
Abstract
Substantial physiologic data point to a significant role for dilator prostanoids in the regulation of cerebral vascular tone in newborn pigs. This study begins to address the hypothesis that multiple dilator systems, including prostanoids, can provide for the initiation and maintenance of cerebral vasodilatation in response to hypotension. Piglets were anesthetized, and cranial windows were implanted. Hypotension was achieved by hemorrhage to 50% of the control arterial pressure and maintained for 10 min. Each piglet served as its own control by the induction of hypotension and return to normotension before treatment. Pial arterioles dilated (38 ± 5% increase in diameter) in response to hypotension. Pretreatment with indomethacin (5 mg/kg i.v.) at 20 min and immediately before hypotension did not block this response (28 ± 7% and 23 ± 9% increase in diameter, respectively). In contrast, when indomethacin was administered 10 min into the hypotensive period, the dilated arterioles (106 ± 6 μm) constricted to the normotensive diameter (75 ± 5 μm) and remained constricted for the duration of the observation. These findings suggest that compensatory mechanisms provide for cerebral vasodilation when prostanoid synthesis and prostacyclin receptors are blocked before hemorrhagic hypotension. However, removal of prostanoids after vasodilation is established in response to hypotension causes constriction without compensation from alternative mechanisms. Awareness of these findings may be important when considering giving indomethacin to sick preterm infants during periods of cardiac instability.
Similar content being viewed by others


Main
Hemorrhagic hypotension in newborn pigs is accompanied by cerebral vasodilatation to maintain a constant CBF. This vasodilatation is sufficient to maintain constant CBF even when the blood pressure drops to 50% of control arterial pressure. The decrease of cerebral vascular resistance in response to hypotension is accompanied by an increase in cerebral prostacyclin production(1, 2).
Indomethacin inhibits prostaglandin synthesis. It is widely used to close a hemodynamically significant patent ductus arteriosus and is reported to protect against germinal matrix and intraventricular hemorrhages in preterm neonates(3–7). The vasodilatory response of the cerebral pial arterioles to hypotension can be reversed by the administration of indomethacin (5 mg/kg i.v.) to acutely hypotensive piglets. The dose of 5 mg/kg indomethacin has been shown to inhibit cerebral prostanoid synthesis and decrease vascular prostanoids to nondetectable levels. The use of 5 mg/kg resulted in an increase in cerebral vascular resistance with a decrease in CBF and cerebral oxygen consumption that produced coma in 75% of the piglets so treated(1). Because severe hypotension is common in very premature neonates, there is concern that the use of indomethacin could cause a severe decrease in CBF without compensation from other dilatory mechanisms.
The purpose of this study was to begin to address the hypothesis that multiple dilator systems, including prostanoids, can provide for the initiation and maintenance of cerebral vasodilatation in response to hypotension.
METHODS
Newborn piglets (1-3 d old) were anesthetized initially with ketamine hydrochloride (33 mg/kg intramuscularly) and acepromazine (3.3 mg/kg intramuscularly) and maintained on α-chloralose (50 mg/kg i.v. initially, plus 5 mg/kg/h). The animals were intubated via tracheotomy and artificially ventilated with a time-cycled, pressure-limited ventilator(Bourns BP 200, Bourns Life Systems, Riverside, CA). Catheters were inserted into both femoral veins for maintenance of anesthesia, 5% dextrose and normal saline maintenance drip, and blood withdrawals. Catheters were inserted in both femoral arteries for continuous blood pressure monitoring and to draw samples for blood gas analysis. Body temperature was maintained at 37-38°C with a servo-controlled heating pad.
Each piglet was equipped with a stainless steel and glass cranial window using a technique for newborn pigs that has been described in detail in previous studies(8, 9). The space under the window(total volume, 500 μL) was filled with artificial CSF through ports incorporated into the sides of the frame. The artificial CSF composition was(in mM) 3.0 KCl, 1.5 MgCl2, 1.5 CaCl2, 132 NaCl, 6.6 urea, 3.7 dextrose, and 24.6 NaHCO3. The artificial CSF was warmed to 37°C and bubbled with 6% CO2 and 6% O2 in N2 and typically showed a pH in the range of 7.33-7.44, and partial pressure of arterial CO2 and O2 in the range of 5.4-6.0 kPa (41-46 mm Hg) and 5.7-6.6 kPa (43-50 mm Hg), respectively. Pial arteriolar diameter was measured with a video micrometer coupled to a television camera mounted on the microscope and a video monitor.
Experimental design. Before each experiment, the space under the cranial window was flushed several times with artificial CSF. In each piglet, two arterioles differing in size were selected for observation. To determine control diameter values, arterioles were measured simultaneously(within 1 min of each other) over a 10-min period during basal normotensive conditions.
The role of prostanoids in determining cerebral vascular tone during hypotension in newborn piglets was tested in four separate groups. In the first group of piglets (n = 7), indomethacin (5 mg/kg i.v.) was given 20 min before hypotension. Hypotension was produced by withdrawing sufficient blood (anticoagulated with 3.8% citric acid) from each piglet to decrease arterial pressure to 50% of the control value. Hypotension was maintained in this group for a 10-min period, and pial arteriolar diameters were measured every 2 min. The blood was returned to the piglets via the femoral vein, and the blood vessels and blood pressure were allowed to return to control conditions. A repeat dose of indomethacin was given to the piglet, and repeat control and hypotension measurements were performed 20 min after the second dose of indomethacin. Blood gas analysis was obtained before each control and after each hypotensive episode.
A second group of piglets was studied to serve as a control for the third experiment. In this group, after a control measurement, piglets were made hypotensive for a 20-min period, and pial arteriolar measurements were obtained every 3 min. The blood was returned to the animal via the femoral vein, and blood pressure and vessels were given time to recover to control values. A repeat control and hypotensive episode was performed with blood gas analysis before and after each hypotensive period. The only difference between the second group of piglets and the third group was that indomethacin was given at 10 min into the first hypotensive period in the third group.
The fourth group of piglets received indomethacin immediately before the initiation of hypotension. Each piglet served as its own control by the induction of hypotension and the return to normotension before treatment with indomethacin.
Statistical analysis. Values are presented as means ± SE of absolute values. Analyses included analysis of variance for repeated measurements followed by Fischer protected least significant differences to isolate differences between groups. A level of p < 0.05 was considered significant in all statistical tests.
RESULTS
Table 1 shows the effect of hemorrhage before and after indomethacin on the arterial pressure, blood gases, and pH in the four experimental groups. Hemorrhage decreased arterial pressure in all four groups before and after treatment with indomethacin. Neither hemorrhage nor treatment with indomethacin affected arterial blood gases or pH.
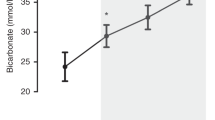
Hemorrhagic hypotension causes an increase in pial arteriolar diameter. In experiment 1, indomethacin, 5 mg/kg, was given 20 min before hemorrhagic hypotension (n = 7). Pretreatment with indomethacin did not block vasodilation of the pial arterioles during hemorrhage (Fig. 1). Furthermore, another subsequent dose of indomethacin during normotension did not block vasodilation to hemorrhagic hypotension. In experiment 2 (n = 5), hemorrhagic hypotension was maintained for a 20-min period, and the vessels remained dilated throughout the hypotensive period. Upon reinfusion of stored blood, pial arteriolar diameters returned to baseline levels. The vessels were able to dilate to a repeat episode of hemorrhagic hypotension at the end of the experiment (Fig. 2). This experiment was done to serve as a control for experiment 3.

Effects of hemorrhage on pial arteriolar diameters when indomethacin is given 20 min before induction of hypotension. *p< 0.05 compared with control; n = 7.

Effects of hemorrhage on pial arteriolar diameters and mean arterial pressure. *p < 0.05 compared with normotension;n = 5.
In experiment 3 (n = 13), indomethacin was given at 10 min into a 20-min period of hemorrhagic hypotension. The pial arterioles, which were previously dilated in response to the hemorrhagic hypotension, constricted when indomethacin was administered and remained constricted throughout the remaining 10 min of hemorrhagic hypotension. During this time period after administration of the indomethacin, the blood flow under the cranial window was obviously decreased, and the area under the window appeared ischemic. After the 20-min hypotension, blood was returned to the piglet, and time was given to allow the vessels to return to previous control values. Once this was achieved a repeat episode of hemorrhagic hypotension was performed, which resulted in dilation of the pial arterioles (Fig. 3).

Effects of hemorrhage on pial arteriolar diameters and mean arterial pressure. Indomethacin was given were indicated. *p< 0.05 compared with normotension; n = 13.
In experiment 4 (n = 5), pial arterioles dilated in response to hypotension, stored blood was reinfused, and vessels were allowed to return to steady state control conditions. Then indomethacin was given immediately before the initiation of a second hypotension, and vasodilation proceeded normally (Fig. 4).

Effects of hemorrhage on pial arteriolar diameters when indomethacin is given immediately before induction of hypotension.*p < 0.05 compared with control; n = 5.
DISCUSSION
The present study provides evidence that prostanoids are important in the maintenance of cerebral vasodilation during hypotension but are not essential for the initiation and maintenance of that vasodilation when prostanoid synthesis is blocked before hemorrhagic hypotension. The major novel finding produced by the present study is that indomethacin given 20 min before and immediately coincident with the onset of hypotension does not block pial arterial vasodilation to hemorrhagic hypotension. However, when indomethacin is given 10 min into a hypotensive episode, pial arterioles constricted to control values and remained constricted for the remaining 10 min of hypotension without compensation from alternative dilators. Because of these observations, there is reason for concern about the safety of the routine use of indomethacin in the hypotensive critically ill preterm infant.
Prostanoids play an important role in the regulation of cerebral hemodynamics in the neonatal animal. Previous studies have shown that, when 5 mg/kg indomethacin is given i.v., it crosses the blood-brain barrier of newborn pigs in sufficient quantity to prevent detectable prostanoid synthesis, resulting in a substantial decrease in CBF when given in the steady state(10). In addition, Parfenova et al.(11) demonstrated that indomethacin directly inhibits prostacyclin receptor-mediated cerebral vascular responses. The dose of indomethacin in this series of experiments was chosen to cause the inhibition of cerebral prostacyclin production and block prostacylin receptors.
In this series of experiments, indomethacin given 20 min before hypotension did not significantly alter pial arteriolar diameters in the normotensive controls. Although indomethacin given at this dose is known to decrease CBF 18-28% in newborn pigs(10), this decrease in flow may not necessarily be reflected by a detectable decrease in measured diameter, because resistance is inversely proportional to a power function of the radius. Therefore, the decrease in diameter may be too modest to be detected by looking at pial arteriolar diameters under the cranial window. Thus, the present study does not confirm or reject the numerous clinical and animal studies that show, in the steady state, various doses of indomethacin cause an immediate, and relatively sustained, reduction in baseline CBF(10, 12–16).
In the present study indomethacin given 20 min before hypotension did not block pial arteriolar vasodilation. It seemed possible that pial arterioles dilated 20 min after indomethacin because indomethacin was no longer present at sufficient concentration to block the prostacyclin receptors. Therefore, to ensure a sufficient indomethacin level at induction of hypotension for inhibition of the prostacyclin receptors, indomethacin was given immediately before hypotension was induced. However, even when indomethacin was given immediately before hypotension, pial arterioles dilated in response to hemorrhagic hypotension.
A study done in newborn piglets by Louis et al.(13) showed that, when indomethacin (dose of 0.2 mg/kg) was given 1 h before induction of hemorrhagic hypotension, there was an initial decrease in CBF 10 min after the administration of indomethacin that remained throughout the experiment. However, once the reduction in blood flow occurred, acute hemorrhagic hypotension did not further compromise CBF. In our study, we used a much higher dose of indomethacin, 5 mg/kg, to ensure inhibition of cerebral prostanoid production, and we measured pial arteriolar diameter instead of measuring CBF with microspheres. However, our results are similar in that indomethacin did not block vasodilation to hemorrhagic hypotension when given 20 min or immediately before hypotension. It appears that dilators that are not prostanoid-dependent initiated and maintained cerebral vasodilation.
This study does not address what the alternative dilators may be. Eidson et al.(9) showed that hypotensive pial arteriolar vasodilation could be blocked by light dye injury to the microvascular endothelium and proposed that hypotension-induced cerebral vasodilation is endothelial-dependent. Whether the stimulus for vasodilation is nitric oxide, oxygen radicals, or some other endothelial-derived vasoactive factor remains unknown. However, it is apparent that this mechanism is complex and probably involves a combination of factors signaling to the adjacent smooth muscle to produce vasodilation.
Previous studies done in our laboratory show that giving indomethacin to an acutely hypotensive piglets causes constriction of pial arteriolar vessels without compensation from other dilators(2). This is consistent with the findings of our present study. When indomethacin was given to the acutely hypotensive piglet, the vessels constricted and the area of the brain viewed under the cranial window appeared ischemic with an obvious decrease in blood flow to the area being studied. Due to the appearance of the brain it could be inferred that these findings are consistent with a previous study by Leffler et al.(1). In that study, giving indomethacin to unanesthetized piglets produced a 40% decrease in CBF associated with a significant decrease in CMRO2 and an increase in cerebral vascular resistance, inducing coma in six of eight animals treated.
This finding is extremely important, because there was no alternative dilator that compensated for the constriction. Constriction was maintained throughout the remaining 10 min of the experiment during hypotension. This was not a transient effect secondary to an increase in blood pressure with the administration of indomethacin; blood pressure was kept constant by continuous withdrawal of blood via the femoral vein to keep blood pressure at 50% of the mean arterial pressure control values. Whether this finding is secondary to prostaglandin cyclooxygenase inhibition, prostacyclin receptor antagonism, or both, or some other unknown property of indomethacin cannot be stated with certainty. Chemtob et al.(17) have suggested that there may be mechanisms other than prostanoid inhibition that are responsible for some of the actions of indomethacin. Among the actions of indomethacin that have been reported, usually at higher concentrations, are inhibition of cAMP phosphodiesterase(18), cAMP-dependent protein kinases(19), and Ca2+ transport(20). None of these purported actions would appear to explain the effects of indomethacin on the cerebral circulation. If such mechanisms exist they do not inhibit vasodilation when indomethacin is given at 20 min and immediately before hypotension induction. Inhibition was achieved only when given during hypotension.
CONCLUSIONS
These findings suggest that compensatory mechanisms provide for cerebral vasodilation when prostanoid synthesis and prostacyclin receptors are blocked before hemorrhagic hypotension. However, removal of prostanoids after vasodilation is established in response to hypotension causes constriction without compensation from alternative mechanisms. Conclusively, awareness of these findings may be important when considering giving indomethacin to sick preterm infants during periods of cardiac instability.
Abbreviations
- CSF:
-
cerebrospinal fluid
- CBF:
-
cerebral blood flow
References
Leffler CW, Busija DW, Beasley DG, Fletcher AM 1986 Maintenance of cerebral circulation during hemorrhagic hypotension in newborn pigs: role of prostanoids. Circ Res 59: 562–567.
Leffler CW, Busija DW 1987 Prostanoids and pial arteriolar diameter in hypotensive newborn pigs. Am J Physiol 252:H687–H691.
Heymann MA, Rudolph AM, Silverman NH 1976 Closure of the ductus arteriosus in premature infants by inhibition of prostaglandin synthesis. N Engl J Med 295: 530–535.
Ment LR, Duncan CC, Ehrenkranz RA, Kleinman CS, Taylor KJW, Scott DT, Gettner P, Sherwonit E, Williams J 1988 Randomized low-dose indomethacin for prevention of intraventricular hemorrhage in very low birth weight infants. J Pediatr 107: 937–943.
Bandstra ES, Montalvo BM, Goldberg RN, Pacheco I, Ferrer P, Flynn J, Gregarios JB, Bancalari E 1988 Prophylactic indomethacin for prevention of intra-ventricular hemorrhage in premature infants. Pediatrics 82: 533–542.
Bada HS, Green RS, Pourcyrous M, Leffler CW, Korones SB, Magill HL, Arheart K, Fitch CW, Anderson GD, Somes G, Tullis K, Campbell J 1989 Indomethacin reduced the risks of severe intraventricular hemorrhage. J Pediatr 115: 631–637.
Ment LR, Oh W, Ehrenkranz RA, Philip AGS, Vohr B, Allen W, Duncan CC, Scott DT, Taylor JW, Katz KH, Schneider K, Makuch RW 1994 Low-dose indomethacin and prevention of intraventricular hemorrhage: a multicenter randomized trial. Pediatrics 9: 543–549.
Leffler CW, Mirro R, Shibata M, Parfenova H, Armstead WM, Zuckerman S 1993 Effects of indomethacin on cerebral vasodilator responses to arachidonic acid and hypercapnia in newborn pigs. Pediatr Res 33: 609–614.
Eidson TH, Edrington JL, Albuquerque ML, Zuckerman SL, Leffler CW 1995 Light/dye microvascular injury eliminates pial arteriolar dilation in hypotensive piglets. Pediatr Res 37: 10–14.
Leffler CW, Busija DW, Fletcher AM, Beasley DG, Hessler JR, Green RS 1985 Effects of indomethacin upon cerebral hemodynamics of newborn pigs. Pediatr Res 19: 1160–1164.
Parfenova H, Zuckerman S, Leffler CW 1995 Inhibitory effect of indomethacin on prostacyclin receptor-mediated cerebral vascular responses. Am J Physiol 268:H1884–H1890.
Leffler CW, Busija DW, Beasley DG 1987 Effects of therapeutic dose of indomethacin on the cerebral circulation of newborn pigs. Pediatr Res 21: 188–192.
Louis PT, Yamashita Y, Del Toro J, Michael LH, Contant CF, Goddard-Finegold J 1994 Brain blood flow responses to indomethacin during hemorrhagic hypotension in newborn piglets. Biol Neonate 66: 359–366.
Pryds O, Greisen G, Johansen KH 1988 Indomethacin and cerebral blood flow in preterm infants treated for patent ductus arteriosus. Eur J Pediatr 147: 315–316.
Edwards AD, Wyatt JS, Richardson C, Potter A, Cope M, Delpy PT, Reynolds EO 1990 Effects of indomethacin on cerebral hemodynamics in very premature infants. Lancet 335: 1491–1495.
Benders MJNL, Dorrepaal CA, van de Bor M, van Bel F 1995 Acute effects of indomethacin on cerebral hemodynamics and oxygenation. Biol Neonate 68: 91–99.
Chemtob S, Beharry K, Barna T, Varma DR, Aranda JV 1991 Differences in the effects in the newborn piglet of various nonsteroidal antiinflammatory drugs on cerebral blood flow but not on cerebrovascular prostaglandins. Pediatr Res 30: 106–111.
Stefanovich V 1974 Inhibition of 3′,5′-cyclic AMP phosphodiesterase with antiinflammatory agents. Res Commun Chem Pathol Pharmacol 7: 573–581.
Kantor HS, Hampton M 1978 Indomethacin in submicromolar concentrations inhibits cyclic AMP-dependent protein kinase. Nature 276: 841–842.
Burch RM, Wise WC, Halushka PV 1983 Prostaglandin-independent inhibition of calcium transport by nonsteroidal antiinflammatory drugs: differential effects of carboxylic acids and piroxicam. J Pharmacol Exp Ther 227: 84–91.
Acknowledgements
The authors thank Mildred Jackson, Alex Fedinec, and Stanley Lopez for their technical assistance; Danny Morse and Laura Malinick for the illustrations; and Maria Swayze for preparing the final manuscript.
Author information
Authors and Affiliations
Additional information
Supported by grants from the National Institutes of Health.
Rights and permissions
About this article
Cite this article
Hayden, J., Leffler, C. The Effects of Treatment with Indomethacin on the Cerebral Vasculature of Newborn Piglets before and during Hemorrhagic Hypotension. Pediatr Res 41, 78–82 (1997). https://doi.org/10.1203/00006450-199701000-00012
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199701000-00012
