
Abstract
Polymicrogyria, a cortical abnormality usually classified among neuron migration disorders, is characterized by different etiologies and pathogeneses. Recently, it has been proposed that polymicrogyria could be acquired as a consequence of a lasting damage to the developing brain. In this study, we test the hypothesis that an infection in the fetal adnexa may give rise to distant brain defects and eventually polymicrogyria. Thirty-two fetuses spontaneously aborted for extensive ascending chorioamnionitis at 15-26 wk of gestation were evaluated. Control subjects were represented by 8 fetuses aborted at 15-24 wk of gestation. A complete autopsy was carried out between 4 and 12 h after fetal expulsion. We found different histologic alterations in the primitive cortical architecture, both isolated and combined (undulation of the cortical ribbon, untimely cortical folding/molecular layer fusion, and neuronal loss). A total of 25 cases presented one or more of the above-described morphologic alterations in the brain (78%). On the contrary, similar alterations were never observed in any of the control brains (p = 0.019). Our findings indicate that chorioamnionitis significantly impairs brain cortex morphogenesis. Such neuron damage may be caused by an unspecific, indirect mechanism of injury to the developing cortex involving hypoxia and free radical generation. The reported brain abnormalities may even evolve into polymicrogyria in surviving fetuses.
Similar content being viewed by others
Main
The question of whether polymicrogyria represents a primary malformation or rather a destructive lesion with secondary malformation continues to be debated. This abnormality is usually classified among neuronal migration disorders, as Bielschowsky believed that the architectonic features of polymicrogyria indicate an arrest of neuronal migration(1). An opposing hypothesis is that polymicrogyria is acquired by intrauterine laminar destruction(2–4). Identification of the precise pathogenesis of this condition is complicated by the absence of tissue reactions in the brain until the second half of pregnancy(5). Moreover, polymicrogyria may be found in a number of congenital autosomal recessive syndromes associated with multiorgan abnormalities (i.e. Zellweger syndrome, congenital muscular dystrophies)(6), yet many cases appear to be sporadic(7).
Polymicrogyria features different microscopical aspects because it may vary from 1 to 4 layers, and often contains an acellular layer. The patterns vary from case to case, as well as within a single case(5). The four cortical layers classically described include 1) molecular layer, 2) outer cellular layer,3) cell-sparse layer, and 4) inner cellular layer(1). However some cases apparently contain neurons from all cortical laminae, although in a disorganized pattern(8). An unlayered form also exists, and is thought to result from an insult occurring early in development(9). Cortical involvement may be focal (often localized in the Sylvian fissure region) or generalized(10). When generalized, it is often associated with other CNS abnormalities such as enlarged ventricles, neuronal heterotopia, or agenesia of corpus callosum. The partial cortical involvement may be bilateral and symmetrical(7). Brain damage resulting in polymicrogyria seems to occur late in gestation, during the second half of the mid-trimester, after the acquisition of aerobic metabolism by the postmigratory neurons of infragranular layers(3). A cortical insult with nearly complete neuron migration (20-24 wk of gestation) will therefore result in a four-layer polymicrogyria, whereas a cortical insult occuring earlier will result in the unlayered form(11). In the present study, we test the hypothesis that certain cases of sporadic polymicrogyria could be secondary to severe inflammation of the fetal adnexa.
METHODS
The subjects studied included 32 fetuses spontaneously aborted at 15-26 wk of gestation, calculated on the basis of the mothers' last menses. The cause of fetal death and abortion was extensive ascending chorioamnionitis. Eight of the subjects were represented by four pairs of dichorionic diamniotic twins. Thus, the twin in the lower intrauterine position presented a more severe chorioamnionitis than the other twin. The main clinical data are described in Table 1. Controls were represented by 8 fetuses aborted at 15-24 wk of gestation, calculated according to the last menses. The reasons for which abortions were induced are listed in Table 2. These fetuses were judged as lacking any malformations involving the CNS, both before abortion and after autopsy.
A complete autopsy was carried out between 4 and 12 h after fetal expulsion. Cases with brain anomalies aside from those in the pachygyria-microgyria group, malformations in other organs, and chromosomal abnormalities were excluded. The brain was removed from the skull and fixed by immersion in 4% phosphate-buffered formaldehyde for 2-4 wk. After this period, the brain was carefully examined and extensively sampled. Histologic examination concerned all areas with grossly identifiable anomalies, the neocortex with both perisylvian areas, periventricular tissue with basal nuclei, the cerebellum, and medulla oblongata. Samples were paraffin-embedded, and dewaxed sections were stained with hematoxylin and eosin.
Analyzed samples from the fetal adnexa included at least three full-thickness placenta areas and sections of the umbilical cord at three different levels. The amniotic sac was also extensively sampled, including the break point in cases of vaginal delivery(12–15). A diagnosis of histologic chorioamnionitis was made on hematoxylin and eosin-stained sections given the presence of at least 10 polymorphonuclear leukocytes per field in 10 nonadjacent 400-power fields(12). Funisitis was defined as the presence of polymorphonuclear leukocytes within the wall of umbilical cord vessels(13). Results were compared with the two-tail Fischer's exact test.
RESULTS
Placental examination. All placentas and amniotic sacs examined fulfilled the criteria described above for histologic chorioamnionitis. Twenty-three cases (72%) also presented severe funisitis,i.e. numerous granulocytes in cord vessel walls. Results are briefly summarized in Table 1. Fetal death was judged to be caused by this severe inflammation. However, no malformation or other fetal anomaly able to justify fetal demise, nor any other placental anomaly(i.e. placenta previa, velamentous insertion of the umbilical cord, large infarcts, and so forth) was identified. Placental and amniotic sacs from controls did not show the presence of granulocyte infiltration in any of the cases.
Brain findings. The cortical surface was regularly flat, without any identifiable alteration at gross examination. On the other hand, the microscopic examination of most fetuses revealed multiple areas of cortical damage (Table 1). Such areas may present different morphologic aspects, most likely due to varying severity of the damage and the different gestational ages at which it occurred. The extent of lesioned areas may also vary. There were nonaffected areas next to affected areas which tended to be bilateral, yet not fully symmetrical. Such areas may be present in the cortex of any brain region, but most frequently in the perisylvian and frontal regions.
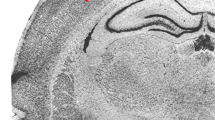
The most striking abnormality was the invasion of the molecular layer by granular layer neurons without any apparent breaking of the internal glial limiting membrane. The result was that the abnormal cortex was markedly undulating, with many small irregular gyri projecting in various planes(Figs. 1 and 2). The inner cellular layers were involved to a lesser degree. In most cases, the molecular layer was not convoluted, thus giving the brain surface a paradoxically flat appearance. In fact, in many places where the deeper layers were markedly redundant, the molecular layer was almost completely uninvolved. Such folded areas could be found in continuity with normal, unchanged cortex (Fig. 2). This alteration of normal brain morphogenesis was found in 19 out of 32 cases(59%). Few cases showed premature folding of the brain surface with molecular layer fusion and evident small vessels (8 out of 32 cases, 25%, yet 8 out of 18 before 20 wk of gestation). It is worth nothing that the folded granular layers remained unmodified and did not appear damaged in these cases(Fig. 3). Eight of our cases (25%) also showed a third type of cortical morphologic abnormality, featuring destruction of granular cells, evident as either thinning of the cortex when compared with adjacent areas or a laminar cell depletion and/or necrosis, often confined to the middle cortical layers (Fig. 4). Two cases presented cortical microhemorrhage as well (Fig. 5). The meninges we observed appeared thick and hypervascularized along the entire abnormal cortex. Hemorrhage in the periventricular area was rarely identified (7 out of 32; 22%) and none of the cases presented intraventricular hemorrhage. The results for each case are briefly summarized in Table 1.

Case 15. Abnormal, marked undulation of the granular layer with invasion of the molecular layer (magnification, 100×).

Case 19. (a) On the left, many irregular small gyri projecting in various planes. On the right, an adjacent normal area (magnification, 50×). (b) Higher magnification(100×) of the same area.

Case 5. An enfolded area with fusion of molecular layers. Small, thin-walled vessels may be identified(arrowheads) (magnification, 210×).

Case 27. Marked reduction in the number of the neuron bodies with evidence of cortical necrosis in the middle layers(magnification, 100×).

Case 18. Microhemorrhage in the middle cortical layers (arrow), in an otherwise undamaged cortex(magnification, 210×).
A comparison between the twin pairs revealed interesting results. In fact, cases 7 and 8 and 22 and 23 presented brain damage, but the first twin of each pair showed severer and larger anomalies. On the contrary, cases 24 and 25, and 30 and 31 presented comparably severe brain damage.
A total of 25 cases presented one or more of the above-described morphologic alterations in the brain (78%). On the contrary, similar alterations were never observed in any of the control brains (p = 0.019). The comparison between controls and cases showing undulation of the granular layer, which was the most frequent alteration observed, also revealed a statistical significance (p = 0.045).
DISCUSSION
In the present report, we observe the association between severe inflammatory lesions of the fetal adnexa and developmental brain damage. Such damage was usually restricted to the cortex, yet some fetuses also presented hemorrhage in the periventricular area. Brain anomalies were found in 78% of the cases, whereas they were never present in healthy controls. In all the present cases, severe chorioamnionitis was the cause of fetal death and abortion; we hypothesize that the brain lesions we describe could have eventually given rise to cortex polymicrogyria if the fetuses had survived.
The above-described cortical lesion represented by "granular invasion of the molecular layer" features striking morphologic similarities to "status verrucosus simplex," an aspect first described by Retzius in 1895(16). He proposed that status verrucosus simplex represents a transient, physiologic stage of the developing cortical cell layout. However, status verrucosus simplex does not present a constant feature, as it should if it were physiologic. If one considers the tectogenic conformity of the development of the cortical plate to a predetermined scheme of cytoarchitectonic organization, it seems difficult to regard the status verrucosus simplex as a normal or physiologic stage of cortical development(17). Other authors have considered status verrucosus simplex as an artifact linked to the practice of supporting fetal brains during formalin fixation on a bag of gauze(18), which was not performed in our study, or to the peeling of the pia arachnoid(19). These mechanisms seem extremely unlikely in our cases; we fixed the whole brains before sampling them, and the molecular layer appeared intact.
An alternative explanation of the cortical lesions originally described as status verrucosus simplex could be that even the fetuses studied by Retzius one century ago were aborted because of chorioamnionitis. In fact, histologic chorioamnonitis represents the leading cause of fetal death and abortion in the second half of pregnancy, when pathologists look for it, followed by lethal fetal malformations and maternal causes(20). Moreover, the presence of polymorphonuclear granulocyte infiltration of the fetal adnexa has been often misinterpreted(13). For instance, it was erroneously accepted that meconium is a chemioattractant for white blood cells(21), whereas such is not the case(13). In our study, we evaluated only cases with huge inflammatory placenta infiltrates and, therefore, chorioamnionitis was not questioned.
The association between chorioamnionitis and several perinatal pathologies has become much clearer in recent years, also in cases that are completely asymptomatic in the mother, but can be identified by histologic examinations of the placenta. It is now widely recognized that subclinical chorioamnionitis is an important entity that leads to premature onset of labor, prematurity, and associated complications(22). Also in the absence of demonstrable fetal colonization, histologic chorioamnionitis is associated with neonatal complications and damage(14,15) and, in mid-pregnancy, with fetal death and abortion(14,15,23). However, it is likely that chorioamnionitis caused by less virulent pathogens may be spontaneously overcome without major fetal damage, as suggested by the high incidence of first trimester vaginal bleeding in cases with histologic chorioamnionitis(24). It is also possible that antibiotic therapy administered to the mother for either unrelated or related problems (i.e. urinary tract infection) may sometimes prevent fetal death.
Reported descriptions of polymicrogyria due to well documented events during the fetal period are limited to a single or few cases, such as intrauterine cytomegalovirus infection(25) or brain perfusion failure(26). It is likely that incomplete ischemia alone may be the cause of developmental cortex damage leading to fully developed polymicrogyria(5). However, the exact timing and the mechanism for the development of this malformation remain controversial in most cases. Since 1974, it has been proposed that intrauterine hypoxia, directly or mediated by perfusion failure, is the etiology of neuron laminar destruction which will give rise to polymicrogyria(27). Moreover, polymicrogyria is often found in well established vascular areas(28).
We propose that a nonlethal infection in the fetal adnexa may eventually give rise to distant brain defects. During midgestation, brain morphogenesis has not yet been completed, and primitive neurons may be damaged by pathogens and their toxins directly or indirectly, possibly by free radical generation. Activation of leukocyte burst and high generation of free radicals also takes place during severe chorioamnionitis(29). It has been recently reported that free radicals are generated in the brain during hypoxia(30,31), and therefore this mechanism is most likely responsible for the pathogenesis at hand(32–34). Placental cytokine production, particularly tumor necrosis factor, could also be involved(35). The hypothesis that unspecific cortical insults result in neuronal death after migration is complete has received experimental support. Ibotenate injection in mice, for example, causes a cortical lesion which closely mimics human four-layered polymicrogyria, via an excitotoxic mechanism(3). Infections may provoke human postmigratory polymicrogyria, producing an excess of free radicals and inducing perfusion failures that are excitotoxicity-mediated. At 20-28 wk of gestation, blood supply to the cerebral cortex in the human fetus appears somewhat critical, because it is supplied by intracortical arteries penetrating from the pia that do not have side branches. Only by 23 wk of gestation does the number of horizontal branches actually increase in the lower cortex(36).
Recently, Marìn-Padilla(37) proposed that acquired polymicrogyria may be due to periventricular hemorrhage, followed by degeneration of radial glial fibers, as studied by rapid Golgi preparation. We cannot confirm such observations, because we found a periventricular hemorrhage in only seven cases. However, we did not study the radial glial fibers and cannot exclude that the cortical disorganization we observed was linked to radial glial fiber damage, or to damage of other tension forces relevant to cerebral cortex folding(38).
In conclusion, many researchers and neuropathologists have proposed that polymicrogyria is an acquired cortical lesion due to unspecific partial necrosis, restricted to primitive postmigratory neurons. This hypothesis seems to receive particular support in sporadic, nongenetic cases of polymicrogyria, in which brain lesions are limited to a portion of the cortex without associated malformations. Here, we propose that unspecific, indirect mechanisms of injury to the developing cortex may result in neuron damage and eventually in polymicrogyria, assuming that the fetus will survive the injury. Thus, prevention of the infectious complications of pregnancy may lead to a reduction in the incidence of this not so rare brain development anomaly.
References
Bielschowsky M 1915 Über Mikrogyrie. J Psichol Neurol 22: 1–47.
Harding B, Copp AJ 1997 Malformations. In: Graham DI, Lantos PL (eds) Greenfield's Neuropathology, 6th Ed. Edward Arnold, London, 397–533.
Marret S, Mukendi R, Gadisseux JF, Gressens P, Evrard P 1995 Effect of ibotenate on brain development: an excitoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol 54: 358–370.
Volpe JJ 1995 Neurology of the Newborn, 3rd Ed. WB Saunders, Philadelphia 53–69.
Norman MG 1996 Malformations of the brain. J Neuropathol Exp Neurol 55: 133–143.
Ferrie CD, Jackson GD, Giannakodimos S, Panayiotopous CP 1995 Posterior agyriapachygyria with polymicrogyia: evidence for an inherited neuronal migration disorder. Neurology 45: 150–153.
Dobyns WB, Truwit CL 1995 Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics 26: 132–147.
deVillemeur TB, Chiron C, Robain O 1992 Unlayered polymicrogyria and agenesis of the corpus callosum: a relevant association. Acta Neuropathol 83: 265–270.
Barth PG 1987 Disorders of neuronal migration. Can J Neurol Sci 14: 1–16.
Rorke LB 1994 A perspective: the role of disordered genetic control of neurogenesis in the pathogenesis of migration disorders. J Neuropathol Exp Neurol 53: 105–117.
Mischel PS, Nguyen LP, Vinters HV 1995 Cerebral cortical dysplasia associated with pediatric epilepsy. Review of neuropathologic features and proposal for a grading system. J Neuropathol Exp Neurol 54: 137–153.
Hillier SL, Witkin SS, Krohn MA, Watts DH, Kiviat NB, Eschenbach DA 1993 The relationship of amniotic fluid cytokines and preterm delivery, amniotic fluid infection, histologic chorioamnionitis and chorioamnion infection. Obstet Gynecol 81: 941–948.
Benirschke K, Kaufmann P 1995 Infectious diseases. In: Benirschke K, Kaufmann P (eds) Pathology of the Human Placenta, 3rd Ed. Springer-Verlag, New York 537–623.
Altshuler G 1996 Role of the placenta in perinatal pathology (revisited). Pediatr Pathol Lab Med 16: 207–233.
Stallmach T, Hebisch G, Joller-Jemelka, Orban P, Schwealler J, Engelmann M 1995 Cytokine production and visualized effects in feto-maternal unit: quantitative and topographic data on cytokines during intrauterine disease. Lab Invest 73: 384–392.
Retzius G 1895 Das Menschenhirn. Stockholm, 17 [Quoted by Larroche (17)]
Larroche JC 1977 Cytoarchitectonic Abnormalities(Abnormalities of Cell Migration). In: Vinken PJ, Bruyn GW (eds) Handbook of Clinical Neurology, Congenital Malformations of the Brain and Skull, Part I. Elsevier, Amsterdam, 479–506.
Friede RL 1975 Developmental Neuropathology, 1st ed. Springer-Verlag, Wein. 307
Streeter G 1912 The cortex of the brain in the human embryo during the fourth month with special reference to the so-called papillae of Retzius. Am J Anat 7: 337–344 [Quoted by Larroche (17)]
Kalousek DK, Neave C 1992 Embryonic and fetal wastage. In: Stocker JT, Dehner LP (eds) Pediatric Pathology. JB Lippincott, Philadelphia, 15–40.
Widholm O, Meyer B, Numers CV 1963 Inflammation of the umbilical cord in cases of foetal asphyxia of unknown clinical etiology. Gynecologia 155: 385–399.
Hack M, Merkatz IR 1995 Preterm delivery and low birth weight a dire legacy [Editorial]. N Engl J Med 333: 1772–1774.
Stallmach T, Karolyi L 1994 Augmentation of fetal granulopoiesis with chorioamnionitis during the second trimester of gestation. Hum Pathol 25: 244–247.
De Felice C, Toti P, Picciolini E, Massafra C, Pecciarini L, Palmeri MLD, Bracci R 1997 High incidence of histologic chorioamnionitis in women with gestational vaginal bleeding. Acta Obstet Gynecol Scand 76: 85–86.
Marques Dias MJ, Harmant-van Rijckervorsel G, Landrieu P, Lyon G 1984 Prenatal cytomegalovirus disease and cerebral microgyria: evidence for perfusion failure, not disturbance of histogenesis, as the major cause of fetal cytomegalovirus encephalopathy. Neuropediatrics 15: 18–24.
Shafrir Y, Latimer ME, France ML 1996 Multifocal neuronal migration as a probable result of a well-documented ischemic event at 18 wk gestation. Ann Neurol (abstr) 40: 296
Richman DP, Stewart RM 1974 Cerebral microgyria in a 27-week fetus: an architectonic and topographic analysis. J Neuropathol Exp Neurol 33: 374–384.
Ferrer I, Català I 1991 Unlayered polymicrogyria: structural and developmental aspects. Anat Embryol 184: 517–528.
Buonocore G, Gioia D, De Filippo M, Picciolini E, Bracci R 1994 Superoxide anion release by polymorphonuclear leukocytes in whole blood of newborns and mothers during the peripartal period. Pediatr Res 36: 619–622.
Yang CS, Lin NN, Tsai PJ, Liu L, Kuo JS 1996 In vivo evidence of hydroxyl radical formation induced by elevation of extracellular glutamate after cerebral ischemia in the cortex of anesthetized rats. Free Radic Biol Med 20: 245–259.
Schmidt H, Grune T, Müller R, Siems WG, Wauer RR 1996 Increased levels of lipid peroxidation products malondialdehyde and 4-hydroxynonenal after perinatal hypoxia. Pediatr Res 40: 15–20.
Hoffman DJ, McGowan JE, Marro PJ, Mishra OP, Delivoria-Papadopoulos M 1994 Hypoxia-induced modification of the N-methyl-D-aspartate receptor in the brain of the newborn piglet. Neurosci Lett 167: 156–160.
Yan SF, Tritto I, Pinski D, Liao H, Huang J, Fuller G, Brett J, May L, Stern D 1995 Induction of interleukin-6 (IL-6) by hypoxia in vascular cells. Central role of the binding site for nuclear factor IL-6. J Biol Chem 270: 11463–11471.
Goel R, Mishra OP, Razdan B, Delivoria-Papadopoulos M 1993 Modification of NMDA receptor by in vitro lipid peroxidation in fetal guinea pig brain. Neurosci Lett 151: 219–223.
Dammann O, Leviton A 1997 Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr Res 42: 1–8.
Norman MG, O'Kusky JR 1986 The growth and development of microvasculature in human cerebral cortex. J Neuropathol Exp Neurol 45: 222–232.
Marin-Padilla M 1996 Developmental neuropathology and impact of perinatal brain damage. I. Hemorrhagic lesions of neocortex. J Neuropathol Exp Neurol 55: 758–773.
Van Essen DC 1997 A tension-based theory of morphogenesis and compact wiring in the central nervous system. Nature 385: 313–318.
Acknowledgements
The authors thank Marie Louise Basso for her editorial assistance in the preparation of this manuscript.
Author information
Authors and Affiliations
Additional information
Supported by a grant from the Italian Ministry of Education MURST (funding for Research of National Interest) and by a grant from the University of Siena.
1Partial data from this study were presented in abstract form at the ESPR meeting, Szeged September 1997.
Rights and permissions
About this article
Cite this article
Toti, P., De Felice, C., Palmeri, M. et al. Inflammatory Pathogenesis of Cortical Polymicrogyria: An Autopsy Study . Pediatr Res 44, 291–296 (1998). https://doi.org/10.1203/00006450-199809000-00005
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199809000-00005
