fluid (60). This was able to identify fetuses at imminent risk of intrauterine death that could be delivered early or given intrauterine, intraperitoneal RBC transfusions to carry them to delivery (61). Finally, Drs. C.A. Clark in Liverpool and V.J. Freda and associates in New York independently showed that primary immunization of Rh negative mother by RBC of their Rh positive fetuses could be prevented by immediate postnatal administration of potent anti-Rh gamma globulin to the mother (62, 63). In most developed countries, erythroblastosis fetalis became rare – a disease of largely historical interest!
With the dramatic decrease in Rh erythroblastosis, other etiologies for neonatal hyperbilirubinemia were recognized. In 1954, Dr. T. Halbrecht pointed out that infants with early hyperbilirubinemia often had an incompatibility with their mother's ABO blood group system (64). The features of ABO hemolytic disease of the newborn were rapidly defined (65). Deficient glucuronidization of bilirubin because of low levels of glucuronyl transferase that contributed to neonatal hyperbilirubinemia, even in the absence of hemolysis, was demonstrated by Drs. Audrey Brown and Wolf Zuelzer (66).
The Red Blood Cell
The mature, circulating RBC is derived from a committed stem cell precursor, whose proliferation and maturation is regulated by a specific cytokine, erythropoietin (EPO). EPO is produced in the fetal liver, and later in life, in the kidneys. The gene for EPO has been identified and cloned enabling the production of recombinant human EPO, which has therapeutic value in the treatment of the aregenerative anemias of end- stage renal disease and malignancy.
Iron deficiency.
Anemia and the therapeutic values of iron was recognized in antiquity and chlorosis or morbus virgineo in adolescent girls as well as its responsiveness to iron therapy were well established in the 1700s. Iron deficiency anemia was prevalent in infants and children, but its acceptance by pediatricians was a long time coming. Diamond and Zuelzer explained that this reluctance was probably because iron deficiency was often associated with severe caloric malnutrition and growth failure, rickets and scurvy, chronic infections, including syphilis and tuberculosis which masked or overshadowed the underlying anemia (23, 42). Another confounding issue was the ineffectiveness of the therapeutic iron medications that were available in the first half of the 20th century, which often resulted in poor therapeutic responses. Increased phosphates in the intestine may have also resulted in malabsorption of iron in rachitic children. Improved nutrition, fortification of milk with vitamin D, the control of infections and better iron medications reduced these confounding factors and iron deficiency as an endemic disorder became increasingly recognized in the United States. This recognition was greatly improved by the definition of the characteristic hypochromia and microcytosis of the red blood cells in iron deficiency and by development of confirmatory blood tests such as serum iron, iron binding capacity and serum ferritin levels.
Dr. George M. Guest (1898–1967), working at the Children's Hospital Research Foundation in Cincinnati between 1932 and 1942, performed extensive studies of the blood of large numbers of normal and anemic children which not only helped define normal age-related standards, but did much to define the prevalence of iron deficiency anemia in infants and children (67). Guest showed that there was a decrease in the MCV and MCH before the development of anemia and these changes could be prevented by iron administration.
Much of the iron deficiency anemia in American children in the mid-20th century reflected changing patterns of infant feeding. Breast-feeding was reduced greatly, and most American babies were fed with whole cow's milk or cow's milk formulas, which are deficient in iron. So-called milk anemia was very common, and 10% to 15% or more of American babies had overt iron deficiency. A major nutritional advance was made possible by the introduction of iron fortified, proprietary infant formulas which were recommended by the Committee on Nutrition of the American Academy of Pediatrics in 1969 (68). Inclusion of iron-fortified formulas by the federal Women, Infants, and Children (WIC) program in 1972 resulted in the virtual eradication of severe iron deficiency in groups of high-risk American children (69). Recognition of nonhematologic effects of iron deficiency including behavioral and learning abnormalities as well as poor performance on psychomotor tests gave an increased importance of iron nutrition (70).
Folic acid deficiency.
Individual cases of a macrocytic anemia in infants fed on goat's milk had been recognized in Europe and the United States early in the 20th century, resulting from the very low folic acid content of goat's milk. However, it was the 1946 report of Drs. Wolf W. Zuelzer and F. Ogden that clearly identified dietary folic acid deficiency anemia by describing 25 children with severe macrocytic anemia and megaloblastic changes in the bone marrow (45). Most of the children were receiving various cows' milk formulas. All responded hematologically to folic acid or liver extract therapy. The American infant formula industry responded by adding folic acid to their products, and megaloblastic anemia became a rare disease.
Vitamin E deficiency
In 1967, Drs. Frank A Oski and Lewis A. Barness reported a new syndrome of hemolytic anemia, thrombocytosis and pretibial edema in premature infants (71).
Vitamin E deficiency was proven by low plasma vitamin E levels, increased hydrogen peroxide induced hemolysis of RBC and by a hematologic response to vitamin E therapy. It was later shown that hemolysis was a result of formulas with low levels of vitamin E, combined with high levels of polyunsaturated fatty acids and iron supplementation (72). The formula industry promptly responded by supplementing of their products with vitamin E and reducing polyunsaturated fat content and this syndrome virtually disappeared.
Hemoglobinopathies
The human hemoglobinopathies illustrate very well the scientific progress of the past 50 years. Following the description of an abnormal Hb in sickle cell disease in 1949, more than 200 mutant hemoglobins were described. Abnormal hemoglobins can be associated with a variety of clinical disorders, including hemolytic anemias, altered oxygen transport, methemoglobinemia and others. Thalassemia major and the sickle cell diseases are the most prevalent and serious hemoglobinopathies, and ones that are predominantly diseases of children.
Thalassemia.
The field of American pediatric hematology was launched by the description of Dr. Thomas E. Cooley in 1925 and 1927 of Erythroblastic or Mediterranean Anemia (34, 35). Following Cooley's original reports, increasing numbers of these patients were recognized in the United States and Europe. Without treatment, they survived only a few years. Regular blood transfusions extended life, but resulted in iron overload and damage to the heart causing death in the second and third decades of life. To attempt to delay hemosiderosis, transfusions were given infrequently, and patients were chronically very anemic with pretransfusion Hb levels <4.0–6.0 Gm/dL. The severe anemia and hemolysis evoked massive compensatory expansion of the erythroid marrow in medullary and extramedullary sites, which caused bone disease and pathologic fractures, distressing cosmetic changes of the face and splenomegaly. In 1964, Dr. Irving Wolman reported that children whose pretransfusion Hb levels were maintained above 8.0–9.9 g/dL had better growth, less organomegaly, less cardiomegaly, fewer fractures, fewer cosmetic deformities and better general health than children kept at lower Hb levels (73). Wolman's hypertransfusion program became widely accepted; but problems of transfusional hemosiderosis remained, perhaps worsened by the increased blood requirements of hypertransfusion. A therapeutic intervention for hemosiderosis became available with the introduction of deferoxamine in the early 1950s. The drug was a specific and safe iron-chelator but it had to be administered parenterally and, because of a short biologic half-life, it was rarely possible to attain a negative iron balance in the face of continuing transfusions. In 1977, Dr. Richard Propper and associates at the Boston Children's Hospital demonstrated that the effectiveness of deferoxamine could be considerably enhanced by prolonged s.c. infusions, making it possible to attain negative iron balance, to reduce the body's iron burden, and to extend life in many patients (74, 75).
The pathophysiology of thalassemia was elucidated by direct measurement of globin chain synthesis by reticulocytes in vivo, which defined both α-thalassemia and β-thalassemia. The severe hemolysis and ineffective erythropoiesis of severe thalassemia were explained by unbalanced globin chain synthesis DNA analysis defined more than 200 genetic mutations that produce the thalassemia phenotype. Prenatal diagnosis, originally accomplished by globin synthetic studies of fetal blood and then by direct gene analysis of fetal DNA, enabled prenatal diagnosis in families at risk of having a child with thalassemia major (76). Increased public awareness and screening of high-risk ethnic groups for the carrier state combined with prenatal diagnosis resulted in many fewer affected births in the United States and elsewhere (77). Bone marrow transplantation, which is curative, has been extensively performed in Italy, but has been done in relatively few cases in the United States (78).
Sickle cell anemia.
Following the original description of sickle cell anemia by Dr. James B. Herrick in 1910, additional cases were slowly reported (79). Dr. Victor E. Emmel showed that the RBC of the hematologically normal father of a patient with sickle cell anemia could be induced to undergo sickling in sealed preparations of his blood (80). The first American children with sickle cell anemia were described by Dr. Virgil Sydenstriker in 1923 who reported that both parents of their patients had latent sickling. Sydenstriker also used the term crisis, and described splenic atrophy at autopsy (81). For many years a confusion between sickle cell anemia and latent sickling delayed recognition of its actual genetic basis, and it was not until 1949 that Dr. JV. Neel, on the basis of pedigree analyses, correctly identified the genetic pattern of inheritance with benign sickle cell trait resulting from heterozygosity and sickle cell anemia associated with homozygosity for the sickle cell gene (82).
The most important scientific understanding of sickle cell anemia came as a result of the work of Dr. Harvey A. Itano, a postdoctoral student in the laboratory of Dr. Linus Pauling. Pauling was told about sickle cell anemia and the sickling phenomenon by Dr. William Castle during a 1945 overnight train ride from Denver to Chicago (83). When Itano joined Pauling's laboratory, he was assigned to study sickle cell and he constructed an electrophoresis apparatus, modified from that of Tiselius, to analyze the Hb. Their results published in 1949 described an altered electrophoretic abnormal of Hb of patients with sickle cell anemia (Hb S) and ascribed the abnormality to a change in the globin (84). The RBC of parents of these patients contained both normal Hb A and Hb S, elegantly confirming the studies of Neel, and advancing for the first time the concept of molecular disease.
The precise abnormality of the Hb S molecule was discovered only seven years later by D. Vernon M. Ingram who studied tryptic digests of Hb A and S by finger printing, an analytic technique involving two dimensional electrophoresis and chromatography (85). Ingram found a single abnormal peptide, which he then isolated and analyzed and showed that the only difference between Hb A and S was the replacement of a single normal glutamic acid by a valine residue (86).
The understanding of the structure of Hb advanced rapidly in the 1960s when it was shown that Hb A is a tetramer of two alpha (α) and two beta (β) polypeptide chains (α2 β2)and that fetal Hb (Hb F) contains a chemically different γ chains. (α2γ2) During fetal development, γ chain synthesis predominates but with approaching term there is a fall off of γ chain synthesis and a reciprocal increase in β-chain production. The mechanisms that regulate this β/γ switch remain to be elucidated. The blood of the newborn contains large amounts of Hb F, averaging 60% to 80%.
Korber, in his doctoral dissertation in 1856 described the resistance of the Hb of the newborn to denaturation by strong alkali (and acid) solutions (87). This became the basis of the Singer one-minute alkali denaturation test for quantitation of fetal Hb (Hb F) as well as the Apt test used for differentiating fetal blood from swallowed maternal blood in newborns with melena (88, 89). The resistance of fetal Hb was also the basis for the RBC acid elution staining procedure developed by Drs. E. Kleihauer, H. Braun and K. Betke in Germany that can be used to quantitate the magnitude of feto-maternal hemorrhages (90).
The high level of Hb F at birth offers temporary protection from hemoglobinopathies such as sickle cell anemia, but may also hamper neonatal diagnosis. Dr. Roland Scott, using sickle
cell preps, demonstrated a much lower frequency of sicklemia in black newborns than was found in older children from the same community (91). The development of methods such as acid agar gel electrophoresis, isoelectric focusing and HPLC permitted an accurate genotypic diagnosis at birth.
In the mid-1970s, a decision was made by the Heart, Blood, and Lung Institute of the National Institutes of Health, to launch a national study of sickle cell anemia. Extensive discussions had identified that understanding the natural history of sickle cell diseases in the United States was a high priority. In 1982 Cooperative Study of Sickle Cell Diseases (CCSCD) in which 23 institutions around the United States entered more than 3000 patients with sickle cell diseases into what became a 15-year longitudinal study. The CSSCD published many studies, which clearly defined American sickle cell diseases and their complications (92).
A cohort of patients diagnosed in the newborn period was included in the CSSCD. It was known that 20% to 30% of children with sickle cell anemia died in the first 5 years of life from overwhelming infections. To address this problem, the Prophylactic Penicillin Study (PROPS) was organized in 1984. Four hundred children less than 4 years of age were randomized to receive either oral penicillin prophylaxis or a placebo. The Study was terminated after only 13 months when it was observed that there was an 84% reduction of pneumococcal sepsis in the penicillin treated group (93). This convincing demonstration of an effective intervention to reduce mortality in very young children led to an National Institutes of Health Consensus Conference, which recommended universal testing of newborns for hemoglobinopathies, so that penicillin prophylaxis could be started in affected infants in the first few months of life (94). Mass neonatal testing for hemoglobinopathies is now routinely performed in 40 of the United States. There has been a striking decrease in childhood mortality and an increased average survival from 17 to more than 40 years, primarily because of the prevention of early deaths.
Despite the fact that the fundamental molecular and genetic defects in sickle cell anemia have been known for more than 50 years, there has been little progress in the day-to-day treatment of these patients and their frequent painful crises. Analgesic therapy and blood transfusions remain the mainstays of management. The recent introduction of hydroxyurea therapy has been of some benefit for many patients, although its mechanism of action is somewhat uncertain (95). Bone marrow or stem cell transplantation can cure sickle cell anemia, but has been infrequently performed in the United States (96). Until problems associated with identifying a suitable donor and the immediate and long-term risks can be reduced, it is unlikely that this procedure will be widely accepted and implemented.
RBC Membrane Abnormalities and Enzymopathies
Inherited abnormalities of the red cell cytoskeletal proteins spectrin, ankyrin, and Band 3 were shown to be the basis of the congenital hemolytic anemias, hereditary spherocytosis (HS), and hemolytic elliptocytosis (97). Management of these congenital disorders often includes splenectomy. Sir Spencer Wells, in 1887 performed a splenectomy on a young woman with jaundice and splenomegaly. When studied 40 years later by Lord Dawson of Penn, the woman was in good health, but her RBC (and also those of her son) had the abnormal osmotic fragility characteristic of HS (98).
The red cells contain all of the enzymes of the glycolytic pathway and inherited deficiencies of these enzymes may result in so-called nonspherocytic hemolytic anemias, the most common of which is pyruvate kinase deficiency (99). Abnormalities of enzymes of the hexose monophosphate shunt, especially glucose-6-phosphate dehydrogenase, affect tens of millions of people throughout the world. G-6-PD deficiency is associated with mutant isozymes and may result in acute hemolytic episodes when affected individuals ingest oxidant drugs or foods such as the fava bean (100).
White Blood Cells
Nonmalignant diseases of the white blood cells are relatively uncommon. Mature white blood cells arise from primitive, multi-potential precursors in the bone marrow whose differentiation and maturation are regulated by a number of cytokines. The genes for some of these cytokines have been cloned and recombinant products have been produced for clinical use.
Neutropenia, with its attendant susceptibility to bacterial infection, is most often a result of chemotherapy treatment for malignancies. Specific, recombinant cytokines such as granulocyte colony stimulating factor (GCSF), have been shown to be effective in reducing chemotherapy related neutropenia, as well as improving inherited neotropenias such as the congenital agranulocytosis of Kostman and cyclic neutropenia (101, 102).
Inherited abnormalities of white blood cell function have also been described. In 1957, Drs. H. Berendes, R.A. Bridges and R.A. Good in Minnesota described a new syndrome in four boys characterized by chronic, suppurative lymphadenitis, hepatosplenomegaly, pneumonia, and dermatitis that was refractory to therapy and had a fatal outcome (103) The disease was named chronic granulomatous disease (CGD). Dr. Paul Quie in Good's laboratory showed that the neutrophils of these boys with CGD could ingest, but not kill, certain bacteria (104). Studies in a number of other laboratories defined the biochemical processes that accompany phagocytosis and showed that in CGD there was a neutrophil defect in oxidase activity. Drs. R. Baehner and D. Nathan developed a rapid diagnostic test based on the inability of CGD neutrophils to reduce the dye, nitroblue tetrazolium (105).
Platelets
Platelets, which have an important role in hemostasis, are derived from committed stem cells, which give rise to megakaryocytes. The proliferation and maturation of megakaryocytes are regulated by a specific cytokine called thrombopoietin. The level of thrombopoietin in the blood appears to be regulated by the peripheral platelet count (106). The gene for thrombopoietin has been identified and cloned enabling production of recombinant thrombopoietin whose clinical use is under active investigation.
The most common pediatric platelet disorder is thrombocytopenia. This can be caused by underproduction of platelets that occurs in bone marrow failure or replacement syndromes. Excessive destruction of platelets by an autoimmune antibody is the basis of idiopathic thrombocytopenic purpura (ITP). ITP in children is usually self-limited and a number of effective therapies are available including corticosteroids, i.v. immunoglobulins and anti Rh immunoglobulin. Splenectomy, usually reserved for children with chronic ITP, was first shown to be effective by Paul Kaznelson, a medical student in Prague who in 1916 persuaded his teacher, Professor Schloffer, to perform the operation on a young woman with profound thrombocytopenia. The operation cured her thrombocytopenia (107). An inherited qualitative platelet abnormality that results in a severe hemorrhagic state was described by Glanzmann (25). Acquired platelet dysfunction occurs when platelets are exposed to aspirin and other drugs that inhibit platelet cyclooxygenase.
Coagulopathies
Hemophilia (love of blood).
More than 2000 years ago, the familial occurrence of severe bleeding after ritual circumcision of boys, who doubtless had hemophilia, was recognized in the Babylonian Talmud (108). A bleeder's disease was described in 1803 by Dr. J.C. Otto of Philadelphia who reported a family with a hemorrhagic disease that was passed by an apparently normal woman to her sons (109). Medical scientists around the world studied coagulation and by the end of the 19th century schemata of the coagulation mechanism were constructed. By 1964, a waterfall cascade model of coagulation was established involving many genetically determined factors (110).
In 1937, Drs. A. Patek and F. Taylor showed. that the defect in hemophilia was a deficiency of a plasma globulin fraction that they called antihemophilic globulin or factor (111) In 1952, the existence of a second type of hemophilia was reported by Drs. I. Schulman and C. Smith who showed that the plasma of a boy with a severe bleeding disorder could correct the coagulation abnormalities of other patients with hemophilia (112). This disorder, called Christmas disease or hemophilia B, was shown to be a deficiency of a clotting factor they designated plasma thromboplastin component or PTC. The nomenclature of these two types of hemophilia as well as the other clotting factors was simplified by a 1962 International Committee of the Society of Thrombosis and Hemostasis which assigned Roman numerals I through XIII for the known coagulation factors. The antihemophilic factor was designated Factor VIII and PTC, Factor IX (113).
Effective treatment of hemophilia evolved very slowly. As early as 1840, Mr. Samuel Lane, a British surgeon, reported that he was able to stop the bleeding of a boy with a hemorrhagic disease by a blood transfusion, but his report went largely unnoticed (114). By 1940, Dr. K.M Brinkhous showed that transfusions of whole blood or plasma temporarily corrected the bleeding abnormalities and infusions of fresh frozen plasma became standard therapy for acute bleeding episodes (115). However, because of the low and variable concentration of Factor VIII in individual plasma donations, adequate levels of Factor VIII were difficult to attain, especially in more severe bleeding episodes. In 1959, Dr. Judith Pool reported that the cold, insoluble precipitate formed during slow thawing of frozen plasma contained a five fold increase in Factor VIII compared with plasma, and that cryoprecipitate could be refrozen and stored for use as replacement therapy. Freeze dried (lyophilized) Factor VIII preparations were developed in the 1960s by fractionating large batches of pooled plasma which increased the Factor VIII concentration 400 fold (116). These products permitted self-administered, home treatment, which greatly simplified the lives of these patients and their families.
In the late 1970s, the HIV virus entered the American blood supply and contaminated the Factor VIII concentrates prepared from large pools of donated plasma. Seventy-five percent or more of patients using Factor VIII concentrates during these years were infected, and most of them ultimately died of AIDS. Methods to eliminate blood transmitted viral infections were developed; but a definitive solution became possible when the gene for Factor VIII was identified and cloned enabling the production of large amounts of recombinant Factor VIII (117, 118). Recombinant Factor VIII became widely available in 1992, and is now the treatment of choice for children.
Hemorrhagic disease of the newborn.
The first definitive description of hemorrhagic disease of the newborn was made by Dr. C.W Townsend of Boston in 1894 (119). Townsend described a generalized, not local, bleeding disorder beginning on the 2nd or 3rd day of life. About 0.6% of newborns had clinical bleeding, usually into the skin and gastrointestinal tract, but occasionally into the brain. There was a 62% mortality, but if not fatal the disease was self-limited with most cases recovering within 5 days. The sexes were equally affected. The onset of transient bleeding in the 1st week of life, as well as the involvement of girls clearly differentiated hemorrhagic disease of the newborn from hemophilia. Dr. Armand Quick postulated that delay of ritual circumcision by Jews until after the 8th day of life may have been based on their empirical observations over centuries that most neonatal bleeding symptoms have waned by that time (120).
Lucas and associates (13) performed clotting times on newborns and showed that during the first 4 days of life: there is a definite and fairly consistent prolongation of the prothrombin time which favors the so called hemorrhagic condition of the newborn. Whipple in 1912 showed that the plasma of a newborn with hemorrhagic disease was deficient in prothrombin (121). More than half of affected babies died of intracranial bleeding or hemorrhagic shock. In 1908 Dr. S.W. Lambert rapidly reversed the bleeding of an affected infant by a transfusion in which the father's radial artery was anastomosed to the baby's popliteal vein (122). In 1923, Dr. J.B. Sidbury, a practicing pediatrician in North Carolina, successfully treated the hypovolemic shock and bleeding disorder of an affected newborn by giving a blood transfusion through the umbilical vein. Sidbury stated that human blood has acted as a specific in this condition (123). In the 1920s continuing into the 1940s, the standard treatment, and in some centers attempts to prevent the condition, used the intramuscular injection of adult blood, often taken from the father. This was before the discovery of the Rh factor and undoubtedly led to Rh isoimmunization of some girls, and erythroblastosis in their offspring (23). In 1929, H. Dam in Denmark identified Vitamin K, and in 1937, Dr. W.W. Waddel showed that Vitamin K administration could prevent coagulation abnormalities in the newborn (124, 125). Routine vitamin K prophylaxis for all newborns was recommended by the Committee on Nutrition of the AAP in 1961 and hemorrhagic disease of the newborn became a rare disease (126).
PEDIATRIC ONCOLOGY
Malignancies in children were believed to be rare and received scant attention in the early pediatric textbooks. L. Emmett Holt's 1897 text The Diseases of Infancy and Childhood had a six-page section on brain tumors and a short description of leukemia, but only brief paragraphs on Hodgkin's disease and other solid tumors (12). In 1940 Dr. Harold W. Dargeon at Memorial Hospital in New York published Cancer in Children.
(127) This 114 page, illustrated book, the first American Pediatric Oncology text, had nine chapters written by physicians from the Memorial Hospital. Twenty years later in 1960, Dr. Dargeon published a more comprehensive textbook, Tumors of Childhood, and a Discussion of Certain Benign Lesions(128). In the mid-20th century, the general pediatric textbooks included sections on pediatric malignancies, usually in their hematology chapters. Beginning with Carl Smith's 1960 Blood Diseases of Infancy and Childhood, pediatric hematology textbooks included large sections on cancer, recognizing that pediatric malignancies were being managed by hematologists (47). A number of comprehensive textbooks on the cancers of childhood have been published in the last 30, including Clinical Pediatric Oncology edited by Drs. W.W. Sutow, D.J. Fernbach and T. Vietti first published in 1973 (129).
Acute lymphoblastic leukemia (ALL).
In 1940, Dr. H. Dargeon stated that the average survival after diagnosis of ALL was less than 3 months. In a survey of hematologists throughout the world in 1965, Drs. Joseph Burchenal and Lois Murphy were able to identify only 71 children in the entire world with ALL who had survived, with or without disease, for more than 5 years (130).
The modern era of leukemia therapy began when Dr. Sidney Farber, head of pathology at the Boston Children's Hospital, observed what he believed was accelerated growth of pediatric tumors, including ALL, induced by of folic acid. Based on this observation (which was probably incorrect!), Farber hypothesized that folic acid antagonists might inhibit tumor growth. He persuaded Y. SubbaRow, a biochemist at the Lederle Company, to synthesize aminopterin, a folate antagonist, which was then used to treat children with ALL. In 1948, Farber and associates reported inducing temporary remissions in 10 of 16 children with ALL with aminopterin (131). (Interestingly, SubbaRow's crucial contribution was not mentioned in this or subsequent publications.) Following Farber's report, new anti-leukemic drugs were introduced. One of the most important was the purine antagonist, 6-mercaptopurine that was shown to be effective in childhood leukemia by Drs. Burchenal, Lois Murphy, and associates in 1953 (132). The armamentarium of antileukemic drugs was enlarged by the introduction of l-asparaginase in 1961, vincristine in 1962 and by other active compounds including corticosteroids, congeners of nitrogen mustard, cytarabidine, and others.
A model of leukemia therapy was developed in experimental animals by Dr. H. Skipper, which showed that drugs with different mechanism of action could be synergistic, and that continuous, combination chemotherapy could theoretically result in cure (133). Based on the Skipper model, various combination drug chemotherapeutic regimens were used in ALL with improved remission rates and survival. In 1965 Dr. E.J Freireich and associates at the National Institutes of Health reported that a quadruple combination of drugs (VAMP) resulted in better survival when compared with sequential treatment with these drugs (134). At the same time Dr. Donald Pinkel at the St. Jude's Hospital in Memphis and the Acute Leukemia Group B consortium reported longer remissions and increased survival with combination chemotherapy (135). A major advance in the treatment of ALL was made by Drs. R.J.A. Auer, D. Pinkel, and associates who hypothesized that relapses in ALL might occur because of persistence of leukemic cells that were protected from systemic chemotherapy in sanctuaries in the CNS and meninges. They used cranio-spinal radiation and later intrathecal methotrexate therapy to destroy these cells (total therapy), which further increased survival (136).
Modern therapeutic protocols that use central nervous system prophylaxis and combination systemic chemotherapy have increased the cure rate of ALL to more than 80%. Immunologic and cytogenetic techniques, have made it possible to identify high-risk cases and to modify therapies to attain the best survival rates with least morbidity (137).
Other malignancies.
Notable successes have occurred in the management of most pediatric malignancies. These improvements have been based upon several principals that have shaped modern therapeutic protocols. These protocols are multimodal and include surgery and radiation to control local disease when possible, integrated with combination chemotherapy given in maximal tolerable doses as adjuvant therapy to control micrometastases.
Many of the advances in the therapy of childhood malignancies have been possible because of the establishment of large, national, multi-institutional cooperative therapy groups, funded by the National Institutes of Health. These groups developed common therapeutic protocols and because of the relatively large numbers of enrolled patients, were able to rapidly assess effectiveness of therapy and treatment toxicities, even in low frequency malignancies. This is well illustrated by the National Wilms' Tumor Study that was organized and funded by the National Cancer Institute in 1969. There are only about 500 new cases of Wilms' tumors each year in the United States so no center had a sufficient number of cases to resolve crucial prognostic and therapeutic questions. Within a decade, a series of National Wilms' Tumor Studies demonstrated that local radiation was not necessary for totally resectable tumors and that surgery combined with multiagent chemotherapy resulted in a >90% survival with a low incidence of treatment related morbidity (138).
The Acute Leukemia Group A was established in 1956 and chaired by Dr. Joseph Burchenal. It later expanded its objectives and was renamed the Children's Cancer Study Group (139). Other national American cooperative groups included the Cancer and Leukemia Group B and the Southwestern Oncology Group, later the Pediatric Oncology Group. The cooperative groups in America have recently been united as the national Children's Oncology Group.
Multi-institutional pediatric oncology groups have also been organized in Europe. In the United Kingdom, there is the Working Committee for Children's Cancer of the Medical Research Council, and in Germany, the Berlin-Munich-Frankfort (BMF) Network.
The spectacular success of these national and international initiatives is evident in the dramatic improvement in survival of children with malignancies (140). In the 1960s the long term survival of all children's malignancies was about 20%; in the 1990s survival and cures had increased to 70%. ALL cures increased from 5% to about 80%; Wilms' tumor cures increased from 30% to 90%; and malignant bone tumor cures increased from 10% to 60%. Cures can be attained even in children with metastatic disease that was previously associated with nearly inevitable death.
Bone marrow and stem cell transplantation is increasingly used in the management of pediatric malignancies that are recurrent or refractory to more conventional therapies. The major toxicity of these procedures: graft rejection, graft versus host disease, and complications of prolonged posttransplantation pancytopenia, are being studied and better controlled. The difficulty of finding a suitable donor, when an HlA matched sibling is not available, is being addressed by national bone marrow registries and the collection and long-term storage of cord blood stem cells.
PEDIATRIC HEMATOLOGY ONCOLOGY IN THE FUTURE
Pediatric Hematology Oncology has increasingly involved other allied medical sciences —genetics, immunology, and molecular biology. Studies to advance the understanding of blood diseases have exploited the technologies of this new biology. The completion of the mapping of the human genome in 2000 holds great promise for the specific tailoring of specific targeted therapeutic agents, especially in Oncology. This has already been accomplished by the design of an inhibitor of a tyrosine-kinase that plays a critical role in chronic myelogenous leukemia (CML) (141). Unlike Dr. Farber's folate antagonists of 50 years ago, this inhibitor does not appear to affect normal cells, but rather, selectively and specifically, destroys CML cells rapidly reversing the hematologic and cytogenetic abnormalities.
The bright promise of gene therapy to correct human diseases is being actively studied in several children's blood diseases. The hemophilias are strong candidates for gene therapy, and the genes for Factor VIII and IX have been successfully transferred into animals using viral vectors. Successful insertion of the Factor VIII gene into the fibroblasts of severe hemophiliacs resulted in small but significant increases in Factor VIII levels which persisted as long as 10 months (142, 143).
Pediatric Hematology Oncology has benefited greatly from advances made by colleagues in Internal Medicine and Pathology, and interactions and communications with them have been mutually beneficial. However, it remains true that there are often significant differences in what appears to be the same disease in pediatric and adult patients. Some pediatric diseases do not occur in adults and vice versa.
As the many areas of Pediatric Hematology Oncology have become ever more complex, there has been an increasing tendency for pediatric hematologists oncologists to concentrate most of their efforts in a specific area. Thus, we now have sub-sub specialists in coagulation, oncology, stem cell transplantation, white cell disorders, red cell disorders, hemoglobinopathies and others. But however fragmented the specialty may seem, we still are able to communicate with each other. One of our basic credos should continue to be that the optimal care of sick infants and children and their families is best provided by physicians trained in pediatrics and pediatric hematology oncology, working together in facilities dedicated to their total care and unique needs.
References
Wintrobe MM 1980 Blood, pure and eloquent. McGraw-Hill, New York, pp 1–31
Leeuwenhoek A 1674 Microscopical observations concerning blood, milk, bones, the brain, cuticula and spittle. Phil Trans (London). ( Cited by Wintrobe Ref. 1, pp. 7–11)
Hewson W 1873 On the figure and configuration of the red particles of the blood, commonly called the red globules. Phil Trans, 63, part 2; 303–323 ( cited by Wintrobe Ref. 1, pp 11–13)
Bizzozaro J 1882 Uber einen neuen Formbestandtheil des Blutes und dessen Rolle bei der Thrombose und der Blutgerinnung. Virchow's Arch Pathol Anat Physiol 90: 261–232. ( Cited by Spaet TH. Platelets; the blood dust. Chapter 16 in Wintrobe Ref.1, pp 533–554
Osler W 1874 An account of certain organisms occurring in the liquor sanguinis. Proc Royal Soc London 22: 391–398
Ehrlich P 1877 Beitrag zur Kenntnis der Amilinfarbungen und ihrer Verwendung in der mikroscopischen Technik. Arch Mikr Anat 13: 263–277. ( Cited by Wintrobe Ref. 1, pp.19–22)
Wintrobe MM 1929 A simple and accurate hematocrit. J Lab Clin Med 15: 287–289
Wintrobe MM Anemia 1934 Classification and treatment on the basis of differences in the average volume and hemoglobin content of the red corpuscles. Arch Int Med 54: 256–280
Coulter WH 1956 High speed automatic blood cell counter and cell size analyzer. Proc National Electron Conf 12: 1034–1040
Dewees WP 1825 A Treatise on the Physical and Medical Treatment of Children. BC Carey & L. Lea, Philadelphia, pp 62, 412
Smith JL 1869 A Treatise on the Diseases of Infancy and Childhood. HC Lea, Philadelphia, p 235
Holt LE 1897 The Diseases of Infancy and Childhood. D Appleton & Co, New York, pp 795–815
Lucas WP, Dearing BF, Hoobler HR, Jones MR 1921 Blood studies on the newborn: morphological, chemical, coagulation, urobilin and bilirubin. Am J Dis Child 22: 524–558
Lippman HS 1924 A morphologic and quantitative study of the blood corpuscles in the new born period. Am J Dis Child 27: 473–526
Lucas WP 1924 Fleichner EC Diseases of the blood. In: Abt IA (ed) Pediatrics. WB Saunders Co, Philadelphia, 406–518
Washburn AH 1934 Blood cells in healthy young infants. Am J Dis Child 47: 993–1010
Guest GM, Brown EW, Wing M 1938 Erythrocytes and hemoglobin of the blood in infancy and childhood. Am J Dis Child 56: 529–549
Kato K 1937 Sternal marrow puncture in infants. Am J Dis Child 54: 209–230
Vogel P, Bassen FA 1939 Sternal marrow of children in normal and in pathologic states. Am J Dis Child 57: 246–268
Sturgeon P 1951 Volumetric and microscopic pattern of bone marrow in normal infants and children. Pediatrics 7: 577–588; 642–650
Hayem G 1928 Die Klinische Haematologie des Kindesalters ( quoted by Baar H, Stransky E). Leipzig, Franz Deuticke, p 9
von Jaksch R 1928 Die Klinische Haematolgie des Kindersalters ( quoted by Baar H, and Stransky E). Leipzig, Franz Deuticke, p 85
Zuelzer WW 1998 Pediatric hematology in historical perspective. In: Nathan DG, Orkin SH (eds) Nathan & Oski's Hematology of Infancy and Childhood, 5th Ed. WB Saunders, Philadelphia, pp 3–17
Pochedley C 1985 Emergence of pediatric hematology as an independent specialty. Am J Pediatr Hematol Oncol 11: 183–189
Glanzmann E 1918 Hereditare hamorrhagische thrombathemia. Jb Kinderheilk 88: 113–120
Glanzmann E 1920 Der Konzeption der Anaphylaktoiden Purpura. Jb Kinderheilk 91: 371–385
Fanconi G 1927 Familiare infantile piriziosartig anamie (Pernizioses blutbild und konstitution). Jb Kinderheilik 117: 257–264
Gasser C 1951 Die Hamolytischen Syndrome des Kindersalters. Stuttgart, Georg Thieme, Verlag
Parsons LG 1938 The haemolytic anaemias of childhood. Lancet 2: 1395–405
Cathie IAB 1950 Erythrogenesis imperfecta. Arch Dis Childhood 25: 313–320
Zibordi F 1925 Ematologia Infantile Normale e Patologia. Milano. Instituto Editoriale Scientifico
Baar HS, Stransky E 1928 Die Klinische Haematologie des Kindersalters. Leipzig, Franz Deuticke
Baar HS, Baar S, Rogers KB, Stransky E 1963 Disorders of Blood and Blood Forming Organs in Childhood. New York, Hafner Publishing Co.
Cooley TB, Lee P 1925 Series of cases of splenomegaly in children with anemia and peculiar bone changes. Trans Am Pediatr Soc 37: 29
Cooley TB, Witwer ER, Lee P 1927 Anemia in children with splenomegaly and peculiar changes in bones. Am J Dis Child 34: 347–363
Zuelzer WW 1985 Reflections of a former pediatrician: acceptance of the Howland Award, 1985. Pediatr Res 19: 1369–1372
Janeway CA 1973 Presentation of the Howland Award to Louis K. Diamond. Pediatr Res 7: 853–857
Diamond LK 1973 Acceptance of the Howland Award. Pediatr Res 7: 857–862
Diamond LK, Blackfan KD, Baty JM 1932 Erythroblastosis fetalis and its association with universal edema of the fetus, icterus gravis neonatorum and anemia of the newborn. J Pediatr 1: 269–309
Diamond LK, Blackfan KD 1938 Congenital hypoplastic anemia. Am J Dis Child 56: 464–475
Blackfan KD, Diamond LK 1944 Atlas of the Blood of Children. Commonwealth Fund, New York
Diamond LK Foreward 1978 In: Miller D, Pearson HA, McMillan (eds) Smith's Blood Diseases of Infancy and Childhood. CV Mosby Co., St Louis pp ix–xiii
Nathan DG, Oski FA 1980 Hematology of Infancy and Childhood. WB Saunders, Philadelphia
Brown AK 1985 Dr. Wolf W. Zuelzer–a unique phenotype, 1985. Presentation of the Howland award. Pediatr Res 19: 1365–1368
Zuelzer WW, Ogden F 1946 Megaloblastic anemia in infancy. Am J Dis Child 72: 211–243
Smith CH, Erlandson ME, Schulman I, Stern G 1957 Hazard of severe infections in splenectomized infants and children. Am J Med 22: 390–404
Smith CH 1960 Blood Diseases of Infancy and Childhood. CV Mosby, Philadelphia
American Board of Pediatrics Minutes of the Sub-committee on Pediatric Hematology Oncology, 1972–1974
Lukens JN, Johnson FL, Lampkin BC 2000 American Society of Pediatric Hematology Oncology: the first 15 years. J Pediatr Hematol Oncol 22: 298–302
Oski FA, Naiman JL 1966 Hematologic Problems of the Newborn. WB Saunders, Philadelphia
Darrow RR 1938 Icterus gravis neonatorum: an examination of etiologic considerations. Arch Pathol 25: 378–417
Landsteiner K, Weiner P 1940 An agglutinable factor in human blood recognized by human serum for rhesus blood. Proc Soc Exp Biol Med 43: 223–234
Landsteiner K Uber Agglutination Serscheiunuingen Normalen Menschlichen Blut. 1901 Wein Klin Wochenschr 14: 1132–1134
Levine P, Burnham L 1941 Isoimmunization in pregnancy: its possible bearing on the etiology of erythroblastosis fetalis. JAMA 116: 825–828
Hart AP 1925 Familial icterus gravis of the newborn and its treatment. Can Med Assoc J 15: 1008–1019
Wallerstein H 1946 Erythroblastosis and its treatment. Lancet 2: 922–924
Weiner AS, Wexler IB 1946 The use of heparin in performing exchange transfusions. J Lab Clin Med 31: 1016–1019
Diamond LK 1948 Replacement transfusion as a treatment of erythroblastosis fetalis. Pediatrics 2: 520–524
Allen FH, Diamond LK 1957 Erythroblastosis fetalis. Little Brown Co, Boston
Liley AW 1954 Liquor amni analysis in the management of the pregnancy complicated by rhesus sensitization. Lancet 1: 1213–1215
Liley AW 1963 Intrauterine transfusion of the foetus in haemolytic disease. BMJ 2: 1107–1011
Clark CA 1967 Prevention of Rh haemolytic disease. BMJ 4: 7–12
Freda VJ, Gorman JG, Pollack W, Robertson JG, Jennings ER, Sullivan JF 1967 Prevention of Rh isoimmunization. Progress report of the clinical trial in mothers. JAMA 199: 390–394
Hallbrecht I 1954 Role of isohemaglutinins anti-A anti-B in the pathogenesis of jaundice of the newborn (icterus neonatorum praecox). Am J Dis Child 68: 248–255
Zuelzer WW, Cohen F 1957 ABO hemolytic disease in heterospecific pregnancy. Pediatr Clin NA NA405–429
Brown AK, Zuelzer WW 1958 Studies on the neonatal development of the glucuronide conjugating system. J Clin Invest 37: 332–340
Guest GM 1947 Hypoferric anemia in infancy. Symposium on Nutrition. Robert Gould Research Foundation, Inc., Cincinnati
Lowe CU, Coursin DB, Filer LJ, Heald FP, Holliday MA, O'Brien D, Owen GM, Pearson HA, Scriver CR 1969 AAP Committee on Nutrition. Iron balance and requirements in infancy. Pediatrics 43: 134–142
Yip R 1990 The epidemiology of childhood iron deficiency: evidence of improving iron nutrition among US children. In: Dobbing J (ed). Brain, Behavior and Iron in the Infant Diet. Springer-Verlag, London, pp 27–42
Parks YA, Wharton B 1990 Iron deficiency and the brain: clinical significance of behavioral changes. In: Dobbing J (ed). Brain, Behavior and Iron in the Infant Diet. Springer-Verlag, London, pp 157–176
Oski FA, Barness LA 1967 Vitamin E deficiency: a previously unrecognized cause of hemolytic anemia in the premature infant. J Pediatr 70: 211–220
Richie JH, Fish MB, McMasters V, Grossman M 1968 Edema and hemolytic anemia in premature infants: a vitamin E deficiency syndrome. N Engl J Med 279: 1185–1190
Wolman IJ 1964 Transfusion therapy in Cooley's anemia. Growth and health as related to long-range hemoglobin levels. Ann NY Acad Sci 119: 736–747
Propper RD, Shurin SB, Nathan DG 1976 Reassessment of the use of desferrioxamine B in iron overload. N Engl J Med 294: 1421–1423
Olivieri NF, Nathan DG, McMillan JH, Wayne AS, Liu PP, McGee A, Martin M, Koren G, Cohen AR 1994 Survival in medically treated patients with homozygous beta thalassemia. N Engl J Med 331: 574–578
Orkin SH, Nathan DG 1998 The thalassemias. In: Nathan DG, Orkin SH (eds) Nathan and Oski's Hematology of Infancy and Childhood, 5th Ed. WB Saunders, Philadelphia
Pearson HA, Cohen AR, Giardina PJV, Kazazian HH 1996 The changing profile of homozygous beta-thalassemia. Pediatrics 97: 352–356
Lucarelli G, Galimberti M, Pochii P, Angelucci E, Baronciani D, Giardina C, Politi P, Durazzi T, Muretto P, Albertini F 1990 Bone marrow transplantation in patients with thalassemia. N Engl J Med 322: 417–421
Herrick JB 1910 Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Trans Assoc Am Physicians 6: 517–521
Emmel VE 1917 A study of the erythrocytes in a case of severe anemia with elongated and sickle-shaped red blood corpuscles. Arch Int Med 20: 5486–598
Sydenstricker VP, Mulherin WA, Houseal RW 1923 Sickle cell anemia: report of 2 cases in children, with necropsy in one case. Am J Dis Child 26: 132–154
Neel JV 1949 The inheritance of sickle cell anemia. Science 110: 64–66
Conley CL Sickle cell anemia–the first molecular disease. ( Cited by Wintrobe, Ref. 1, pp 318–371)
Pauling L, Itano HA, Singer SJ, Wells IC 1949 Sickle cell anemia, a molecular disease. Science 110: 543–548
Ingram VM 1956 A specific chemical difference between the globins of normal human and sickle cell anaemia haemoglobin. Nature 178: 792–794
Ingram VM 1957 Gene mutations in human haemoglobin: the chemical difference between normal and sickle-cell haemoglobin. Nature 180: 326–328
Korber E 1926 Inaugural Dissertation Dorpet, 1886. Cited by Bischoff H. Untersuchungen uber die Resistenz des Hamoglobins des Menchenblut mit besonderer Beruchsichtigung des Sauglingalters. Zeitchr des Exp Med 48: 472–489
Singer K, Chernoff AI, Singer I 1951 Studies on abnormal hemoglobins. 1. Their demonstration in sickle cell anemia and other hematologic disorders by means of alkali denaturation. Blood 6: 413–428
Apt L, Downey WS 1955 Melena neonatorum; the swallowed blood syndrome: a simple test for the differentiation of adult and fetal hemoglobins in bloody stools. J Pediatr 47: 6–12
Kleihauer E, Braun H, Betke K 1957 Demonstration von fetalen Hamoglobin in den Erythrocyten eines Blutstrichsx. Klin Wschr 35: 637–638
Scott RB 1948 Screening for sickle cell in newborn infants. Am J Dis Child 75: 842–846
Gaston MH, Rosse WF 1982 The cooperative study of sickle cell diseases. Am J Pediatr Hematol Oncol 4: 197–200
Gaston MH, Verter JL, Woods G, Pegelow C, Keleher J, Presbury G, Zarlowsky H, Vichinski E, Gill F, Ritchey K, Falletta J 1986 Prophylaxis with oral penicillin in children with sickle cell anemia. N Engl J Med 314: 1593–1599
Whethers D, Pearson H, Gaston M 1989 NIH Consensus Conference. Newborn Screening for Sickle Cell Disease and Other Hemoglobinopathies. Pediatrics 83: 813–914
Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP 1995 Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med 332: 1317–1322
Walters MC 1999 Bone marrow transplantation for sickle cell diseases: where do we go from here?. Am J Pediatr Hematol Oncol 21: 467–472
Gallagher PG, Forget BG, Lux S 1998 Disorders of the erythrocyte membrane. In: Nathan DG, Orkin SH (eds) Nathan and Oski's Hematology of Infancy and Childhood, 5th Ed. WB Saunders, Philadelphia, pp 544–664
Dawson of Penn 1921 The Hume Lectures on haemolytic icterus. Cited by Dacie JV, The lifespan of the red blood cell and circumstances of its premature death. (Cited in Wintrobe, Ref. 1, pp 210–225)
Mentzer WC 1998 Pyruvate kinase deficiency and disorders of glycolysis. In: Nathan DG, Orkin SH (eds) Nathan and Oski's Hematology of Infancy and Childhood, 5th Ed. WB Saunders, Philadelphia, pp 665–703
Luzzato L 1998 Glucose-6-phosphatase deficiency and hemolytic anemia. In: Nathan DG, Orkin SH (eds) Nathan and Oski's Hematology of Infancy and Childhood, 5th Ed. WB Saunders, Philadelphia, pp 704–726
Bonila MA, Gillio AP, Ruggerio M, Kernan NA, Brochstein JA, Abboud M, Fumagalli L, Vincent M, Gabrilove JL, Welte K, Souze LM, O'Reilly RJ 1989 Effects of recombinant human granulocyte colony stimulating factor on neutropenia in patients with congenital agranulocytosis. N Engl J Med 320: 1574–1580
Hammond WP, Price TH, Souza LM, Dale DC 1989 Treatment of cyclic neutropenia with granulocyte stimulating Factor. N Engl J Med 320: 1306–1311
Berendes H, Bridges RA, Good RA 1957 A fatal granulomatosis of childhood: the clinical study of a new syndrome. Minn Med 40: 309–312
Quie P, White JG, Holmes B, Good RA 1967 In vitro bacteriocidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J Clin Invest 46: 668–679
Baehner RL, Nathan DG 1968 Leukocyte oxidase: quantitative nitroblue tetrazolium test in chronic granulomatous disease. N Engl J Med 278: 971–976
Kaushansky K Thrombopoietin. 1998 N Engl J Med 339: 746–754
Crosby WH The spleen. Chapter 5 ( Cited in Wintrobe Ref. 1. pp 97–138)
Garrison FH 1929 An Introduction to the History of Medicine, 4th Ed. WB Saunders, Philadelphia
Otto JC An account of a hemorrhagic disposition existing in certain families. Med Respos 1803 6: 1–4
Davie EW, Ratnoff OD 1964 Waterfall sequence for intrinsic blood clotting. Science 145: 1310–1311
Patek AJ, Taylor FA 1937 Hemophilia II. Some properties of a substance obtained from normal plasma effective in accelerating the coagulation of hemophilic blood. J Clin Invest 16: 113–124
Schulman I, Smith CH 1952 Hemorrhagic disease in an infant due to a previously undescribed factor. Blood 7: 794–807
Wright IS 1962 The nomenclature of clotting factors. Thromb Diath Hemorrh 7: 381–395
Lane S Hemorrhagic diathesis: successful transfusion of blood. Lancet 1840 1: 185–188
Brinkhous KM 1939 A study of the clotting defect in hemophilia: the delayed formation of thrombin. Am J Med Sci 198: 509
Pool JG, Hershgold EJ, Pappenhagen AR 1962 High-potency antihaemophilic factor concentrate prepared from cyroglobulin precipitate. Nature 203: 312
Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker TL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orre C, Amphlett GW, Foster WB, Coe ML, Knutsen GJ, Fass DN, Hewick RM 1984 Molecular cloning of a cDNA encoding human antihemophilic factor. Nature 312: 341–347
White GC, McMillan CW, Kingdon HS, Shoemaker CB 1989 Use of recombinant antihemophilic factor in the treatment of two patients with classical hemophilia. N Engl J Med 320: 166–170
Townsend CW 1894 The hemorrhagic disease of the newborn. Arch Pediatr 11: 559–565
Quick AJ 1942 The Hemorrhagic Diseases and the Physiology of Hemostasis. Charles C Thomas, Springfield, Ill
Whipple GH 1912 Hemorrhagic diseases. Arch Int Med 9: 363–399
Lambert SW 1908 Melena neonatorum with report of a case cured by transfusion. Med Record 73: 885–887
Sidbury JB 1923 Transfusion through the umbilical vein in hemorrhage of the newborn. Am J Dis Child 25: 290–296
Dam H 1929 Cholesterinstoffwechsel in Huhneriern und Huhnchen. Biochem Zeit 215: 475–492
Waddell WW, Guerry DP, Bray WE, Kelly OR 1939 Possible effects of Vitamin K on prothrombin and clotting time in newly born infants. Proc Soc Exp Biol Med 40: 432–434
AAP Committee on Nutrition 1961 Vitamin K in the newborn. Pediatrics 28: 501–507
Dargeon HW 1940 Cancer in children and a discussion of certain benign tumors. CV Mosby, Philadelphia
Dargeon HW 1960 Tumors of Childhood. Paul B. Hoeber, New York
Suttow WW, Fernbach DJ, Vietti T (eds) 1977 Clinical Pediatric Oncology. CV Mosby, St. Louis 126:
Burchenal HH, Murphy ML 1965 Long term survivors in acute leukemia. Cancer Res 25: 1491–1495
Farber S, Diamond LK, Mercer RD, Sylvester RA, Wolff JA 1948 Temporary remissions in acute leukemia of children produced by folic acid antagonist 4-aminopteroyl-glutamic acid (aminopterin). N Engl J Med 238: 787–793
Burchenal HH, Murphy ML, Ellison RR, Sykes MP, Tan TC, Leone LA, Karnofsky DA, Craver LF, Dargeon HW, Rhodes CP 1953 Clinical evaluation of a new antimetabolite, 6-mercapto purine in the treatment oif leukemia and allied diseases. Blood 8: 965–999
Skipper HE, Schabel F, Wilcox WS 1964 Experimental evaluation of potential anticancer agents. On the criteria and kinetics associated with “curability” of experimental leukemia. Cancer Chemother Rep 35: 1–111
Freireich EJ, Karon M, Frei E 1964 Quadruple combination therapy (VAMP) for acute lymphoblastic leukemia of childhood. Proc Am Assoc Cancer Res 5: 20
Acute Leukemia Group B 1965 New treatment schedule with improved survival for acute lymphocytic leukemia of childhood. JAMA 194: 75–81
Auer RJA, Simone J, Hustu O, Walters T, Borella L, Pratt C, Pinkel D 1971 Central nervous system therapy and combination chemotherapy of childhood lymphocytic leukemia. Blood 37: 272–281
Pui CH, Evans WE 1998 Drug therapy: acute lymphoblastic leukemia. N Engl J Med 339: 605–614
D'Angio GJ, Breslow N, Beckwith JB, Evans A, Baum H, deLorimier A, Fernbach D, Hrabovsky E, Jones B, Kelalis P et al 1989 Treatment of Wilms' Tumor: Results of the Third National Wilms' Tumor Study. Cancer 64: 349–360
Pochedly C 1986 M. Lois Murphy. Am J Pediatr Hematol Oncol 8: 60–64
Boring CC, Squires TS, Tong MS, Montgomery BA 1944 Cancer statistics, 1994. CA 44: 7–30
Goldman JA, Melo JV 2001 Targeting the BCR-ABI tyrosine kinase in chronic myelogenous leukemia. N Engl J Med 344: 1031
Roth DA, Tawa NE, O'Brien JM, Treco DA, Selden RF 2001 Non-viral transfer of the gene encoding coagulation Factor VIII in patients with severe hemophilia. N Engl J Med 344: 1735–1742
Miller DG, Stamatoyannopoulos G 2001 Gene therapy for hemophilia. N Engl J Med 344: 1782–1783
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pearson, H. History of Pediatric Hematology Oncology. Pediatr Res 52, 979–992 (2002). https://doi.org/10.1203/00006450-200212000-00026
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200212000-00026
