
Abstract
Cystic fibrosis (CF) is caused by mutations of the gene encoding for the CFTR (CF transmembrane conductance regulator) protein. The most frequent mutation, the ΔF508 mutation, results in a defective cAMP-regulated chloride transport in the epithelial cells. The spectrum of clinical manifestations in patients bearing homozygous ΔF508 mutations can vary considerably, suggesting that, in the patients with a mild disease, CFTR could be partly functional. To test this hypothesis, we explored in nasal ciliated epithelial cells (NCC) of 9 control subjects and 23 ΔF508 homozygous patients the anion conductive pathway by a halide sensitive fluorescent dye assay SPQ (6-methoxy-N-3′-sulfopropylquinolinium) and the CFTR transcript levels by RT-PCR. As 50% represented the lowest fraction of the control subjects NCC demonstrating a cAMP-dependent conductance, a CF patient was considered as “cAMP responder” if at least 50% of the NCC tested displayed a cAMP-dependent conductive pathway. According to these criteria, 8 of the 23 patients were considered as cAMP responders. They had a significantly less severe disease considering the respiratory function and infectious status. The amount of CFTR mRNA did not differ between the control subjects and the patients. No statistical correlation could be found between the transcript level and the expression of a cAMP conductive pathway. This cAMP-dependent Cl− conductance detected in homozygous NCC could be due to a residual CFTR activity and may explain the mild phenotypes observed in some ΔF508 homozygous patients.
Similar content being viewed by others
Main
Cystic fibrosis (CF) is caused by mutations in a single gene on the long arm of chromosome 7 encoding for the CF transmembrane conductance regulator (CFTR) protein (1). CFTR acts as a chloride channel activated by cyclic AMP (cAMP) (2). The most common mutation is the deletion of a single phenylalanine residue at position 508 in the CFTR protein, called ΔF508 mutation. This mutation has deleterious effects on the processing of the ΔF508 CFTR protein through the endoplasmic reticulum causing drastically reduced levels of protein to be expressed on the plasma membrane of epithelia. One of the consequences is a defective cAMP-regulated chloride transport in the epithelial cells (3). The resultant clinical manifestations classically include high sweat chloride concentrations, pancreatic insufficiency, and a suppurative progressive obstructive pulmonary disease that leads to respiratory failure (4). However, the spectrum of clinical manifestations of patients bearing homozygous ΔF508 mutation can vary considerably, from a late-onset, mild, nearly asymptomatic pulmonary disease to an extremely severe neonatal and rapidly lethal form (5). To 4test whether this clinical heterogeneity is linked to functional heterogeneity, we investigated the cAMP-dependent chloride conductive pathways by a halide sensitive fluorescent dye assay SPQ (6-methoxy-N-3′-sulfopropylquinolinium) and the CFTR transcript levels in nasal epithelial cells (NCC) of ΔF508 homozygous patients with varying severity of pulmonary disease.
METHODS
Patients.
We investigated NCC obtained from both healthy control subjects and ΔF508 homozygous patients. The control subjects had a normal sweat test and had been negatively screened for the 30 most frequent CFTR mutations. All the patients were pancreatic insufficient. Exclusion criteria were recent upper respiratory tract infection, smoking, and nasal polyps.
Clinical status was assessed with the following criteria: sputum bacteriology and number of antibiotic cures in the preceding year indicative of infectious status; chest radiographic score; forced expiratory volume (FEV1) and forced vital capacity (FVC) indicative of pulmonary function and Shwachman score indicative of global disease severity (6). FEV1 and FVC were expressed as percent of predicted values for age and sex. Maximal value of Shwachman score was 100.
The study was approved by the Necker-Enfants Malades Ethics Committee, and informed consent was obtained from each subject.
Cell sampling.
Cells were obtained from below the middle turbinate using a cytology brush (approximately 1 × 106 cells per subject). Brushes were immediately placed in cold Dulbecco Modified Eagle's Medium (DMEM). The cells were transported to the laboratory on ice. An aliquot was cytocentrifuged on a slide to check the quality of the sample under the light microscope and the remaining cells were divided into 2 aliquots that were pelleted (600 g, 2 min, 4°C). The first pellet was suspended in 500 μL Trizol™ reagent (GIBCO-BRL, Les Ulis, France) for RNA extraction. The second aliquot was used for functional assay.
Each sample was assessed for quality according to the presence of necrotic, squamous, inflammatory (polymorphonuclear cells), and ciliated cells (Table 1). Samples without NCC and those with necrotic or more than 20% inflammatory cells were discarded (quotation 1 to 5).
Functional assay.
The transmembrane anion conductive pathway was measured by a halide sensitive fluorescent dye assay SPQ according to the method described by Verkman, Stern, et al., and Tondelier et al.(7–9). This method allows measuring the rate of Cl− transport as the rate of fluorescent changes in response to the exchange of extracellular Cl− with NO3−, an anion that passes through CFTR but, unlike Cl−, does not quench indicator fluorescence. After addition of the cAMP agonists, activation of CFTR results in Cl− efflux and NO3− influx, producing an increase in fluorescence. In our experimental design, Cl− was replaced by iodide (I−) as CFTR is permeable to I− and SPQ fluorescence is more strongly quenched by I− than by Cl−.
Briefly, the cells of the second aliquot were loaded with 10 mM SPQ (Molecular Probe, Eugene, OR) in a hypotonic buffer during 2 min (1:1 dilution of DMEM with sterile deionized water). Aliquots of cells were then mounted on a glass coverslip coated with Cell-Tak (Collaborative Biomedical Products, Becton Dickinson, Bedford, MA, U.S.A.) to improve cell adherence and left to settle for 30′ at 37°C. The coverslip was then placed in a perfusion chamber on the stage of an inverted microscope (Diaphot-TMD, Nikon, Champigny-sur-Marne, France) where they were continuously perfused at 37°C with the control isotonic NaI solution (in mM: 135 NaI, 2.4 K2HPO4, 0.6 KH2PO4, 1 CaSO4, 1 MgSO4, 10 glucose, 10 HEPES, pH = 7.4). Intracellular SPQ was excited at 350 nm by a 100 W halogen lamp. Cells were viewed with a Nikon × 40 fluorescence objective and emitted fluorescence (>410 nm) which was collected using a low-light charge coupled device video camera (Photomic Science, Grenoble, France) interfaced with a computer containing a digital imaging system (Imstar, Paris, France).
Intracellular SPQ fluorescence was measured at 15-s intervals in cells with actively beating cilia. Basal SPQ fluorescence was measured for 2 min in the presence of NaI solution. NaI solution was then replaced equimolarly by NaNO3 (NaNO3 solution) to unmask a basal anion conductance. The further addition of the “cAMP cocktail” (in μM: 25 forskolin (Sigma Chemical Co., St. Louis, MO, U.S.A.), 100 isobutylmethylxanthine - IBMX (Sigma Chemical Co.), 500 8-(4-chlorophenylthio)-adenosine 3′:5′-cyclic monophosphate (Sigma Chemical Co.) - pCPT-cAMP) allowed measuring the cAMP-dependent anion conductive pathway. Two experimental protocols were used to test the sensitivity of the assay (Fig. 1). In protocol 1, the cells were superfused sequentially with NaI, NaNO3, and cAMP cocktail solution while in protocol 2, NaNO3 was first replaced by NaI solution before adding the cAMP cocktail. At the end of both experimental designs, NaNO3 solution was replaced by NaI solution again to test if the fluorescence changes were reversible to the original background. The background signal due to cell and instrument autofluorescence was considered during all the procedure. The experiment was considered for further analysis if results were obtained in more than five cells per patient.

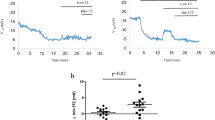
SPQ fluorescence measurement of halide anion conductance in NCC with protocol 1 and protocol 2. In protocol 1, the cells are superfused with NaI, NaNO3, and cAMP cocktail solution. In protocol 2, NaNO3 is first replaced by NaI solution before adding the cAMP cocktail. The NaNO3 solution is replaced by NaI solution at the end of both experimental designs. Basal anion conductance is assessed by ΔFbasal/Δt. cAMP conductance is assessed by ΔFcAMP/Δt in protocol 1 and ΔFcAMP/ΔFbasal in protocol 2.
Fluorescence changes were measured during the first 45 s after replacement of the solutions. Basal anion conductance was defined as the maximal fluorescence change after replacement of I− by NO3− (ΔFbasal/Δt). The cell was considered to display a basal conductance if ΔFbasal/Δt was >0.1. cAMP anion conductance was defined as the maximal fluorescence change after addition of the cAMP cocktail in the NaNO3 solution. cAMP anion conductance was assessed by ΔFcAMP/Δt in protocol 1 and ΔFcAMP/ΔFbasal in protocol 2, as NaNO3 was replaced by NaI before adding the cAMP cocktail (Fig. 1). The cell was considered to display a cAMP-stimulated anion conductive pathway if ΔFcAMP/Δt was >0.1 in protocol 1, and if ΔFcAMP/Δt was >0.1 and ΔFcAMP/ΔFbasal >1 in protocol 2. Responses of a “responding” wild type and a “non-responding” ΔF508 cell are shown in Fig. 2. We verified in preliminary experiments on the human pulmonary Calu-3 cell-line (American Type Culture Collection, Rockville, MD, U.S.A.), that both protocols provided similar responses (data not shown). Two patients and two control subjects NCC were successively tested with both protocols and showed equivalent results (data not shown).

SPQ fluorescence measurement of halide anion conductance in a wild type “responder” NCC and in a ΔF508 “non-responder” cell according to protocol 1.
Semi-quantitative analysis of CFTR transcripts.
Total RNA was isolated using Trizol™ reagent by acid guanidine thiocyanate-phenol-chloroform and precipitated by isopropanol at −20°C. RNA (2 μg) was placed in a final volume of 20 μL containing 100 units of MMLV Superscript II reverse transcriptase (GIBCO-BRL, Les Ulis, France), 500 μM dNTP, 10 mM DTT, and 200 ng of random hexanucleotide primers (Pharmacia Biotech, Saint Quentin en Yvelines, France). cDNA was then diluted 5-fold in sterile water.
cDNA (5 μL) was added to a 25 μL final volume containing 2 U of Taq DNA polymerase (Boehringer Mannheim, Meylan, France), 500 μM dNTP, 1 μCi of [αP32]dCTP (Amersham, Les Ulis, France), 2.5 mM MgCl2 and 20 pmoles of CFTR upstream (5′-GCC TTC CGA GTC AGT TTC AG-3′) and downstream (5′-CTG CCT TCT GTG GAC TTG GT-3′) primers selected on exon 6 to exon 7 or 10 pmoles of β2microglobulin upstream (5′-ACC CCC ACT GAA AAA GAT GA-3′) and downstream (5′-ATC TTC AAA CCT CCA TGA TG-3′) primers. The samples were then submitted to 35 cycles of amplification for CFTR or 30 cycles for β2microglobulin consisting of 1 min denaturation at 94°C, 1 min annealing at 55°C, and 1 min 30 elongation at 72°C. Amplification products were then submitted to electrophoresis on 8% polyacrylamide gels at 750 V for 3.5 h. The gels were then exposed to X-OMAT films (Eastman Kodak, Rochester, NY, U.S.A.) overnight at −20°C. Autoradiographs were scanned using a Bio-Rad densitometer (Bio-Rad, Ivry sur Seine, France) and Molecular Analyst Software (Bio-Rad). CFTR mRNA content was expressed as the ratio of the densitometric value of CFTR amplification product to that of β2microglobulin amplification product. The positive control used was the human pancreatic carcinoma cell line Capan 1 obtained from the American Type Tissue Collection.
Statistics.
Statistical analyses were performed using the BMDP software package (University of California, Los Angeles, CA, U.S.A.) (10). A single value was derived from the results of all the cells tested in a given subject. Data are presented as mean (SD) of the fluorescence change and mean (range) percent of cells responders per subject. The comparison tests were assessed by the correlation test for quantitative measurements and by ANOVA tests for qualitative measurements. The null hypothesis was rejected at p < 0.05.
RESULTS
Subjects
Nine healthy control subjects (Table 2) and 23 ΔF508 homozygous patients (Table 3) were included in the study. All the control subjects were asymptomatic for respiratory or gastrointestinal symptoms and had normal chest radiography and pulmonary function tests.
Baseline characteristics of the patients are presented in Table 3. Mean patient's age was 14.9 y (5–30). Eleven patients had a chronic colonization with Pseudomonas aeruginosa. Disease severity was highly variable as assessed by the range of FEV1 (19–115) and FVC (50–116), the number of antibiotic courses for bronchial exacerbations in the preceding year (0–13), and the Shwachman score (40–90).
Functional Assay
Control subjects.
The NCC of the control subjects was tested with protocol 1 (Table 2). Ninety-two percent (63–100) NCC per control subject displayed a basal anion conductive pathway with a mean increase ΔFbasal/Δt of 0.55 (0.1–1.35). A mean of 75% NCC per control subject displayed a cAMP-stimulated anion conductive pathway with broad range from 50% to 100%. In this group, the mean increase of ΔFcAMP/Δt was 1.03 ± 1.11.
Considering the high variability of these results, we derived the following criteria for the analysis of the patients' data. For a given patient, we considered that a basal anion conductance or a cAMP-dependent conductive pathway may be involved in the ion transport process if the proportion of cells displaying this pathway was at least equal to the lowest proportion found in the control subjects e.g. 63% of the tested NCC for the basal conductance (see control subject 7 in Table 2) and 50% for the cAMP dependent pathway (see control subject 8 in Table 2).
Patients.
Basal anion conductance. Seventeen patients (group 1) had more than 63% NCC, with a mean of 86 (69–100) % NCC displaying a fluorescence increase ΔFbasal/Δt >0.1 and were therefore considered as having a basal conductance (Table 3). The remaining 6 patients (group 2) had less than 63% NCC, with a mean of 37% (0–61), displaying a basal conductance and were considered as lacking a basal Cl− conductance. The mean quality cell quotation was similar in the two groups.
The respiratory function tests were better in the group of patients displaying a basal conductance but this difference did not reach the significant level (Table 4). There was no correlation between the presence of a basal anion conductance and the Shwachman score or the number of the preceding antibiotic cures.
cAMP-dependent anion conductance. Eight patients (group 1) had a mean of 75% (56–100)% NCC per patient displaying a fluorescence increase after addition of the cAMP cocktail and were therefore considered as “cAMP responders” (Table 3). Fifteen patients (group 2) had less than 50% of NCC, with a mean of 21.9% (0–46) NCC displaying a significant variation of the fluorescence signal during cAMP stimulation and were considered as “cAMP non-responders.” Three out of these 15 patients had no cells at all demonstrating a cAMP-mediated pathway. The cell quality quotation was similar in the 2 groups.
There was no correlation between the proportion of NCC displaying a cAMP conductive pathway and the intensity of the fluorescence change after the addition of the cAMP cocktail (r = 0.117; NS).
The cAMP conductive pathway was not correlated with the presence of a basal conductance, considering the percent of NCC (r = 0.304; NS) or the intensity of the fluorescence change (r = 0.266; NS). However, six of the seven patients with a cAMP conductance had also a basal conductance and the basal conductance was higher in the group of patients displaying a cAMP conductive pathway (ΔFbasal/Δt = 0.397 (0.322)) than in the patients without (ΔFbasal/Δt = 0.196 (0.094);p = 0.03).
Patients bearing a cAMP dependent Cl− conductance had a significantly less severe disease, considering the respiratory function tests, the number of bronchial exacerbations requiring antibiotics, and the index of global disease severity (Table 5). The FEV1 and the Shwachman score were positively correlated with the proportion of NCC displaying a cAMP conductance (Fig. 3). The number of antibiotic courses in the preceding year was inversely correlated (r = 0.509;p = 0.02). This relation disappeared when the fluorescence change was considered instead of the proportion of the NCC. The 2 groups did not differ for age, Pseudomonas aeruginosa colonization, or concomitant nasal or pulmonary treatment.

Correlation between FEV1 and Shwachman score and the percentage of patient's NCC having a cAMP-dependent Cl− conductive pathway.
CFTR Expression
The amount of CFTR mRNA did not differ between the control subjects (mRNA = 0.584 (0–1.369)) and the patients (mRNA = 0.535 (0.39)). For the patients, CFTR mRNA expression did not correlate with FEV1 (r = 0.35; NS); FVC (r = 0.25; NS), the number of antibiotic courses, (r = 0.003; NS) and the Shwachman score (r = 0.18; NS). There was no correlation between the percent of NCC demonstrating a cAMP conductive pathway or the cAMP induced-fluorescence change and the transcript levels. However, levels of CFTR gene transcripts were higher in samples with NCC demonstrating a basal conductance or a cAMP-dependent conductance but this did not reach a significant level (Table 4 and 5).
DISCUSSION
Our results provide evidence for a cAMP-regulated pathway in ΔF508 NCC, similar to that described in cells expressing wild type CFTR channel and in healthy subjects. The presence of this cAMP- mediated anion conductance in more than 50% NCC tested for a given patient is associated with a better clinical status. This suggests that the mild phenotypes encountered in some ΔF508 homozygous patients could be linked to a residual function of CFTR in airway tissues. This study also highlights a great heterogeneity in the cAMP conductive pathway in NCC of both CF and non-CF control subjects.
The SPQ fluorescence assay is used extensively in cell culture systems to measure the Cl− transporting function of CFTR with both a high sensitivity and specificity (7, 8). This study demonstrates that this assay may be routinely applied for studying cAMP-mediated Cl− conductance in freshly obtained, noncultured primary NCC of CF patients. The sampling is easy, rapid, and painless; it can be performed at the bedside. The tissue requirement is modest, making it particularly suited to measurement in children. It avoids potential alterations in phenotype resulting from cell culture technique. However, several technical problems must be overcome to obtain valuable and reproducible results. First, the experiments have to be done as soon as possible. A waiting of more than 3 h, even on ice, damages the cells. Second, primary ciliated cells, even when covered with mussel collagen (Cell-Tak, Bedford, MA, U.S.A.), adhere poorly to the glass surface. This results in movements or even loss of the cells during the serial perfusion protocol. This problem can be overcome by lowering the perfusion rate and the waste suction. Third, SPQ cell loading may vary from one sample to another and even from one cell to another. This can be limited by the loading of the total sample at once and studying of at least 5 cells or cell clumps per patient. Moreover, the experiment must not last for more than 2 h to avoid ulterior dye leak. After nasal brushing, the cells occur predominantly in clumps. This makes it difficult to assess the type of epithelial cell within each clump and, in turn, the relative contribution of each cell type to Cl− efflux. We decided to study the cell clumps only when surrounding cells with actively beating cilia could be seen. This was based on the results of Stern and collaborators who showed that single ciliated cells and cell-clumps containing at least some ciliated cells with actively beating cilia did not show any significant difference in the intensity of fluorescence nor in the rate of change of fluorescence (8). Moreover, as preliminary experiments showed that the NCC from inflammatory mucosa was not suitable for SPQ experiments, the inflammatory samples were eliminated.
Even taking into account all the above listed problems, the interpretation of fluorescence data has been particularly challenging because of the variability of the fluorescence changes within the same patient and the necessity to derive a final conclusion for a single patient from the results obtained in the NCC of the whole sample. An endpoint was derived from the results obtained in control subjects to class the patient as “responder” or “non-responder”e.g. the lowest proportion of the control subjects NCC demonstrating a significant fluorescence change in the presence of cAMP. Finally, all individual ciliated cells or clump-cells in a series of images were considered and the results were displayed as percentages of cells having different Cl− pathways. This pattern of qualitative classification was highly reproducible and allowed to limit the day-to-day intrasubject and intersubject variability.
We measured the cAMP conductance using 2 protocols. In protocol 2, NaNO3 was replaced by NaI before adding the cAMP cocktail to sensibilize the measure. The cAMP-dependent conductance was therefore calculated relatively to the basal conductance (ΔFcAMP/ΔFbasal). However, it must be underlined that the magnitude of the initial fluorescence partly depends on the initial loading and the intracellular dye concentration and may vary from cell to cell. This modality of calculation (ΔFcAMP/ΔFbasal) may thus underestimate the cAMP variation if the basal conductance is strong and conversely overestimates the cAMP conductance if the basal conductance is low. Our final conclusion is that protocol 2 is less sensitive than protocol 1 which considers the absolute fluorescence variation due to cAMP agonists.
Our results demonstrate that the cAMP-dependent Cl− conductance pathway can be low in NCC of non-CF subjects. This is consistent with the study of McLachlan, which revealed considerable variation of the cAMP dependent conductance pathway within the control subjects (11). This may result from a low amount of protein at the apical membrane and therefore a low CFTR-mediated conductance. This suggests that even low level of functional protein is sufficient to offer protection from symptoms of CF disease. These results are consistent with previous studies demonstrating that in normal individuals the CFTR gene is expressed at low levels of 1–2 transcripts per cell in the epithelium of the nose, trachea, and bronchi (12) and that a low level of normal CFTR is sufficient for restoration of normal airway epithelial function in gene transfer experiments (13, 14).
The main finding of this study is the existence of a c-AMP-regulated pathway in NCC from ΔF508 patients, similar to that described in cells expressing wild-type CFTR channel. It is generally recognized that the inability of the ΔF508-CFTR nascent chain to mature and to be exported from the endoplasmic reticulum to the cell membrane is the basis of the abnormal fluid secretion in CF epithelia (3). However, if misprocessing is overcome, ΔF508-CFTR has a functional activity (15). A few studies have demonstrated that ΔF508-CFTR may be appropriately located in CF epithelia of homozygous ΔF508 patients (16–18). A recent study, based on immunocytochemistry, has evidenced a gradient for the apical localization of CFTR in freshly obtained nasal cells from non-CF subjects (56%), carriers (42%), and ΔF508 homozygous patients (22%) (18). This suggests that a modification of the cell quality control could account for a possible correct CFTR location in ΔF508 airway cells. Therefore, the cAMP-dependent anionic conductance observed in our study in NCC from ΔF508 homozygous patients could be linked to the presence of a functional ΔF508 CFTR at the apical membrane of NCC. This results in a better hydration of mucous secretions, a better pulmonary clearance and therefore a less severe pulmonary disease. This could explain the overall better clinical status observed in the “cAMP responders” patients of our study.
Evidence that some ΔF508 CFTR may be functionally expressed in the apical membrane has been shown in several studies. In mice, the combination of forskolin and milrinone hyperpolarizes the nasal epithelium of ΔF508 mice but not of cftr(−/−) mice, which is indicative of a CFTR-induced Cl− secretory response in ΔF508 mice (19). In humans, nasal potential difference measurements have evidenced a significant hyperpolarization in response to isoproterenol in ΔF508 patients (20–22). This residual CFTR activity seems to be clinically relevant as the FEV1 is significantly better in the patients with the greater ability to secrete Cl− in response to isuprel (21, 22). Similarly, short circuit current studies in rectal suction biopsies have shown that Cl− secretion in response to cAMP agonists can be present in intestinal tissue of ΔF508 homozygous patients (22–23) and correlates with a better nutritional status and a milder CF lung disease (22).
However, it should be underlined that numerous immunocytochemical focusing on the sweat glands (24), the bronchial tissues (25), or primary cultures of CF airway epithelia (26) did not detect any amount of ΔF508 CFTR at the apical cell membrane. We cannot formally exclude that the response observed in the ΔF508 NCC cells could also be explained by alternative cAMP-dependent Cl− channels. It has been indeed demonstrated that the airway epithelium contains at least three stimulus-activated Cl− conductance pathways, i.e. a Ca2+-activated conductance, a volume-activated conductance, and the CFTR conductance that is activated by cAMP (27). In cftr−/− mice, it has been speculated that the prominent Ca2+-mediated Cl− secretory pathway detected in the airways protects the tissue from the absence of CFTR-mediated Cl− conductance and prevents these animals from CF lung disease (28). In humans, it has been suggested that the variable electrophysiological characteristics observed in ΔF508 CF respiratory and intestinal tissues might be explained by these alternative Ca2+-regulated Cl− channels (29). Other modifier genes or environmental factors could play an additional role (30). In our experiments, we did not test the Ca2+-mediated conductance but focused on cAMP-dependent conductance to investigate CFTR function. Volume-dependent conductance may be activated after preincubation of the cells in hypotonic buffer leading to cellular swelling and generation of intracellular signals including intracellular cAMP and [Ca2+]. This conductance might contribute to the basal and cAMP conductance (31). However, in our protocol, the cells are left to settle for 30 min after loading and are studied at least 45 min after preincubation in the hypotonic buffer. This time is probably enough for the cell to return to its basal status and for the cAMP-stimulated anion conductance to be deactivated. Besides, it should be underlined that the experimental conditions are always similar. If there is any activation, this should be present in all the samples. This therefore does not explain the differences observed between the CF samples. We thus hypothesize that this cAMP-stimulated Cl transport could be due to CFTR. But the formal proof can only be done after patch clamp experiments.
In contrast to our results, two previous studies with SPQ imaging in freshly isolated NCC did not find any cAMP-regulated pathway in ΔF508 NCC (4, 11). This could be explained by the fact that in both studies only a single mean value was derived from the values from all cells from a given subject. This procedure may underestimate the physiologic heterogeneity in response and mask therefore the response of cells displaying a cAMP-conductive pathway. However, one of these studies underlines considerable variation and indeed some overlap between the CF patients and the control group, suggesting that some ΔF508 cells might have a cAMP-dependent conductive pathway (11).
In our study, the cAMP conductance was not correlated with the basal conductance, considering the proportion of cells or the intensity of the fluorescence change. However, 6 of the 7 patients displaying a cAMP conductance had also a basal conductance and a higher ΔFbasal/Δt. This is not due to a better sampling as the quality quotation and the number of cells studied per patient did not differ between the patients. Interestingly, the respiratory function tests were better in the group of patients displaying a basal conductance. However, the difference did not reach the significant level. This may be due to the lack of power of the study because of the small number of patients. Basal Cl− current have been recently described in native murine airways and it has been hypothesized that they may be responsible for normal respiratory function in CF mice (32). Further experiments are needed to assess if human NCC basal conductance is linked with CFTR or could in itself cause dysregulation of Cl− absorption and be therefore a factor of disease severity.
We report variable levels of CFTR mRNA in NCC of both CF ΔF508 patients and healthy volunteers, with no significant reduction in ΔF508 patients in comparison with the healthy subjects. These results are similar to those of other studies (12, 33, 34). No statistical correlation could be found between CFTR transcript levels and the presence of a cAMP dependent Cl− conductance. This confirms the results of a previous study, based on techniques of microelectrodes that did not establish a relationship between CFTR mRNA expression and electrophysiological properties (34).
CONCLUSION
In conclusion, we demonstrated that the SPQ assay might be routinely applied for studying cAMP-mediated anion transport in freshly isolated, non-cultured human NCC. Considering the cellular heterogeneity in airway samples, we developed a qualitative analysis that could clearly distinguish between the “cAMP responder” and the “non cAMP responder” subjects. Although our results provide evidence for a considerable variability of cAMP Cl− conductance pathway in NCC from both CF and non-CF subjects, ΔF508 homozygous CF patients with residual CFTR activity appear to have a less severe disease. This could be a part of the explanation for the clinical heterogeneity in CF disease. This test could be applied to gain more insight into the patient's electrophysiological characteristics in correlation with disease severity. Novel treatment to maximize this pathway could be a new target for pharmacological therapy in patients with a severe pulmonary disease.
Abbreviations
- CFTR:
-
cystic fibrosis transmembrane conductance regulator
- CF:
-
cystic fibrosis
- cAMP:
-
cyclic AMP
- FEV1:
-
forced expiratory volume
- FVC:
-
forced vital capacity
- SPQ:
-
6-methoxy-N-3′-sulfopropylquinolinium
References
Kerem BS, Rommens JM, Buchanan JA, Markiewicz D, Cox RTK, Chakravarti A, Buchwald M, Tsui LC 1989 Identification of the cystic fibrosis gene: genetic analysis. Science 245: 1073–1080
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL 1989 Identification of the cystic fibrosis gene: cloning characterization of complementary DNA. Science 245: 1066–1073
Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE 1990 Defective intracellular transport processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63: 827–834
Stern RC 1997 The diagnosis of cystic fibrosis. N Engl J Med 336: 487–491
Kerem E, Corey M, Kerem BS, Rommens J, Markiewicz D, Levison H, Tsui LC, Durie P 1990 The relationship between genotype phenotype in cystic fibrosis -analysis of the most common mutation (ΔF508). N Engl J Med 323: 1517–1522
Shwachman H, Kulczycki LL 1958 Long term study of one hundred five patients with cystic fibrosis with cystic fibrosis. Am J Dis Child 96: 6–15
Verkman AS 1990 Development biological applications of chloride-sensitive fluorescent indicators. Am J Physiol 259: C375–C388
Stern M, Munkonge FM, Caplen NJ, Sorgi F, Huang L, Geddes DM, Alton EWFW 1995 Quantitative fluorescence measurements of chloride secretion in native airway epithelium from CF non-CF subjects. Gene Ther 2: 766–774
Tondelier D, Brouillard F, Lipecka J, Labarthe R, Bali M, Coste de Beauregard MA, Torossi T, Cougnon M, Edelman A, Baudouin-Legros MY 1999 Aspirin some nonsteroidal antiinflammatory drugs inhibit cystic fibrosis transmembrane conductance regulator protein gene expression in T-84 cells. Mediators Inflamm 8: 219–227
Dixon WJ 1990 BMDP Statistical Software manual. University Press of California, Berkeley
McLachlan G, Ho L-P, Davidson-Smith H, Samways J, Davidson H, Stevenson BJ, Carothers AD, Alton EWFW, Middleton PG, Smith SN, Kallmeyer G, Michaelis U, Seeber S, Naujoks K, Greening AP, Innes JA, Dorin JR, Porteous DJ 1996 Laboratory clinical studies in support of cystic fibrosis gene therapy using pCMV-CFTR-DOTAP. Gene Ther 3: 1113–1123
Trapnell BC, Chu C-S, Paakko PK, Banks TC, Yoshimura K, Ferrans VJ, Chernick MS, Crystal RG 1991 Expression of the cystic fibrosis transmembrane conductance regulator gene in the respiratory tract of normal individuals individuals with cystic fibrosis. Proc Natl Acad Sci USA 88: 6565–6569
Dorin JR, Farley R, Webb S, Smith SN, Farini E, Delaney SJ, Wainwright BJ, Alton EW, Porteous DJ 1996 A demonstration using mouse models that successful gene therapy for cystic fibrosis requires only partial gene correction. Gene Ther 3: 797–801
Zhang Y, Jiang Q, Dudus L, Yankaskas JR, Engelhardt JF 1998 Vector-specific complementation profiles of two independent primary defects in cystic fibrosis airways. Hum Gene Ther 9: 635–648
Canhui L, Ramjeesingh M, Reyes E, Jensen E, Jensen T, Chang X, Rommens JM, Bear CE 1993 The cystic fibrosis mutation (delta F508) does not influence the chloride channel activity of CFTR. Nat Genet 3: 311–316
Dray-Charier N, Paul A, Scoazec JY, Veissière D, Mergey M, Capeau J, Soubrane O, Housset C 1999 Expression of Delta F508 Cystic Fibrosis Transmembrane Conductance Regulator Protein related chloride transport properties in the gallbladder epithelium from cystic fibrosis patients. Hepatology 29: 1624–1634
Kälin N, Claaß A, Sommer M, Puchelle E, Tümmler B 1999 ΔF508 CFTR expression in tissues from patients with cystic fibrosis. J Clin Invest 103( 10): 1379–1386
Penque D, Mendes F, Beck S, Farinha C, Pacheco P, Nogueira P, Lavinha J, Malho R, Amaral MD 2000 Cystic Fibrosis ΔF508 patients have apically localized CFTR in a reduced number of airway cells. Lab Invest 80: 857–868
Kelley TJ, Thomas K, Milgram LJH, Drumm ML 1997 In vivo activation of the cystic fibrosis transmembrane conductance regulator mutant ΔF508 in murine nasal epithelium. Proc Natl Acad Sci USA 94: 2604–2608
Ho LP, Samways JM, Porteous DJ, Dorin JR, Carothers A, Greening AP, Innes JA 1997 Correlation between nasal potential difference measurements, genotype clinical condition in patients with cystic fibrosis. Eur Respir J 10: 2016–2022
Thomas SR, Jaffe A, Geddes DM, Hodson ME, Alton EWFW 1999 Pulmonary disease severity in men with ΔF508 cystic fibrosis residual chloride secretion. Lancet 353: 984–985
Bronsveld I, Mekus F, Bijman J, Ballmann M, De Jonge HR, Laabs U, Halley DJJ, Ellemunter H, Mastella G, Thomas S, Veeze HJ, Tummler B 2001 Chloride conductance genetic background modulate the cystic fibrosis phenotype of ΔF508 homozygous twins siblings. J Clin Invest 108: 1705–1715
Bronsveld I, Mekus F, Bijman J, Ballmann, Greipel J, Hundrieser J, Halley DJJ, Laabs U, Busche R, De Jonge HR, Tümmler B, Veeze HJ, and the European CF twins and siblings study consortium 2000 Residual chloride secretion in intestinal tissue of ΔF508 homozygous twins siblings with cystic fibrosis. Gastroenterology 119: 32–40
Kartner N, Augustinas O, Jensen TJ, Naismith AL, Riordan JR 1992 Mislocalization of deltaF508 CFTR in cystic fibrosis sweat gland. Nat Genet 1: 321–327
Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, Boucher RC, Cohn JA, Wilson JM 1992 Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat Genet 2: 240–248
Denning GM, Ostedgaard LS, Welsh MJ 1992 Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J Cell Biol 118: 551–559
Wagner JA, Cozens AL, Schulman H, Gruenert DC, Stryer L, Gardner P 1991 Activation of chloride channels in normal cystic fibrosis airway epithelial cells by multifunctional calcium/calmodulin-dependent protein kinase. Nature 349: 793–796
Clarke LL, Grubb BR, Yankaskas JR, Cotton CU, McKenzie A, Boucher RC 1994 Relationship of a non-cystic fibrosis transmembrane conductance regulator-mediated chloride conductance to organ-level disease in Cftr(−/−) mice. Proc Natl Acad Sci USA 91: 479–483
Schwiebert EM, Cid-Soto LP, Stafford D, Carter M, Blaisdell CJ, Zeitlin PL, Guggino WB, Cutting GR 1998 Analysis of ClC-2 channels as an alternative pathway for chloride conduction in cystic fibrosis airway cells. Proc Natl Acad Aci USA 95: 3879–3884
Drumm ML 2001 Modifier genes variation in cystic fibrosis. Respir Res 2: 125–128
Watson PA 1991 Function follows form: generation of intracellular signals by cell deformation. FASEB J 5: 2013–2019
Tarran R, Gray MA, Evans MJ, Colledge WH, Ratcliff R, Argent BE 1998 Basal chloride currents in murine airway epithelial cells: modulation by CFTR. Am J Physiol 274: C904–C913
Dupuit F, Kälin N, Brezillon S, Hinnrasky J, Tümmler B, Puchelle E 1995 CFTR differentiation markers expression in non-CF delta F-508 homozygous CF nasal epithelium. J Clin Invest 96: 1601–1611
Beck S, Kühr J, Schütz VV, Seydewitz HH, Brandis M, Greger R, Kunzelmann K 1999 Lack of correlation between CFTR expression, CFTR Cl−currents, Amiloride-sensitive Na+ conductance, cystic fibrosis phenotype. Pediatr Pulmonol 27: 251–259
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by INSERM and grants from ABCF Proteines Association and Assistance Publique des Hôpitaux de Paris.
Rights and permissions
About this article
Cite this article
Sermet-Gaudelus, I., Vallée, B., Urbin, I. et al. Normal Function of the Cystic Fibrosis Conductance Regulator Protein Can Be Associated with Homozygous ΔF508 Mutation. Pediatr Res 52, 628–635 (2002). https://doi.org/10.1203/00006450-200211000-00005
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200211000-00005
