
Abstract
Studies in humans and animals indicate that peripheral insulin sensitivity is decreased after puberty. Although glucose, after its uptake and phosphorylation, will be diverted to either the glycolytic or glycogen synthesis pathway, these pathways have not been characterized after the transition to puberty. Thus, we examined the changes in the pathways of glucose utilization in conscious (n = 22) prepuberty (81 ± 3 g), and postpuberty (258 ± 9 g) Sprague-Dawley rats. Insulin stimulated(by insulin clamp 18 mU/kg/min) glucose uptake [rate of glucose disappearance(Rd)] was decreased by ≈30% postpuberty (from 339 ± 22 to 239 ± 28 μmol/kg/min; p < 0.001). Although glycolysis (estimated by the rate of conversion of [3H]glucose to3 H2O) decreased by ≈15% (p < 0.05), glycogen synthesis decreased by ≈40% (from 200 ± 17 prepuberty to 122± 22 μmol/kg/min postpuberty; p < 0.001), and accounted for ≈80% of the decrease in Rd postpuberty. Decrease in the capacity to store glycogen in response to insulin was also confirmed by≈40% decrease in both glycogen levels, and in 3H accumulation into glycogen (from 3H-glucose) at the end of the clamp study. This occurred in the absence of any changes in either the Km or the Vmax of glycogen synthase nor in the activity of glycogen phosphorylase. We conclude that the postpubertal decrease in insulin responsiveness is characterized by decreased ability to store muscle glycogen. We propose that high capacity for muscle glycogen synthesis may be required to sustain the increased metabolic requirements during peripubertal growth.
Similar content being viewed by others
Main
Human puberty is normally associated with a decline in tissue sensitivity to insulin(1). Recent studies in pubertal children using the euglycemic clamp technique(2) have shown that the ability of insulin to promote glucose disposal is reduced compared with that in healthy preadolescents(1–3). Despite the decline in peripheral insulin sensitivity, normal glucose homeostasis is maintained by compensatory increases in basal and in glucose-stimulated insulin secretion(4–6). This insulin resistance has been demonstrated in peripheral tissue but not in the liver during puberty(3). Interestingly, insulin resistance in adolescents going through puberty is more severe than that of young adults(7). However, young adults compared with prepubertal children still have decreased insulin sensitivity. Although increased GH may be responsible for the insulin resistance observed during puberty, the exact mechanisms for postpubertal decrease in peripheral insulin action is unclear.
The effective evaluation of glucose metabolism includes the combined assessment of in vivo glucose fluxes and in vitro tissue enzyme activities to correlate between these physiologic and biochemical alterations in insulin action during puberty. Human studies in adults have used tracer infusion in combination with the insulin clamp techniques to study factors influencing insulin-mediated glucose metabolism. Indirect calorimetry was used in combination with insulin clamps to establish the relative roles of oxidative and nonoxidative glucose disposal of metabolic fuels(8–11). However, this technique lacks normative data for children, and an affect of the technique itself on resting metabolic rates cannot be ruled out. Furthermore, muscle biopsies have not been obtained from healthy children pre- and postpuberty for the assessment of the glycogen synthetic pathway. We studied an established homogeneous rodent model(12) to evaluate the changes in glucose metabolism after puberty. Indeed, development and aging in rodents has been associated with an increase in plasma glucose and insulin levels, comparable to that seen in humans(13, 14). We carefully selected rats which were pre- and postpuberty, to avoid studying rats going through puberty and its associated hormonal changes and their effects. Most specifically, we studied the changes in muscle intracellular glucose pathways during insulin-mediated glucose metabolism in pre- and postpuberty rats. Our data support the notion that the capacity to store muscle glycogen is impaired postpuberty.
METHODS
Animals. Two age groups were studied: one group of 3-wk-old prepuberty (n = 10) and 8-wk-old postpuberty (n = 12) male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA). Postweaning(≈2 d), prepubertal animals were identified by their age (<3 wk), body weights (<100 g), and lack of testicular descent(12). Rats were housed in individual cages and subject to a standard light (0600 to 1800 h)-dark (1800 to 0600 h) cycle. Three to five days before the in vivo study, rats were anesthetized with an intraperitoneal injection of pentobarbital (50 mg/kg of body weight), and indwelling catheters were inserted in the right internal jugular vein and in the left carotid artery. The venous catheter was extended to the level of the right atrium, and the arterial catheter was advanced to the level of the aortic arch(15–18). Rats were studied when their body weight was within 5% of their preoperative weight.
Body composition. Lean body mass and fat mass were calculated from the whole body volume of distribution of water, estimated by tritiated water bolus injection in each experimental rat(18). The distribution space of water was obtained by dividing the total radioactivity injected by the steady state specific activity of plasma water, which was assumed to be 93% of the total plasma volume, and from the whole body water distribution space divided by 0.73. Because carcass protein was not changed in pre- and postpubertal Sprague-Dawley rats, we assumed a similar distribution volume of water in all rats. Briefly, 20 μCi of 3H2O (Dupont NEN, Boston, MA) were injected intraarterially on the morning of the study. In our experience, steady state for 3H2O-specific activity in rats is generally achieved within 30-45 min, and so five samples were collected between 1 and 3 h after injection. Epididymal fat pads were dissected and weighed after the completion of the study, as an additional index of adiposity.
Hyperinsulinemic euglycemic clamp. To study insulin responsiveness (glucose uptake at maximally stimulated insulin levels), rats from each group were studied by using the euglycemic clamp technique. Studies were performed in awake, unstressed, chronically catheterized rats(16–18). Briefly, all rats received a primed-continuous (15-40-μCi bolus, 0.4 μCi/min) infusion of HPLC purified [3H-3]glucose (Dupon NEN) throughout the study. After 120-min measurements of basal glucose turnover, primed continuous infusion of insulin(18 mU/kg·min) was administered, and a variable infusion of a 25% glucose solution was started and periodically adjusted, to clamp the plasma glucose concentration at the basal level (120 mg/dL) for the additional 120 min of the clamp.
Plasma samples for determination of [3H]glucose-specific activity were obtained at 0, 60, 80 90, and 100 min, and every 5 min through the last 20 min of the insulin infusions. Plasma samples for determination of plasma insulin, lactate, and FFA concentrations were obtained at time 0 (before insulin infusion) and at 60 and 120 min of the study. The total volume of blood withdrawn was ≈2.0 mL/study; to prevent volume depletion and anemia, a solution (1:1 vol/vol) of ≈2.0 mL of fresh blood (obtained by heart puncture from a littermate of the test animal) and heparinized saline (10 U/mL) was infused. At the end of the insulin infusion, rats were anesthetized(pentobarbital 60 mg/kg body weight, i.v.), the abdomen was quickly opened, and the rectus abdominal muscle was freeze-clamped in situ with aluminum tongs precooled in liquid nitrogen(16–18). The time from the injection of the anesthetic until freeze-clamping of the liver was less than 45 s. All tissue samples were stored at -80°C for subsequent analysis.
The study protocol was reviewed and approved by the Animal Care and Use Committee of the Albert Einstein College of Medicine.
Whole body glycolysis. The rate of glycolysis was estimated from the rate of conversion of [3H-3]glucose to 3H2O as previously described(16, 18). Briefly, plasma-tritiated water-specific activity was determined by liquid scintillation counting of the protein-free supernatant (Somogyi filtrate) before and after evaporation to dryness. Because tritium on the C-3 position of glucose is lost to water during glycolysis, it can be assumed that plasma tritium is present either in 3H water or glucose. Thus, the rate of appearance of tritiated water in the plasma, multiplied by the whole body water space, should predict the 3H2O infusion rate.
Glycogen formation in vivo. Indices for muscle glycogen synthesis were obtained by three independent methods. One was quantitated by subtracting the glycolytic rate from the Rd. It was also quantitated by measuring the incorporation of [3H]glucose counts into muscle glycogen divided by the specific activity of [3H]glucose in the plasma. In addition, rats (n = 8) from each group were studied without insulin infusion and then killed after 4 h of saline infusion. Thus, the increase in muscle glycogen after insulin infusion was determined. Muscle glycogen concentration was determined after digestion with amyloglucosidase as previously described(16–18). The intraassay and the interassay coefficients of variation were <10% (at 0.25 g/100 ml tissue weight) when a muscle homogenate was assayed as multiple aliquots. Glycogen was precipitated by washing in 10 volumes of absolute ethanol and by incubation for 1 h at -20°C. The procedure was repeated three times, and then the precipitate was collected, dried down, and dissolved in water before scintillation counting.
GS activity. Muscle GS activity was measured by a modification(16–18) of the method of Thomas et al.(19) and is based on the measurement of the incorporation of radioactivity into glycogen from UDP-[U-14C]glucose. Tissue samples (20-30 mg) were homogenized in 2.0 mL of Tris/HCl buffer, pH 7.8, containing 10 mmol/L EDTA, 5 mmol/L dithiothreitol, 50 mmol/L NaF, and 2.5 g/L rabbit liver glycogen type III. The homogenate was centrifuged at 2000× g for 15 min (at 4°C), and the supernatant was used for GS assay by measuring the incorporation of UDP-[U-14C]glucose into glycogen at 30°C. To approximate the in vivo conditions, synthase activity was measured in the presence of physiologic 0.11 mmol/L glucose 6-phosphate. Total GS D activity was measured in the presence of 7.2 mmol/L glucose 6-phosphate. For the kinetic analysis, the assay was conducted at final concentrations of 0.003, 0.017, 0.033, 0.09, 0.33, and 1.4 mM uridine diphosphoglucose data were linearized as Eadie-Hofstee plots and fit using linear regression. The Km for uridine diphosphoglucose is the reciprocal of the slope, whereas Vmax is the y-intercept divided by the slope.
GP activity. Muscle GP activity was measured as previously described(16–18). This assay is based on the measurement of the incorporation of 14C into glycogen from labeled glucose 1-phosphate. GP-α, the active phosphorylated enzyme, was assayed in the absence of AMP in the tissue homogenates (20-30 mg), which were prepared as described above. The supernatant was used for GP assay by measuring the incorporation of [14C]glucose 1-phosphate into glycogen at 30°C in a mixture containing 33 mM morpholinoethanesulfonic acid, 200 mM KF, 0.45% mercaptoethanol, 15 mM glucose 1-phosphate (50 μCi/mmol), and 3.4 mg/mL glycogen.
Analytical procedures. Plasma glucose was measured by the glucose oxidase method (Glucose Analyzer II, Beckman Instruments, Palo Alto, CA) and plasma insulin by RIA using rat and procine insulin standards. Plasma[3H]glucose radioactivity was measured in duplicate on the supernatants of Ba(OH)2 and ZnSO4 precipitates of plasma samples after evaporation to dryness to eliminate tritiated water. Muscle glycogen was determined as previously described(16–18). Plasma nonesterified fatty acid concentrations were determined by an enzymatic method with an automated kit according to the manufacturer's specifications(Waco Pure Chemical Industries, Osaka, Japan).
Calculations. Data for total body glucose uptake and HGP represent the mean values during the last 30 min. The HGP was calculated as the difference between the tracer-derived rate of appearance and the infusion rate of glucose. Whole body glycogen synthesis was estimated by subtracting whole body glycolysis from whole body glucose uptake (Rd). Rates of whole body glycolysis were estimated from the increment per unit time in tritiated water (disintegrations/min/mL) × body water mass(mL)/[3-3H]glucose specific activity (disintegrations/min/g). All values are presented as the mean + SE. Comparisons between groups were made using repeated measures analysis of variance where appropriate. Where F ratios were significant, further comparisons were made using t tests.
RESULTS
Experimental rat fat mass and biochemical characteristics at the basal and insulin-stimulated state. The prepubertal group weighed 81± 1 g and the postpubertal group weighed 258 ± 9 g (p< 0.001) at the time of the study. Fat mass determined by3 H2O accounted for 32 ± 3 g or 12 ± 1% of body weight in postpubertal rats, whereas fat mass was very small in the prepubertal rats accounting for 2 ± 5 g or 0.5 ± 2%. Epididymal fat (the largest fat pad in male rats) was 1.79 ± 0.18 g in postpubertal rats, whereas that of the prepubertal rats weighed 0.35 ± 0.07 g.
At the basal state plasma glucose levels were comparable in both groups; however, basal plasma insulin and FFA levels were ≈2-fold increased postpuberty (Table 1). Although insulin infusion rates were similar, plasma insulin levels were ≈20% higher in the postpubertal group (reflecting a decrease in the metabolic clearance rate of insulin). Plasma FFA levels were similar during the hyperinsulinemic clamps.
HGP. Basal HGP was decreased by ≈40% from 142 ± 17μmol/kg/min in prepuberty to 78 ± 6 μmol/kg/min in postpubertal rats (p < 0.001). Hyperinsulinemia suppressed HGP by 78% in the prepubertal and by 73% in postpubertal rats. However, HGP during hyperinsulinemia was still significantly increased in the prepubertal rats (31± 5 in the prepubertal and 21 ± 1 μmol/kg/min in the postpubertal rats, p < 0.001).
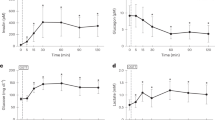
Glucose-uptake (Rd), glycolysis, and glycogen synthesis Insulin responsiveness was decreased by≈30% from 339 ± 22 μmol/kg/min in prepubertal to 239 ± 28μmol/kg/min in the postpubertal rats (p < 0.001)(Fig. 1). When Rd is expressed per lean body mass (assuming that most of glucose uptake is in muscle), it is still decreased by ≈25% after puberty (p < 0.001). Glycolysis was estimated from the rate of conversion of [3H]glucose to3 H2O, and was at similar rates of 139 ± 11 μmol/kg/min in prepubertal to 117 ± 11 μmol/kg/min in the postpubertal rats(p < 0.05). Glycogen synthesis calculated by subtracting the glycolytic rate from the Rd, was decreased by ≈40% from 200 ± 17 in prepubertal to 122 ± 22 μmol/kg/min in the postpubertal rats (p < 0.001). Thus, although the decrease in glycolysis accounted for ≈20 μmol/kg/min decrease in Rd, glycogen synthesis accounted for ≈80 μmol/kg/min in the decrease in Rd.

Glucose disposal and intracellular Pathways in prepubertal and postpubertal Sprague-Dawley rats. (A) Glucose disposal measured during maximally stimulating insulin levels. (B) Glycolysis measured by generation of 3H2O from tritiated glucose. (C) Glycogen synthesis measured by subtracting glycolytic rates from Rd. *p < 0.001 vs prepuberty; **p <0.05 vs prepuberty.
Glycogen synthesis was determined by other methods (Table 2). The increment in muscle glycogen concentrations above fasting level, was decreased by ≈45% in prepubertal rats (p < 0.005). The incorporation of [3H]glucose counts into muscle glycogen was also decreased by ≈40% in the postpubertal rats (p < 0.005).
Muscle GS and GP activities. No differences were noted in the Vmax for UDP-glucose of GS in prepubertal and postpubertal rats in the basal state (Table 3). Insulin-activated(decrease in Km for UDP-glucose) GS was similar in prepubertal and postpubertal rats. Also, the independent from of GP was similar and unchanged with insulin in prepubertal and postpubertal rats.
DISCUSSION
In the present study we used the rodent model to evaluate peripheral insulin responsiveness and the intracellular fate of phosphorylated glucose after puberty. A marked decrease in insulin mediated glucose disposal is observed after puberty is completed. Our data suggests that most (≈80%) of this evolving defect was accounted for by a decrease in the ability to store glycogen, whereas glycolysis was only mildly decreased.
Although basal glucose levels were similar, postpubertal rats were characterized by a 2-fold increase in plasma insulin and FFA levels from that of prepubertal rats. These increases were associated with development of fat mass in the postpubertal rats. Indeed, plasma insulin and FFA levels increase with age and correlate best with fat mass in both humans and rats(18, 20). Thus, although low plasma insulin levels could result in increased lipolysis and plasma FFA levels, the small amount of total fat mass results in lower plasma FFA levels in prepubertal rats. The suppression of HGP by insulin may be accounted for, to a large extent, by the lower plasma FFA levels(21, 22), suggesting a potential role for FFA in regulating also basal HGP. Thus, the higher basal HGP in prepubertal rats may reflect the low plasma insulin and the preference of carbohydrates rather than lipids as the major substrate for metabolism, and may also reflect an increase in basal metabolic rates, as was demonstrated by indirect calorimetry in children(9, 11). Additionally, with hyperinsulinemia the decrease in plasma FFA levels and the percent decrease in HGP were similar in pre- and postpubertal rats. Lack of defect in suppression of HGP was also demonstrated in peripubertal humans(4).
The hyperinsulinemic euglycemic clamp technique is considered the best for estimation of insulin action, and human studies demonstrated a decrease of glucose uptake at a variety of insulin levels before and during puberty(2, 3). However, the appropriate interpretation of the data depends on the plasma insulin levels achieved. Thus, the hyperinsulinemia achieved in this study was designed to overcome defects, which may exist in glucose transport and/or phosphorylation, and to accurately measure the rates of glycolysis and glycogen synthesis(17). Significantly less exogenous glucose was required to maintain euglycemia during the insulin clamp in the postpubertal rats as reflected by the ≈30% decrease in glucose uptake (Rd). The plasma insulin levels achieved were significantly higher in the postpubertal rats. This may be explained by a decrease in the metabolic clearance rate of insulin in postpubertal rats. As is the case in this study, we have previously shown that insulin levels greater than 2000 pmol/L exert a maximally stimulated effect on glucose uptake in conscious rats(16). If this is not the case in prepubertal rats, the high glucose uptake demonstrated in this study may even be underestimated. Interestingly, puberty appears to selectively impair glucose metabolism with relative sparing of fat and protein metabolism(7). Thus, the hyperinsulinemia seen in puberty serves a beneficial role by amplifying anabolic effects of insulin on protein metabolism when an increase in growth velocity is expected.
Several lines of evidence support decreased glycogen synthesis rather than glycolysis as accounting for most of the decrease in insulin responsiveness after puberty. First, glycogen synthesis rates, calculated from Rd glycolysis was significantly decreased at rates which accounts for ≈80% of the decrease in Rd(Fig. 1). Second, during this study [3H]glucose incorporation in muscle glycogen was decreased in postpubertal rats in a similar magnitude. A third way was to estimate insulin-induced increase in muscle glycogen, which was also decreased to a similar magnitude in postpuberty rats. Although the former methods obtained results from the same rats, the latter was obtained by subtraction from other rats treated with saline for the duration of the study. In support of our findings is the increase in the basal nonoxidative glucose disposal, which was observed in prepubertal humans by indirect calorimetry(10, 12). Although these data support the notion that decreased glycogen synthesis accounts for the development of insulin resistance in postpubertal rats, we demonstrated no changes in the kinetics of muscle GS nor in the activity of GP. This suggests that, rather than changes in enzyme activity, other regulation such as hormones or substrates may regulate the storage of glycogen in postpuberty muscle.
Puberty is hormonally characterized by a surge of a variety of hypothalamic/pituitary peptides, resulting in an increase in peripheral hormones. Many of these, such as GH and IGFs have complex effects on glucose metabolism(1). Because both acute and chronic increases in plasma GH levels may result in peripheral insulin resistance and increased HGP, this hormone is a major candidate for an explanation of our observations. Recently, administration of GH in rats for 3 d induced insulin resistance and decreased incorporation of [3H]glucose into glycogen(23). Although short-term GH administration (5 h) increased lypolysis and decreased fat mass, GH administration for 3 d increased fat mass significantly, suggesting chronic anabolic effects on fat mass(23). Because GH levels decrease significantly after puberty is completed, it may not be directly responsible for the insulin resistance observed postpuberty. Rather, GH may the mediator of increased fat mass during puberty in humans and animals(24). Once fat mass is increased, an increased plasma FFA level and hyperinsulinemia are associated with the development of insulin resistance in humans(25) and rats(18), regardless of GH levels. The impact of fat mass on fuel metabolism was demonstrated by the negative correlation between resting energy expenditure and fat mass in pre- and postpubertal humans(9, 11). Also, Arslanian and Satish(26) demonstrated negative significant correlation between fat oxidation and glucose disposal, and oxidative disposal in children before and after puberty. These studies support the role of fat mass and FFAs on the insulin resistance that develops postpuberty. Increase in plasma FFA levels was believed to compete with glucose for energy oxidation, thus impairing peripheral glucose metabolism(27). However, although glucose oxidation is decreased during FFA and insulin infusion, insulin-stimulated glucose uptake is unchanged. Only when FFA is maintained elevated for >3-6 h, a defect in glucose uptake and glycogen synthesis will develop(28). The decrease in glucose uptake and in glycogen synthesis may be explained by increased flux through the hexosamine pathway(29), which regulates the function of the insulin-dependent glucose transporter (GLUT 4). It remain to be seen if this pathway is involved in the development of defects in insulin action and storage postpuberty. However, to the extend that lipoprotein(a) is an index for increased lipid metabolism, lipoprotein(a) levels were not correlated with insulin sensitivity in different tanner stages(30). Thus, the correlation between lipids and glucose metabolism before and after puberty may be more complex.
Thus, we suggest that the transition to puberty results in an increase in fat mass, which determines through FFA, glycerol, or other bioactive peptide(such as tumor necrosis factor-α secreted by fat cells) the development of peripheral insulin resistance. This insulin resistance is characterized by marked decrease in the ability to store glycogen and a minor decrease in the glycolytic pathway. Thus, increase in muscle glycogen synthesis may be required to sustain the increased metabolic requirements during peripuberty growth.
Abbreviations
- GS:
-
glycogen synthase
- GP:
-
glycogen phosphorylase
- HPG:
-
hepatic glucose production
- Rd:
-
rate of diffusion
REFERENCES
Bloch CA, Clemons P, Sperling MA 1987 Puberty decreases insulin sensitivity. J Pediatr 110: 481–487
Amiel SA, Caprio S, Simonson DC, Lauritano AA, Tamborlane WV 1986 Impaired insulin action in puberty: contributing factor to poor glycemic control in adolescents with diabetes. N Engl J Med 315: 215–219
Amiel SA, Caprio S, Sherwin RS, Plewe G, Haymond M, Tamborlane WV 1991 Insulin resistance of puberty: a defect restricted to peripheral glucose metabolism. J Clin Endocrinol Metab 72: 277–282
Caprio S, Plewe G, Diamond MP, Simonson DC, Boulware SD, Sherwin RS, Tamborlane WV 1989 Increased insulin secretion in puberty: a compensatory response to reduction in insulin sensitivity. J Pediatr 114: 963–967
Smith CP, Archibald HR, Thomas JH, Tarn AC, Williams AJK, Gale EAM, Savage MO 1988 Basal and stimulated insulin levels rise with advancing puberty. Clin Endocrinol 28: 7–14
Rosenbloom AL, Wheeler L, Bianchi R, Chin FT, Tiwary CM, Gorgic A 1975 Age adjusted analysis of insulin responses during glucose tolerance tests in normal children and adolescents. Diabetes 24: 280–288
Caprio S, Cline G, Boulware S, Permenente C, Shulman RS, Tamborlane WV 1994 Effect of puberty and diabetes on metabolism of insulin sensitive fuels. Am J Physiol 266:E885–E891
Tappy L, Paquot N, Tounian P, Shneiter P, Jaquier E 1995 Assessment of glucose metabolism in humans with simultaneous use of indirect calorimetry and tracer techniques. Clin Physiol 15: 1–12
Goran MI, Kaskoun M, Johnson R 1994 Determinants of resting energy expenditure in young children. J Pediatr 125: 362–367
Scott CB 1993 Resting metabolic rate variability as influenced by mouth-piece and noseclip practice procedures. J Burn Care Rehabil 14: 573–577
Goran MI, Carpenter WH, Poehlman ET 1993 Total energy expenditure in 4- to 6-yr-old children. Am J Physiol 264:E706–E711
Winter JSD, Faiman C 1990 Comparative aspects of animal and human puberty. In: Grumbach MM, Sizonenko PC, Acbert ML (eds) Control of the Onset of Puberty. Williams & Wilkins, Baltimore, pp 211–230
Goodman MN, Dluz SM, McElaney MA 1983 Glucose uptake and insulin sensitivity in rat muscle: changes during 3-96 wk of age. Am J Physiol 244:E93–E100
Brancho-Romero E, Reaven GM 1977 Effect of age on plasma glucose and insulin response in rat. J Am Geriatr Soc 25: 299–302
Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA 1987 Correction of hyperglycemia with phlorozin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest 79: 1510–1515
Rossetti L, Giaccari A 1990 Relative contribution of glycogen synthesis and glycolysis to insulin-mediated glucose uptake. A dose-response euglycemic clamp study in normal and diabetic rats. J Clin Invest 85: 1785–92
Rossetti L, Hu M 1993 Skeletal muscle glycogenolysis is more sensitive to insulin than is glucose transport/phosphorylation: relation to the insulin-mediated inhibition of hepatic glucose production. J Clin Invest 92: 2963–2974
Barzilai N, Rossetti L 1995 The relationship between changes in body composition and insulin responsiveness in models of the aging rat. Am J Physiol 269:E591–E597
Thomas JA, Schlender KK, Larner J 1968 A rapid filter paper assay for UDPG-glycogen glucosyltransferase, including an improved biosynthesis of UDP[14C]glucose. Anal Biochem 25: 486–499
Fraze E, Chiou M, Chen Y, Reaven GM 1987 Age related changes in postprandial plasma glucose, insulin, and FFA concentrations in non-diabetic individuals. J Am Geriatr Soc 35: 224–228
Lewis GF, Zinman B, Groenewoud Y, Vranic M, Giacca A 1996 Hepatic glucose production is regulated both by direct hepatic and extrahepatic effects of insulin in human. Diabetes 45: 415–462
Ader M, Bergman RN 1990 Peripheral effects of insulin dominate suppression of fasting hepatic glucose production. Am J Physiol 258:E1020–E1032
Hettiarachchi M, Watkinson A, Jenkins AJ, Theos V, HO KKY, Kraegen EW 1996 Growth hormone-induced insulin resistance and its relationship to lipid availability in the rat. Diabetes 45: 454–421
Forbes GB 1974 Stature and lean body mass. Am J Clin Nutr 27: 595–602
Boden G, Jadali F, White J, Liang Y, Mozzoli M, Chen E 1991 Effects of fat on insulin-mediated carbohydrate metabolism in normal men. J Clin Invest 88: 960–966
Arslanian SA, Kalhan SC 1994 Correlation between fatty acid and glucose metabolism: potential explanation of insulin resistance of puberty. Diabetes 43: 908–914
Randle PJ, Garland PB, Hales CN, Newsholmes EA 1963 The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1: 785–789
Boden G, Chen X, Ruiz J, White LV, Rossetti L 1994 Mechanisms of fatty acid-induced inhibition of glucose uptake. J Clin Invest 93: 2438–2446
Rossetti L, Hawkins M, Chen W, Gindi J, Barzilai N 1995 In vivo glucosamine infusion induces insulin resistance in normoglycemic but not in hyperglycemic conscious rats. J Clin Invest 96: 132–140
Suprasongsin C, Arslanian SA 1995 Is there a relationship between lipoprotein(a) level and in vivo insulin sensitivity in childhood?. Diabetes Care 18: 138–139
Author information
Authors and Affiliations
Additional information
Supported by grants from the National Institutes of Health (R29-DK 45024 and R01-DK 48321), the American Federation for Aging Research (AFAR), the American Diabetes Association, the Core Laboratories of the Albert Einstein Diabetes Research and Training Center (DK 20541), and by a fellowship grant from Eli Lilly Research Laboratories. N.B. is supported by a grant from the National Institutes of Health (KO8-AG00639). S.R. was a fellow in the division of Pediatric Endocrinology.
Presented in part at the Annual Meeting of the Society for Pediatric Research, San Diego, 1995.
Rights and permissions
About this article
Cite this article
Raghavan, S., Saenger, P., Hu, M. et al. Intracellular Pathways of Insulin-Mediated Glucose Uptake before and after Puberty in Conscious Rats. Pediatr Res 41, 340–345 (1997). https://doi.org/10.1203/00006450-199703000-00006
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199703000-00006
